
Abstract
Historically, the diving duck, Baer’s Pochard (Aythya baeri) was widely distributed in East and South Asia, but according to a recent estimate, its global population is now less than 1000 individuals. To date, the mitochondrial genome of A. baeri has not been deposited and is not available in GenBank. Therefore, we aimed to sequence the complete mitochondrial genome of this species. The genome was 16,623 bp in length, double stranded, circular in shape, and contained 13 protein-coding genes, 22 tRNA genes, two rRNA genes, and one non-coding control region. Many structural and compositional similarities were discovered between A. baeri and the other three Aythya mitochondrial genomes. Among 13 protein-coding genes of the four Aythya species, the fastest-evolving gene was ATP8 while the slowest-evolving gene was COII. Furthermore, the phylogenetic tree of Anatidae based on Bayesian inference and maximum likelihood methods showed that the relationships among 15 genera of the Anatidae family were as follows: Dendrocygna was an early diverging lineage that was fairly distant from the other ingroup taxa; Cygnus, Branta, and Anser were clustered into one branch that corresponded to the Anserinae subfamily; and Aythya, Asarcornis, Netta, Anas, Mareca, Mergus, Lophodytes, Bucephala, Tadorna, Cairina, and Aix were clustered into another branch that corresponded to the Anatinae subfamily. Our target species and three other Aythya species formed a monophyletic group. These results provide new mitogenomic information to support further phylogenetic and taxonomic studies and genetic conservation of Anatidae species.
Similar content being viewed by others


Introduction
The mitochondrion is a type of organelle. It has the ability to convert organic materials directly into energy to power the cell’s biological functions1. In multicellular animals, the mitochondrial genome (mitogenome) is a circular, double-stranded molecule with a closed structure. It usually has a length of ~ 16 kb and is made up of a heavy strand (H-strand) and a light strand (L-strand) containing a total of 37 genes: 13 protein-coding genes (PCGs), two ribosomal RNA genes (rRNAs), and 22 transfer RNA (tRNA) genes2,3. It also has one or two non-coding control regions (CR), known as A + T-rich regions4. Among the abovementioned genes in most birds, one PCG (ND6) and eight tRNA genes (tRNA-Ala, tRNA-Cys, tRNA-Glu, tRNA-Gln, tRNA-Asn, tRNA-Pro, tRNA-Ser2, and tRNA-Tyr) are located on the L-strand, whereas the CR and the remaining 28 genes are located on the H-strand5,6. Owing to its simple structure, maternal inheritance, small length, conserved gene sequences, and high evolutionary rate7,8 and its easy isolation, amplification, sequencing9,10, mitochondrial DNA (mtDNA) may be preferred to nuclear DNA for some applications that help elucidate population genetic diversity and molecular phylogenetic relationships11,12. Identifying and managing genetic diversity of a threatened species can remarkably help execute species recovery plans13,14,15. Thus, a basic step in analyzing variable molecular tags for conservation research is sequencing the complete mtDNA.
Baer’s Pochard (Aythya baeri) is a diving duck that belongs to the order Anseriformes, subfamily Anatinae, family Anatidae. Historically, it was distributed in the Amur and Ussuri River basins of far eastern Russia and northeastern China during the breeding season and in eastern and southern China, India, Bangladesh, and Myanmar following migration in winter16. It mainly feeds on insects and aquatic plants and animals17. In 2008, after a sharp decline in its population, it was classified as Endangered by the International Union for Conservation of Nature (IUCN), after which its conservation level increased to Critically Endangered in 201218. The global population of Baer’s Pochard has been estimated to be less than 1000 individuals, but it could also be considerably lower19. Until the 1990s, major threats to its populations included habitat loss and degradation; recently, illegal hunting and disturbance may have become more significant factors causing its decline20. To protect this rare species from extinction, urgent and coordinated actions are needed.
The mitochondrial genome of A. baeri has not been deposited in GenBank so far, and molecular studies on this species are limited. In the current study, the complete mitogenome of this species was successfully sequenced. Based on the new sequence and previous data of other species, this study’s objectives were to address: (1) the characteristics of the mitogenome of A. baeri, (2) a comparison of the mitogenome of A. baeri and its relatives, (3) phylogenetic relationships of the family Anatidae on the basis of the combined mitochondrial gene set. These findings will help us better understand the A. baeri mitogenome and provide important information on the conservation and restoration of this endangered bird. Furthermore, the results could serve as a foundation for phylogenetic study as well as molecular biological data for taxonomic research on Anatidae species.
Results and discussion
Nucleotide composition and structure information
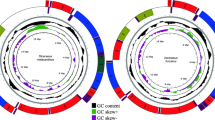
The entire mitochondrial genome of A. baeri was sequenced. It was deposited in GenBank under the accession number MT129533. Its size is 16,623 bp (Table 1). The lengths of the sequences discovered for other three Aythya mitogenomes were comparable at 16,616 bp. All the sizes were within the range of avian mitochondrial genomes21. As with the other animals22,23, mitogenome of A. baeri displayed a typical circular structure, including 13 PCGs, 22 tRNA genes, two rRNA genes, and one CR (Fig. 1). The arrangement and orientation of genes were similar to those found in other Anatidae species that had been determined24.

Circular map of the Aythya baeri mitochondrial genome. Different types of genes are indicated by squares of various colours. Genes encoded on the heavy or light strand are displayed on the outer or inner side of the circle, severally. The GC content of the mitogenome is represented by the black inner circles.
The parameters A + T content, AT-skew, and GC-skew are often employed to study the base composition pattern of mitogenomes25. The A + T content for A. baeri was 51.95%. It was slightly higher than that of other Aythya species genomes, which ranged from 51.61% to 51.62% (Table 1). In the full genome of A. baeri, the AT- and GC-skews were 0.143 and − 0.362, respectively. It demonstrated that the genome sequence was skewed away from T and G in favor of A and C, showing no difference from that of A. americana, A. ferina, and A. fuligula (Fig. 2). There were 35 bp of overlaps across the complete A. baeri mitogenome, with the greatest overlap having 10 bp between ATP8 and ATP6. The A. baeri mitogenome had 12 intergenic spacers ranging in length from 1 to 10 bp, totaling 46 bp of non-coding nucleotides. The two longest spacers were located between tRNA-Thr and tRNA-Pro and tRNA-Pro and ND6, respectively (Table 2).

Nucleotide compositions of each gene and several genomic regions of four Aythya mitogenomes. The panels represent the AT- and GC-skews and the A + T content for every gene and several genomic areas within the mitogenome that are specified in the X-axis.
Protein-coding genes (PCGs) and codon usage
Among the four mitogenomes studied, only one gene, ND6, was located on the L-strand, whereas the remaining 12 genes were located on the H-strand (Fig. 1). Moreover, the entire size of the 13 PCGs of the four Aythya species ranged from 11,400 bp (A. ferina) to 11,406 bp (A. americana; Table 1). Furthermore, the average A + T content of the PCGs in each of the four Aythya species varied from 50.51% (A. fuligula) to 50.94% (A. baeri; Table 1). The AT- and GC-skew values of each PCG were similar among the four Aythya species (Fig. 2). Notably, all 12 genes located on the H-strand had biases with a positive AT-skew and a negative GC-skew, whereas the ND6 gene located on the L-strand-encoded had the opposite biases.
Among the PCGs of A. baeri, the shortest PCG was ATP8 that was found between tRNA-Lys and ATP6. In contrast, the longest PCG was ND5 that was found between tRNA-Leu1 and Cytb. Except for COI, COII, and ND5, all of which had GTG as the start codon, all PCGs in the A. baeri mitogenome had a standard ATG start codon (Table 2). Seven genes (COII, ATP6, ND3, ND4L, ND5, ATP8, and Cytb) terminated with TAA. ND1 and COI terminated with AGG, ND2 and ND6 terminated with TAG. Among the stop codons, TAA was the most common. COIII and ND4 both had an incomplete T-stop codon.
Besides the stop codons, the PCGs contained 3800 to 3802 codons (Supplementary Tables 1–4) and showed similar codon distribution among the four Aythya species. In these mitogenomes, it can be found that codons encoding Leu1, Ala, Thr, Pro, and Ile were the most common. On the contrary, those encoding Cys were the least commonly observed (Fig. 3). In order to gain an insight into the genetic codon bias of the four Aythya species, the relative synonymous codon usage (RSCU) in PCG is shown in Fig. 3 and Supplementary Tables 1–4. The highly consistent RSCU was observed in the four birds, which could be due to their tight association as members of the same genus. It was also observed that for most amino acids, the use of synonymous codons was skewed. For example, in these species, the GCC codon for Ala was utilized frequently, whereas the GCG codon was rarely used for the same amino acid.

Codon distribution (left) and relative synonymous codon usage (right) in the mitochondrial protein-coding genes of four Aythya species.
Transfer and ribosomal RNA genes
The A. baeri mtDNA contained 22 tRNA genes (Fig. 1 and Table 2) that ranged from 66 bp (tRNA-Cys) to 76 bp (tRNA-Trp). Among these tRNA genes, 14 genes were located on the H-strand and eight on the L-strand. As shown in Fig. 2, the tRNA genes located on the H-strand had positive and negative AT- and GC-skew values, respectively. However, the contrast was true for genes on the L-strand. Additionally, secondary structures of the 22 tRNAs were predicted (Fig. 4). Except for tRNA-Ser1 lacking the dihydrouridine arm, almost all of the tRNA genes were found to possess a typical cloverleaf secondary structure. It is a characteristic generally observed in vertebrate tRNA genes26. In the mechanism of structural compensation between the other structures, it is possible that the missing arm in tRNA-Ser1 is functional27.

Secondary structures of Aythya baeri tRNA.
Ribosomal RNA genes in the A. baeri mitochondrial genome include 12S rRNA and 16S rRNA. The two genes were found on the H-strand between tRNA-Phe and tRNA-Leu2, and separated by tRNA-Val, as in other animals28,29. The A + T content of these two genes among the four Aythya species varied from 56.09% to 56.54%. The AT- and GC-skew values for these two rRNA genes in each Aythya species were positive and negative, respectively (Fig. 2).
Control region
The CR is thought to have transcription initiation sites as well as be involved in replication regulation in the mitochondrial genome30. In the field of phylogeny, this area has become a hot spot of research because of its rapid evolution and the largest variation in the whole mtDNA31. This CR was found to be located between tRNA-Glu and tRNA-Phe. The size of this region in A. baeri was 1071 bp (Table 1). This result was similar to the sequence lengths of the other three Aythya birds, ranging from 1054 bp (A. fuligula) to 1066 bp (A. americana). The AT skews for the four species in CR varied from 0.0490 to 0.075 (Fig. 2). The GC skews varied from − 0.3412 to − 0.3573.
Nucleotide diversity and selection pressures
For the four ducks of Aythya, the sliding window analysis demonstrated that the nucleotide diversity levels differed dramatically among the PCGs and rRNA genes. As shown in Fig. 5, ND3 (Pi = 0.0793) has the most polymorphism among the PCGs genes, followed by ND4L (Pi = 0.07351), ND1 (Pi = 0.04243), and ND5 (Pi = 0.0403). In contrast, ATP8 (Pi = 0.0129), COII (Pi = 0.0254), and COI (Pi = 0.0418) had the lowest values and were the most conserved genes across the PCGs. Compared with most PCGs, 16S rRNA (Pi = 0.01154) and 12S rRNA (Pi = 0.01511) had relatively low variability.

Sliding window analysis of PCGs and rRNA genes between four Aythya aves. The blue curve displays the value of nucleotide diversity (Pi). Each gene’s Pi value is displayed under the squares.
The ratio of Ka/Ks is an effective parameter for indicating the selection pressure32,33. In order to analyze the pressure on mitochondrial PCGs, Ka/Ks ratios were evaluated for the four Aythya aves. As shown in Fig. 6, all the Ka/Ks ratios were lower than 1, demonstrating that the 13 PCGs are undergoing purifying selection. The Ka/Ks ratios were varied for each of the 13 PCGs, indicating differing amounts of functional constraints among the genes34. Specifically, the highest value was observed for ATP8 (Ka/Ks = 0.10578), suggesting that ATP8 faces the least amount of selection pressure and is the gene with the fastest rate of evolution. The lowest value was observed for COII (Ka/Ks = 0), reflecting that COII is subjected to the highest selective pressure and is the gene with the slowest evolution.

The ratios of Ka/Ks and genetic distances of each PCG among four Aythya aves.
In order to estimate the average divergence among the four Aythya mtDNAs, the overall mean distances were analyzed based on 13 PCGs. As shown in Fig. 6, ND3, and ND4L appear to possess high distances of 0.087 and 0.080, followed by ND1, ND5, and ND2, while ATP8, COII, and COI have low distances of 0.013, 0.025, and 0.025, respectively. This tendency was similar to the nucleotide diversity levels of each PCG.
Mitogenome phylogeny
The mitochondrial sequence is a widely used molecular marker for inferring phylogenetic estimations among animals35. In the present study, to further elucidate the phylogenetic interrelationships within Anatidae, concatenated nucleotide sequences of 13 PCGs and two rRNA genes were obtained from 37 Anatidae aves, concerning four Aythya species, one Netta species, one Asarcornis species, one Mareca species, eight Anas species, three Mergus species, one Lophodytes species, three Bucephala species, one Cairina species, one Aix species, two Tadorna species, four Cygnus species, one Branta species, five Anser species, and one Dendrocygna species.
In both the Bayesian inference (BI) and maximum likelihood (ML) analyses, the phylogenetic trees had almost identical topologies (Fig. 7, Supplementary Figs. 1 and 2). Both trees from the two analyses were supported by good statistical values. In general, the genera Aythya, Netta, Asarcornis, Anas, Mareca, Mergus, Lophodytes, Bucephala, Cairina, Tadorna, and Aix clustered within the subfamily Anatinae, whereas the subfamily Anserinae had a sister relationship with Anatinae, including Cygnus, Branta, and Anser. Dendrocygna javanica, whose erect posture, elongated neck and legs, and conspicuous perching-tree habit distinguish it from most other Anseriformes species, diverged earlier from the main lineage and represents a sister clade to Anatinae-Anserinae36. Our topology was highly consistent with the results of studies based on the analysis of concatenated mitochondrial nucleotide sequences of ND2 and Cytb genes, as well as with morphological data36,37,38.

Molecular phylogenetic analysis of 37 Anatidae species constructed by MrBayes and IQ tree. Numbers beside the nodes represent posterior probabilities (left) and bootstrap values (right).
Within Anserinae, species of the genus Anser are related to Branta canadensis closely. These taxa had a sister relationship with the Cygnus species assemblage. This is in accordance with previous morphology-based analyses39. The situation is more complex for the larger subfamily Anatinae. In this subfamily, we found four consistent clades: Aythyini (Aythya + Netta + Asarcornis), Anatini (Anas + Mareca), Mergini (Mergus + Lophodytes + Bucephala), and Cairinini (Cairina + Aix + Tadorna). Cairinini was the sister group of all other Anatinae tribes, which was strongly supported by the results of the present study. In the Anatinae tribes, Anatini and Aythyini have closer phylogenetic relationships, and these results are supported by morphological studies and phylogenetic analysis based on two PCGs37,38. Within the genus Aythya, A. americana and A. ferina were clustered together with well-supported values. Meanwhile, A. fuligula and the clade (A. americana + A. ferina) formed a high-supported value branch. Our target species, A. baeri, and the clade (A. fuligula + (A. americana + A. ferina)) were clustered into one branch, suggesting the four species form a monophyletic group.
Conclusions
The current study provided the mitogenome of A. baeri (16,623 bp, accession number MT129533). This circular mtDNA included 13 PCGs, two rRNAs, 22 tRNAs, and one CR. Through comparison, it was found that the genome of A. baeri displayed characteristic sequence structures and was quite comparable to those of Aythya aves. Among the PCGs, ND3 had the most polymorphism and highest genetic distance. Our phylogenetic analysis based on combined mitochondrial gene set (13 PCGs and two rRNAs) of 37 Anatidae species using BI and ML algorithms indicated that A. baeri is closely related to A. fuligula, A. americana, and A. ferina. In fact, we deduced that these species were monophyletic. Moreover, all the genera for which multiple species were studied were also discovered to be monophyletic with the exception of Anas, as that included Mareca. The present study provides an essential reference for the conservation of the studied critically endangered species as well as valuable information for further phylogenetic and evolutionary research on the family Anatidae.
Materials and methods
Sampling and DNA extraction
A sample of leg muscular tissue was obtained from a dead bird seized by the police in Sihong County, China, on October 27, 2019, no living animal was involved in this study. The sample was kept at minus 80 °C until DNA extraction. We extracted genomic DNA from the tissue using the Universal Genomic DNA Extraction Kit (Takara Biomedical Technology Co. Ltd., Beijing, China). The DNA quality was measured using agarose gel electrophoresis. DNA purity and concentration were assessed by NanoDrop 2000 (NanoDrop Technologies, Wilmington, NC, USA).
Mitogenome sequencing and assembly
Following DNA extraction, we fragmented 1 μg of purified DNA and used it to set up 200 bp short-insert libraries. These qualified libraries were sequenced with PE150 bp on an BGISEQ-500 sequencer according to the manufacturer’s instructions detailed in the previous literature40. As a result, we obtained 5.2 Gb of clean data and trimmed all the raw reads using FASTQ41. Subsequently, we mapped the high-quality reads using default parameters to reference Aythya mitogenome datasets (accession numbers: AF090337, KJ710708, KJ722069), which we obtained from GenBank through Bowtie42. The sequence of the coding gene having the maximum coverage was then utilized as a seed sequence for de novo assembly of the mitochondrial genome NOVOPlasty43. Once the assembly was completed, we re-mapped all the sequencing reads to the candidate mitochondrial genome to re-confirm the assembly of the mitochondrial genome.
Mitogenome annotation and analysis
All PCGs were first identified utilizing the NCBI website’s open reading frame finder44. Then, MEGA 7.0 was used to translate the sequences into putative proteins45. Verifications of tRNA genes were performed using the MITOS WebServer46, and secondary structures were inferred using tRNAscan-SE47. Sequences of the identified genes were re-checked by comparing them with that of other Aythya species manually. A map of the A. baeri mitochondrial genome was generated by CGView48. MEGA 7.0 was used to analyze the base compositions and RSCU values of 13 PCGs45. To estimate the bias in nucleotide composition across the entire sequence, AT- and GC-skews were used. This was obtained based on the formulas: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C)49. For the four Aythya aves, the nucleotide diversity (Pi) of each PCG and rRNA, a sliding window analysis in 500 bp windows every 100 bp of 13 PCGs and two rRNAs, the ratios of non-synonymous (Ka) to synonymous (Ks) substitutions rates of each PCG were conducted by DnaSP 5.050. MEGA 7.0 was used to compute the overall mean distances among the four Aythya species, using the Kimura-2-parameter model45.
Phylogenetic analysis
The phylogenetic status of A. baeri was assessed by comparing the combined mitochondrial gene set (13 PCGs and two rRNA genes) with that of 36 other Anatidae species across three subfamilies: Anatinae, Anserinae, and Dendrocygninae, and 15 genera: Aythya, Asarcornis, Netta, Anas, Mareca, Mergus, Lophodytes, Bucephala, Tadorna, Cairina, Aix, Cygnus, Branta, Anser, and Dendrocygna (Table 3). In addition, Phasianus colchicus and Chrysolophus pictus were set as outgroups for rooting the tree. GenBank was used to obtain all the mitogenome sequences.
The sequences of the gene set of mitogenomes of 37 species were combined, and MAFFT was used to generate the alignment of the concatenated genes51. Gblocks was used to select conserved sequences within the database. Using the method described by Xia et al.52, DAMBE was used to perform a substitution saturation analysis for dataset.
The results showed that the dataset was suitable for further analyses. BI and ML methods were applied to phylogenetic analysis. Using ModelFinder53, it was found that the GTR + F + I + G4 model was the best-fit model for the BI and ML methods based on the AIC54 and BIC55, respectively. The BI was performed by MrBayes56 with two simultaneous MCMC chains, running for twenty million cycles, sampling every 2000 generations, and discarding the initial 0.25 of sampled data as burn-in. Trace plot in Tracer v1.7 was used to assess convergence. The effective sample size of model parameters was much more than 200. The ML analysis was run with standard bootstrap for 1000 replicates using IQ-TREE57. Both resulting phylograms were visualized and edited in FigTree 1.4.458.
Data availability
The mitochondrial genome data has been submitted to NCBI GenBank under the following Accession Numbers MT129533.
References
van der Bliek, A. M., Sedensky, M. M. & Morgan, P. G. Cell biology of the mitochondrion. Genetics 207, 843–871 (2017).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780 (1999).
Taanman, J. W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1410, 103–123 (1999).
Zhang, D. X. & Hewitt, G. M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 25, 99–120 (1997).
Mindell, D. P., Sorenson, M. D. & Dimcheff, D. E. Multiple independent origins of mitochondrial gene order in birds. Proc. Natl. Acad. Sci. U. S. A. 95, 10693–10697 (1998).
Slack, K. E., Janke, A., Penny, D. & Arnason, U. Two new avian mitochondrial genomes (penguin and goose) and a summary of bird and reptile mitogenomic features. Gene 302, 43–52 (2003).
Gissi, C., Iannelli, F. & Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity (Edinb.) 101, 301–320 (2008).
Subramanian, S. et al. High mitogenomic evolutionary rates and time dependency. Trends Genet. 25, 482–486 (2009).
Kocher, T. D. et al. Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. U. S. A. 86, 6196–6200 (1989).
Clayton, D. A. Mitochondrial DNA replication: What we know. IUBMB Life 55, 213–217 (2003).
Schmidt, C. & Garroway, C. J. The conservation utility of mitochondrial genetic diversity in macrogenetic research. Conserv. Genet. 22, 323–327 (2021).
Ballard, J. W. O. & Rand, D. M. The population biology of mitochondrial DNA and its phylogenetic implications. Annu. Rev. Ecol. Evol. S. 36, 621–642 (2005).
Jang, J. E. et al. Genetic diversity and genetic structure of the endangered Manchurian trout, Brachymystax lenok tsinlingensis, at its southern range margin: Conservation implications for future restoration. Conserv. Genet. 18, 1023–1036 (2017).
Li, C. et al. Native bighead carp Hypophthalmichthys nobilis and silver carp Hypophthalmichthys molitrix populations in the Pearl River are threatened by Yangtze River introductions as revealed by mitochondrial DNA. J. Fish Biol. 96, 651–662 (2020).
Moritz, C. Applications of mitochondrial DNA analysis in conservation: A critical review. Mol. Ecol. 3, 401–411 (1994).
BirdLife International. Species factsheet: Aythya baeri http://www.birdlife.org (2020).
Li, C. K. et al. Study on the breeding ecology of Aythya baeri in Henan Chenqiao Wetland. J. Henan Agric. Univ. 54, 91–97 (2020).
IUCN. International Union for Conservation of Nature of red list of threatened species: Baer’s Pochard http://www.iucnredlist.org (2020).
Wang, X., Barter, M., Cao, L., Lei, J. & Fox, A. D. Serious contractions in wintering distribution and decline in abundance of Baer’s Pochard Aythya baeri. Bird Conserv. Int. 22, 121–127 (2012).
Hearn, R. The troubled Baer’s Pochard Aythya baeri: cause for a little optimism?. BirdingASIA 24, 78–83 (2015).
Shields, G. F. & Helm-Bychowski, K. M. Mitochondrial DNA of birds. In Current Ornithology (ed. Johnston, R. F.) 273–295 (Springer, New York, 1988).
Prabhu, V. R., Singha, H. S., Kumar, R. G., Gopalakrishnan, A. & Nagarajan, M. Characterization of the complete mitochondrial genome of Barilius malabaricus and its phylogenetic implications. Genomics 112, 2154–2163 (2020).
Xu, N. et al. The mitochondrial genome and phylogenetic characteristics of the Thick-billed Green-Pigeon, Treron curvirostra: The first sequence for the genus. ZooKeys 1041, 167–182 (2021).
Liu, G., Zhou, L. Z. & Gu, C. M. Complete sequence and gene organization of the mitochondrial genome of scaly-sided merganser (Mergus squamatus) and phylogeny of some Anatidae species. Mol. Biol. Rep. 39, 2139–2145 (2012).
Hassanin, A., Léger, N. & Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst. Biol. 54, 277–298 (2005).
Luiz, P. Mitochondrial genome organization and vertebrate phylogenetics. Genet. Mol. Biol. 23, 745–752 (2000).
Steinberg, S. & Cedergren, R. Structural compensation in atypical mitochondrial tRNAs. Nat. Struct. Biol. 1, 507–510 (1994).
Wolstenholme, D. R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 141, 173–216 (1992).
Singh, B., Kumar, A., Uniyal, V. P. & Gupta, S. K. Complete mitochondrial genome of northern Indian red muntjac (Muntiacus vaginalis) and its phylogenetic analysis. Mol. Biol. Rep. 46, 1327–1333 (2019).
Stoneking, M., Hedgecock, D., Higuchi, R. G., Vigilant, L. & Erlich, H. A. Population variation of human mtDNA control region sequences detected by enzymatic amplification and sequence-specific oligonucleotide probes. Am. J. Hum. Genet. 48, 370–382 (1991).
Clayton, D. A. Nuclear gadgets in mitochondrial DNA replication and transcription. Trends Biochem. Sci. 16, 107–111 (1991).
Hurst, L. D. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486 (2002).
Yang, Z. & Bielawski, J. P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15, 496–503 (2000).
Muse, S. V. Examining rates and patterns of nucleotide substitution in plants. Plant Mol. Biol. 42, 25–43 (2000).
Zardoya, R. & Meyer, A. Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Mol. Biol. Evol. 13, 933–942 (1996).
Livezey, B. C. A phylogenetic analysis of the whistling and white-backed ducks (Anatidae, Dendrocygninae) using morphological characters. Ann. Carnegie. Mus. 64, 65–97 (1995).
Donne-Goussé, C., Laudet, V. & Hänni, C. A molecular phylogeny of anseriformes based on mitochondrial DNA analysis. Mol. Phylogenet. Evol. 23, 339–356 (2002).
Gonzalez, J., Düttmann, H. & Wink, M. Phylogenetic relationships based on two mitochondrial genes and hybridization patterns in Anatidae. J. Zool. 279, 310–318 (2009).
Livezey, B. C. A phylogenetic analysis of geese and swans (Anseriformes: Anserinae), including selected fossil species. Syst. Biol. 45, 415–450 (1996).
Huang, J. et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 6, 1–9 (2017).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. https://doi.org/10.1093/nar/gkw955 (2017).
Pombert, J. F., Otis, C., Lemieux, C. & Turmel, M. The complete mitochondrial DNA sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) highlights distinctive evolutionary trends in the chlorophyta and suggests a sister-group relationship between the Ulvophyceae and Chlorophyceae. Mol. Biol. Evol. 21, 922–935 (2004).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Bernt, M. et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57 (2016).
Stothard, P. & Wishart, D. S. Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539 (2005).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Xia, X., Xie, Z., Salemi, M., Chen, L. & Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 26, 1–7 (2003).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Yamaoka, K., Nakagawa, T. & Uno, T. Application of Akaike’s information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J. Pharmacokinet. Biopharm. 6, 165–175 (1978).
Volinsky, C. T. & Raftery, A. E. Bayesian information criterion for censored survival models. Biometrics 56, 256–262 (2000).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Rambaut A. FigTree, a graphical viewer of phylogenetic trees http://tree.bio.ed.ac.uk/software/figtree (2007).
Acknowledgements
This research was supported by the Excellent scientific and technological innovation team of higher education in Jiangsu Province (2019-29), Qing Lan Project of higher education of Jiangsu province, and Fundamental Research Funds for Central Universities (LGYB2021004).
Author information
Authors and Affiliations
Contributions
D.L. and S.H. designed the work, analyzed the data and wrote the paper, Y.Z. and Y.F. prepared the figures and tables, C.X. reviewed drafts of the paper and supervised and directed the work. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, D., Zhou, Y., Fei, Y. et al. Mitochondrial genome of the critically endangered Baer’s Pochard, Aythya baeri, and its phylogenetic relationship with other Anatidae species. Sci Rep 11, 24302 (2021). https://doi.org/10.1038/s41598-021-03868-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03868-7
This article is cited by
-
Complete mitochondrial genomes of Lycosa grahami and Lycosa sp. (Araneae: Lycosidae): comparison within the family Lycosidae
International Journal of Tropical Insect Science (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
