
Abstract
The leaves and seed oil of Perilla crop (Perilla frutescens L.) have attracted interest as health foods in East Asia. This crop has been traditionally cultivated and used for a long time as a folk plant, especially in Korea. In our study, the 22 SSR markers and eight morphological traits were used to assess the genetic diversity and population structure, to select a core collection of 400 Perilla accessions conserved in the RDA-Genebank of South Korea. A total of 173 alleles were detected and the number of alleles per locus ranged from 4 to 15 (average = 7.9). Gene diversity and polymorphic information content ranged from 0.138 to 0.868 (average = 0.567) and 0.134 to 0.853 (average = 0.522), respectively. The 400 accessions were not clearly distinguished geographically by STRUCTURE and UPGMA analyses. A core collection (44 accessions) was selected from the entire collection by using PowerCore. The core collection accounted for 11.0% of the entire Perilla collection, including 100% of the number of alleles maintained in the whole collection and with similar or greater Shannon–Weaver and Nei diversity indices than the whole collection. The core collection selected by SSR markers was evenly distributed in three clusters on a scatter plot by eight morphological traits. The first core collection of Perilla accessions was constructed, and it maintained allelic richness. Further modification of the core collection is expected with the continuous addition of new accessions of the two cultivated types of Perilla crop and their weedy types.
Similar content being viewed by others
Introduction
Plant genetic resources (PGRs) mean individuals or populations of wild species, genetic stocks, and cultivars, which are maintained as types of plants, seeds, and tissues1. These PGRs are utilized to provide new plant varieties with better resistance, adaptation, and nutritive qualities. Conservation and collection of PGRs is necessary to maintain higher genetic diversity for food security and use in the future2. Moreover, the conservation and collection of PGRs in most crops has been made possible by the establishment of genebanks in countries around the world. The fundamental role of a genebank is to increase genetic diversity and prevent genetic erosion by collecting, conserving, and documenting PGRs. In South Korea, the Genebank of the National Agrobiodiversity Center (NAAS) of the Rural Development Administration (RDA-Genebank) plays an important role in supporting genetic resource conservation and utilization. The RDA-Genebank currently preserves a total of 266,649 genetic resources for 3,083 species, including food, horticulture, industrial, and forage crops (http://genebank.rda.go.kr). Although a large number of PGRs have been collected in genebanks, the large sample size and lack of adequate information about population structure and genetic diversity of these collections hinders the successful utilization of the genetic potential for PGRs3. Efficient management and utilization of the existing collections could be greatly enhanced by reducing the number of accessions from large collections4. Frankel (1984) suggested the concept of a core collection, which is a subset of accessions with minimum redundancy and maximum genetic diversity from an entire collection. Various studies have proposed subsampling proportions ranging from 5 to 30% for a core collection3,6,7,8. A core collection is commonly established to conserve phenotypic and genetic diversity and enable the selection of candidate alleles associated with important agronomic traits9. Development of a core collections has been promoted as the most important activity in conservation and utilization of PGRs since 19962. Therefore, core collections have already been reported for many crops, such as rice10, soybean11, and wheat12. The development of core collections was based at first on passport data, geographical distribution, and phenotypic data13,14,15. Subsequently, the advance of molecular markers, which are an efficient means of confirming genetic diversity, has allowed the development of more powerful core collections in many crop species, either alone16 or in combination with phenotypic data4,17. Among various molecular markers, simple sequence repeats (SSRs) are often selected for genetic studies, such as for genetic diversity and the development of core collections, because of their advantages such as high reproducibility, polymorphism, abundance, and codominance12,18,19. Although there are a number of useful methods for selection of a core collection, the power core method for the development of a core collection utilizes the advanced M (maximization) strategy implemented through a modified heuristic algorithm by Kim et al. (2007). Using this program, core collections have been developed in various crops, such as rice21, soybean11, barnyard millet22, ragi23, and cassava24.
Perilla crop (Perilla frutescens L., 2n = 40) is an annual self-fertilizing species of the Lamiaceae family. This species has been traditionally cultivated and used for a long time as a folk plant, especially in East Asian countries such as Korea, Japan, and China. It is divided into two different varieties (or cultivated types), P. frutescens var. frutescens and var. crispa, based on morphology and use in East Asia. For example, P. frutescens var. frutescens, which is called “dlggae” in Korea, “egoma” in Japan, and “ren” in China, is tall with a large seed size (more than 2 mm), green leaf and stem color, and non-wrinkled leaf and used as an oil and leafy vegetable crop25,26. In contrast, P. frutescens var. crispa, which is called “cha-jo-ki” in Korea, “shiso” in Japan, and “zisu” in China, is short with a small seed size (less than 2 mm), red or green leaves and stems, and wrinkly or non-wrinkly leaves and is used as a vegetable and herbal medicine crop25,26.
P. frutescens var. frutescens has become a cash crop in South Korea. The leaves of var. frutescens are abundant in vitamin B and C, and it is a favored salad vegetable eaten with meat and used for pickles. Furthermore, Perilla seed oil of cultivated P. frutescens var. frutescens has a high content of polyunsaturated fatty acids, such as linoleic acid (C18:2) and α-linolenic acid (C18:3), which comprise approximately 80% of Perilla seed oil. The Perilla seed oil, similarly to soybean, rapeseed, corn, and sesame seed oils, has been used for foods such as cooking oils27,28. Recently, the leaves and seed oil have attracted interest as health foods in South Korea; therefore, the cultivation area of P. frutescens var. frutescens has increased greatly. Although approximately thirty commercial varieties have been registered in Korea (http://www.seed.go.kr), there have been few efforts to develop new cultivars for Perilla crop. Most farmers still cultivate landraces of their own regions29. The RDA-Genebank in South Korea maintains 2,368 genetic resources of cultivated and weedy types of Perilla crop, which are available to researchers and farmers (http://www.genebank.go.kr/).
To maximize the utilization of Perilla resources and for more efficient breeding programs, the morphological and genetic characteristics of the collected resources should be defined before use. Although many Perilla accessions in the RDA-Genebank have already been evaluated for phenotypic characteristics and diversity in the field, these characteristics are easily affected by environmental effects and evaluations are time and labor-intensive30. To overcome these limitations, molecular marker systems could be used to mitigate against the environmental effects and provide a better explanation of the variation than phenotype-based evaluation31. However, there has been a lack of information about genetic diversity and relationships at the molecular level for the RDA-Genebank accessions. Most of the Perilla accessions of the RDA-Genebank have been used rarely or not at all in Perilla crop breeding programs. Moreover, there are no reports of a core collection of Perilla based on DNA molecular markers.
Therefore, in our study, we performed population structure and genetic diversity analyses of 400 accessions of the Perilla germplasm collection of the RDA-Genebank using 22 Perilla SSRs. This allowed us to 1) examine the level of genetic diversity and the population structure within accessions of cultivated P. frutescens var. frutescens of the RDA-Genebank and 2) select a Perilla core collection that represents the entire collection without redundancy. This core group will provide useful information for efficient conservation and the utilization of genetic resources as well as for the selection of useful genetic resources for Perilla breeding programs.
Results
Genetic variation in the whole collection determined using SSR markers
A total of 22 SSR loci were used to evaluate the GDI, population structure, and genetic relationships among the 400 accessions of cultivated P. frutescens var. frutescens (Table 1). In this study, the 22 SSR primer sets were selected in a preliminary study using about 120 SSR primer sets to identify polymorphic SSR primer sets. Finally, a total of 22 SSR loci were confirmed within 173 alleles in the 400 accessions. The number of alleles per locus ranged from 4 (KNUPF36, KNUPF37, KNUPF59, KNUPF74) to 15 (KNUPF89), and the average number of alleles per locus was 7.9 (Table 1). The average GD was 0.567, with a range from 0.138 (KNUPF59) to 0.868 (KNUPF10). The average PIC value was 0.522, with a range of 0.134 (KNUPF59) – 0.853 (KNUPF10). The average MAF was 0.566, with a range of 0.195 (KNUPF10) – 0.928 (KNUPF59) (Table 1).
To confirm the geographical difference for genetic diversity for accessions collected from central (Group I, 148 accessions) and southern (Group II, 211 accessions) regions of South Korea and foreign or unknown (Group III, 41 accessions), our study compared the number of alleles and the GDI of the three groups (Table 2). The average number of alleles was 6.5, 6.9, and 5.1 alleles for accessions of Group I, II, and III, respectively. In the results for GDI, the average GD values were 0.562, 0.554, and 0.578 for Group I, II, and III, respectively. The average PIC values were 0.521, 0.506, and 0.533 for Group I, II, and III, respectively. The average MAF values was 0.576, 0.571, and 0.541 for Group I, II, and III, respectively (Table 2). Among the 173 alleles, 23 private alleles (13.3%) were only detected in one of the 400 accessions of cultivated P. frutescens var. frutescens. The percentage of rare alleles (frequency < 0.05) was 56.1% (97 alleles) among the 173 alleles, whereas intermediate-frequency alleles (frequency of 0.05–0.5) and abundant alleles (frequency > 0.5) represented 36.4% (63 alleles) and 7.5% (13 alleles), respectively, of the 173 alleles (Fig. 1).

Histogram of allele frequencies in 400 accessions of cultivated type of var. frutescens based on 22 SSR markers.
Population structure and genetic relationships among 400 accessions of cultivated P. frutescens var. frutescens
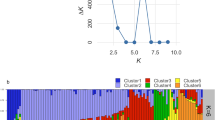
To understand the population structure among the 400 accessions of cultivated P. frutescens var. frutescens, our study used a model-based approach in STRUCTURE to divide the accessions into subgroups. We applied the ad hoc measure ΔK using the method developed by Evanno et al. (2005) to clearly determine the number of subgroups in interpreting the actual K values. The highest value of ∆K for the 400 accessions of cultivated P. frutescens var. frutescens was found at K = 2 (Fig. 2). Although we confirmed K = 2 by the ∆K method, some accessions were admixed between these two groups. Thus, we divided the accessions into two main groups and an admixed group in accordance with the method of Wang et al. (2008) based on a threshold of 0.8 (Fig. 3). Based on K = 2, Group I included 70 accessions from the central region, 104 accessions from the southern region, and 18 accessions from foreign or unknown. Group II comprised 24 accessions from the central region, 45 accessions from the southern region, and 12 accessions from foreign or unknown. The admixed group included a total of 127 accessions, which consisted of 54 accessions from the central region, 62 accessions from the southern region, and 11 accessions from foreign or unknown.

Magnitude of ΔK as a function of K. The peak value of ΔK was at K = 2 in 400 accessions of cultivated type of var. frutescens of Perilla crop.

Population structure pattern for the highest ΔK value (K = 2) of 400 accessions of cultivated type of var. frutescens of Perilla crop.
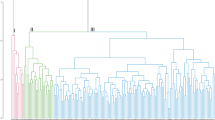
To confirm the genetic relationships by distance-based analysis among the 400 accessions of cultivated P. frutescens var. frutescens, a dendrogram was constructed using UPGMA. This showed that all Perilla accessions were clustered into ten major groups with a genetic similarity of 45.7% (Supplementary Fig. 2). Group IV (182,549, foreign), VI (215,256, central region), VIII (104,486, central region), IX (215,257, central region), and X (214,479, unknown) each included only one accession. Meanwhile, Group I contained 33 accessions (3 accessions from the central region and 30 accessions from the southern region). Group II consisted of 325 accessions (132 accessions from the central region, 167 accessions from the southern region, and 26 accessions from foreign or unknown). Group V and VII included four accessions (3 accessions from the central region and 1 accession from the southern region) and six accessions (1 accession from the central region, 2 accessions from the southern region, and 3 accessions from foreign or unknown), respectively (Supplementary Fig. 2). In addition, Group II, which contained 81.8% of the 400 accessions, was further subdivided into ten sub-clusters with a genetic similarity of 51.8% (Supplementary Fig. 2). The first sub-cluster contained 261 accessions, which divided again into eight sub-groups with a genetic similarity of 56.9% (95 accessions from the central region, 147 accessions from the southern regions, and 19 accessions from foreign or unknown). The second sub-cluster consisted of 13 accessions (10 accession from the central region and 3 accessions from the southern region). The third sub-cluster (2 central and 3 southern region) and fifth sub-cluster (4 central and 1 southern region) each contained five accessions. The fourth sub-cluster consisted of 12 accessions (7 accessions from the central region, 2 accessions from the southern region, and 3 accessions from foreign or unknown). The sixth sub-cluster contained 16 accessions (9 accessions from the central and 7 accessions from the southern region). The seventh sub-cluster (2 accessions from the southern region) and eighth sub-cluster (2 accessions, one collected from the central and one from the southern region) each consisted of two accessions. The ninth sub-cluster included eight accessions (3 accessions from the central region, 1 accession from the southern region, and 4 accessions from foreign or unknown). Finally, the tenth sub-cluster of Group II contained only one accession, collected from the central region (Supplementary Fig. 2).
Development and evaluation of the core collection
To establish a core collection, data of 22 SSR genotypes with a total of 173 alleles were used to construct a core set from the 400 accessions of cultivated P. frutescens var. frutescens using PowerCore software20. For the core collection, 44 accessions were selected. The core collection contained 16 representatives from the central region, 21 from the southern region, and 7 foreign or unknown (Table 3). The core collection accounted for 11.0% of the whole Perilla collection, including 100% of the number of alleles maintained in the whole collection. To compare the allelic richness between the core and whole collections, our study confirmed the diversity by the Shannon–Weaver (Sh.W.) and Nei calculation (Fig. 4, Table 4). The distributions of the Sh.W. and Nei indices in the 22 SSR markers for the core and the whole collections represented a high similarity or those of the core collection were greater than those of the whole collection (Fig. 4, Table 4). The results of a Student’s t test using SPSS software showed that there was no significant difference in the average genetic diversity (Sh.W. and Nei indices) between the core collection and the whole collection (p = 0.102 for Sh.W. and 0.192 for Nei indices). This indicates that the core collection selected herein sufficiently represents allelic richness and genetic diversity of the entire accessions.

Comparison of a Shannon–Weaver diversity index (A) and Nei’s diversity index (B) using 22 SSR markers between the core collection and the whole collection.
Morphological variation and principal component analysis among 372 accessions of cultivated P. frutescens var. frutescens
The results of examining eight morphological traits of the 372 accessions of cultivated P. frutescens var. frutescens are summarized in Supplementary Table 3.
In the survey of color of leaf surface (QL1), 59 accessions showed light green color, 207 showed green, and 104 showed deep green. The remaining 2 accessions showed light purple color. For color of reverse side leaf (QL2), 144 accessions showed light green color, 203 accessions showed green, and 24 showed deep green. One accession only showed light purple. For stem color (QL3), 58 accessions showed light green, 207 accessions showed green, and 105 accessions showed deep green. Only 2 accessions had light purple. For leaf shape (QL4), 161 accessions showed lanceolate shape, 146 accessions showed heart shape, and 65 accessions showed oblong shape. For degree of pubescence (QL5), 90 accessions showed slightly pubescent, 222 accessions showed normal pubescent, and 58 accessions showed heavily pubescent. The remaining 2 accessions showed more heavily pubescent. In the case of flowering time (QL6), 179 accessions showed intermediate flowering (flowering days from August 15 to September 5) and 193 accessions showed late flowering (flowering days from September 6 after September 25). In the case of the two seed characteristics, for seed color (QL7) of the 372 Perilla accessions, 86 accessions showed dark brown, 212 accessions showed brown, 24 accessions showed gray, 37 accessions showed white, the remaining 13 accessions showed mixed colors. For seed hardness (QL8) of the 372 Perilla accessions, 305 accessions showed soft seeds and the remaining 67 accessions showed hard seeds (Supplement Table 3).
This study used this morphological data to perform PCA analysis. The analysis results showed that the first and second principal components accounted for 25.5% and 16.8%, respectively, of the total variance (Table 5). In this study, QL1 and QL3 contributed in the positive direction on the first axis, and QL5 and QL6 contributed in the positive direction on the second axis. Thus, the traits that contributed in the positive direction on the first or second axes are considered to be useful for discrimination between accessions of cultivated P. frutescens var. frutescens (Table 5). Based on the first axis, most accessions of cultivated P. frutescens var. frutescens were clearly separated into three groups on the positive and negative sides of the first axis by color of leaf surface and stem (Fig. 5). Thus, the traits (QL1, QL3, QL5, QL6) that contributed in the positive or negative direction on the first or second axis (Table 5) are considered to be useful for discrimination between accessions of cultivated P. frutescens var. frutescens.

Projection of 372 accessions of cultivated type of var. frutescens in the first and second principal components.
In addition, to validate the core set selected by molecular markers, PCA analysis was used to confirm the distribution for the core collection selected by SSR markers. Two accessions (157,474 and 104,421) of the core collection could not be investigated for morphological characteristics (Table 3), but the remaining 42 accessions were evenly distributed in the three parts on a scatter plot by the PCA analysis (Fig. 5).
Discussion
Information about the genetic diversity of PGRs, which provide useful alleles associated with plant development and improvement, is very important for both the conservation and the utilization of germplasm that has been collected in a genebank34,35. With the development of molecular biology, DNA molecular marker technology provides useful information for the analysis of genetic diversity, genetic relationships, population structure, and core collections in the germplasm of many crop species4,9,16,21,22,23,24.
In the case of Perilla crop, much analysis has been performed on genetic diversity, genetic relationships, and population structure using amplified fragment length polymorphisms (AFLP)29, random amplification of polymorphic DNAs (RAPD)26,36, and SSR markers37,38,39,40. Unfortunately, and in contrast with other major crop species, other molecular marker technologies in Perilla species have not yet been developed. Among these marker systems, as already explained in the Introduction, SSR marker technology is highly polymorphic and reproducible, generally co-dominant and abundant in the plant genome, and it has provided useful information for the analysis of genetic diversity, genetic relationships, population structure, etc. in the germplasm of Perilla species37,41,42,43,44. Recently, SSR primer sets have been developed for Perilla crop by many researchers37,43,44,45,46 and used successfully for the analysis of genetic diversity, genetic relationships, population structure, and association mapping among the accessions of cultivated and weedy types of Perilla crop28,38,39,40,43,47,48. Therefore, this study used SSR markers to identify genetic diversity and relationships, population structure, and a core collection of RDA-Genebank Perilla germplasm. The GD and PIC values determined in this study of 0.567 and 0.522, respectively (Table 1), were compared with those of previous studies of Ma et al. (2019) and Park et al. (2019) that contained more weedy Perilla accessions and showed values of 0.577 and 0.625 for GD and 0.537 and 0.582 for PIC with 21 and 25 SSR markers, respectively. These findings reveal a lower level of genetic diversity in the collection of this study, which mainly consisted of cultivated P. frutescens var. frutescens. Although wild species have not yet been found in Perilla crop, many accessions of the weedy type of Perilla crop have been reported in East Asia, particularly in South Korea and China, and they show higher genetic diversity than accessions of the cultivated type of Perilla crop25,26,29,38,39,40,43,49,50.
Meanwhile, polymorphism of loci can be considered high, medium, or low with GDI > 0.5, GDI < 0.5 and > 0.25, or GDI < 0.25, respectively, according to a report by Vaiman et al. (1994). The population in the current study consisting of the 400 accessions of cultivated P. frutescens var. frutescens has average GD and PIC values of over 0.5 in the 22 SSR markers, indicating that this population has a relatively high genetic diversity (Table 1). Moreover, the 14 (based on GD) and 12 (based on PIC) SSR markers among the entire SSR markers showed a high level of polymorphism based on GDI (each > 0.5) (Table 1). The SSR markers named KNUPF used in this study were recently developed by our previous studies for Perilla crop43,44,46. Although only cultivated P. frutescens var. frutescens of Perilla crop, which has relatively lower diversity, was used as material in this study, many SSR markers showed relatively high GDI29,38,39,40,49. Therefore, these SSR markers were considered useful for identifying genetic diversity and population structure and for selecting a core collection for accessions of Perilla crop. Furthermore, these SSR markers will be very useful for genome-wide association study (GWAS) or quantitative traits loci (QTL) analysis, because these markers were developed by using the results of transcriptome analysis for Perilla crop43,44,45,52.
In East Asia, although China is considered the origin of Perilla crop, South Korea is assumed to be the secondary center of biodiversity of Perilla crop because of extensive cultivation, various uses, and high morphological and genetic diversity as well as the existence of weedy types25,26,49,50. Recently, cultivated P. frutescens var. frutescens of Perilla crop has become a cash crop in South Korea, and the cultivation area has expanded significantly. To maximize the use of genetic resources of cultivated P. frutescens var. frutescens of Perilla crop preserved in the RDA-Genebank, the genetic characteristics of the collected resources should be analyzed for efficient conservation and utilization in South Korea. Therefore, this study compared the average allele numbers and GDI values among the central (Group I, 148 accessions) and southern region (Group II, 211 accessions) accessions of South Korea and foreign or unknown (Group III, 41 accessions) accessions (Table 2). The highest allele number was revealed in Group II, followed by Group I and Group III, while the highest GDI values were confirmed in Group III, followed by Group I and Group II (Table 2). Although Group II had the highest allele number, genetic diversity for Group II was lower than that of Group I and II. This result suggests that Group II has the highest number of accessions with more Group II-specific alleles, while this group consists of more accessions with similar genetic characteristics than the other two groups. Moreover, when comparing the South Korea (Group I and II) and foreign accessions (Group III), the South Korea accessions show lower genetic diversity than the foreign accessions (Table 2). This result indicates that the cultivated type of var. frutescens of South Korea has a narrower genetic diversity than the foreign accessions, even though South Korea is the secondary center of Perilla crop. It may be that the environmental variation in the central and southern regions of South Korea is not severe and that farmers for Perilla crop want uniform properties for cultivated type of var. frutescens, such as green leaves and seeds with higher oil yield. However, in South Korea, many accessions of weedy type of var. frutescens with high genetic and phenotypic diversity were found throughout the region25,29,38,43,49.
Although numerous PGRs are currently conserved in genebanks around the world, the large amount of PGRs makes their accessibility and application difficult3,8. Moreover, management of these PGRs requires significant effort and expense. It is essential to select and manage a core collection, which can represent the entire collection. The evaluation of genetic distance or population structure among genotypes helps in the selection of parental combinations for generating new segregating populations, which preserves genetic diversity in breeding programs53. The identification of the genetic relationships and population structures of an entire collection may provide useful information for core collection selection and the management of PGRs. In this study, to understand the genetic relationships and population structure of 400 accessions of cultivated type of var. frutescens from central and southern regions of South Korea, we used two different methods: a model-based approach with STRUCTURE and a distance-based approach with a UPGMA dendrogram (Fig. 3, Supplementary Fig. 2). The STRUCTURE results revealed that the 400 accessions of cultivated var. frutescens could be divided into two major groups and an admixed group at K = 2 (Figs. 3, 4), while the UPGMA dendrogram results showed that the 400 accessions of cultivated var. frutescens were divided into ten major groups with 45.7% of genetic similarity. As mentioned above in the Results section, there was no clear geographical classification by STRUCTURE and UPGMA analysis among the 400 accessions of cultivated var. frutescens from the central and southern regions of South Korea and foreign regions (Fig. 3, Supplementary Figs. 1, 2). Xie et al. (2008)54 mentioned that population structure and genetic relationship patterns of many accessions are affected by many factors, such as gene flow, selection by environment or human, and breeding systems. Perilla crop has a long history of cultivation in East Asia including in South Korea. In South Korea, because many native landraces of Perilla crop are still widespread, these seeds might be frequently exchanged between diverse regions by farmers or animals and birds, as previously reported by Lee et al. (2002)29.
Meanwhile, because of failure to select a core set by model and distance-based methods, this study utilized PowerCore to construct a core collection with maximum genetic diversity from the entire initial collection and with a minimal number of germplasm resources. In particular, this study used molecular data rather than phenotypic data to construct the core collection. This is because molecular data using molecular markers is more accurate for ensuring genetic diversity of the initial collection and preventing missing data or environmental interactions that typically exist in phenotypic data55. PowerCore in this study captured 100% of the alleles with a sampling percentage of 11%, based on 22 SSR markers throughout the collection (Table 4). The percentage of selected samples identified in this study was similar to the percentage (~ 10%) proposed by Brown (1989)56, while lower than the suggested percentage (20 ~ 30%) by Yonezawa et al. (1995)7. Sh.W. and Nei diversity indices were used for validation of the core collection, and the averages of the Sh.W. index (1.306) and Nei index (0.639) of the Perilla core collection were higher than those of the entire initial collection (Sh.W. = 1.059, Nei = 0.569), indicating increased genetic diversity of the core collection. This may be because of the removal of genetic redundancy in the core collection compared with the entire initial collection. It is obvious that this core collection is an exact representation of the diversity of the entire collection (Fig. 4, Table. 4). Although some accessions were absent and only a small number of traits were investigated in the PCA analysis, the accessions of the core collection selected by SSR markers were well reflected in three clusters based on the first axis of the PCA scatter plot by eight morphological traits (Fig. 5). In detail, the morphological analysis revealed all types of color of leaf surface (QL1) and stem color (QL3) in the core collection. Although no accession with light purple for color of reverse side leaf (QL2) was included in the core collection, the remaining three types were contained in the core collection (Supplementary Tables 2, 3). All three types of leaf shape (QL4) were included amongst the 42 accessions in the core collection. Moreover, all four types of degree of pubescence (QL5) were included in the 42 accessions of the core collection. In the case of flowering time (QL6), there were no accessions for early flowering type in the morphological evaluation of the 42 accessions, but the intermediate and late flowering types were contained in the core collection. All five types of seed color (QL7) were included in the 42 accessions of core collection. In addition, most of the 42 accessions in core collection had soft seeds, while seven accessions showed hard seeds (Table 3).
This study constructed the first core collection of Korean Perilla accessions and maintained allelic richness. It can be considered as germplasm for identifying useful genes for important agricultural traits. Further modification of the core collection is expected by the continuous addition of new Perilla accessions, such as accessions of the two cultivated types of Perilla crop and their weedy types. Further analysis of phenotypic and agronomic traits for the core collection is necessary to provide more valuable information for the development and utilization of Perilla accessions in breeding programs.
Materials and methods
Plant materials and DNA extraction
A total of 400 accessions of cultivated P. frutescens var. frutescens collected in South Korea and other regions (foreign or unknown accessions) were obtained from the RDA-Genebank of the Republic of Korea (http://genebank.rda.go.kr). The accessions of Perilla germplasm preserved by the RDA-Genebank of the Republic of Korea have been collected for decades with the approval of farmers for the Korean native Perilla accessions. The IT number and location information for these materials is shown in Supplementary Table 1. South Korea is geographically divided into central and southern regions (Supplementary Fig. 1). The central region includes Gangwon-do, Gyeonggi-do, and Chungcheong-do; while the southern region includes Gyeongsang-do, Jeolla-do, and Jeju-do. A total of 359 accessions were collected from the central and southern regions of South Korea, whereas the remaining 41 accessions were either collected from foreign countries (20 accessions) or the collection region is unknown (21 accessions) (Supplementary Table 1). In the central region, 148 accessions were collected from Gangwon-do (46 accessions), Gyeonggi-do (53 accessions), and Chungcheong-do (49 accessions); while in the southern region, 211 accessions were collected from Gyeongsang-do (125 accessions), Jeolla-do (85 accessions), and Jeju-do (1 accession). The accessions used in this study were selected from the RDA-Genebank as breeding materials for the development of leaf vegetable cultivars of cultivated var. frutescens. Total DNA was extracted from the young leaf tissue of individual representative plants of each accession according to Plant DNAzol Reagent protocols (GibcoBRL Inc., Grand Island, NY, USA). Our studies were complied with local and national regulations and following Kangwon National University (Chuncheon, Korea) and National Agrobiodiversity Center, National Institute of Agricultural Sciences, RDA (Jeonju, Korea) regulations.
Evaluation of morphological characteristics
For evaluation of morphological variation among the 400 accessions of cultivated P. frutescens var. frutescens, 20 seeds of each accession were sown in a nursery bed in early May 2020, and kept in a glass house for a month. After that, seven seedlings of each accession were then transplanted to the fields of the experimental farm of Kangwon National University, Chuncheon, Gangwon-do in early June 2020. We examined eight morphological characteristics related to leaf and seed quality and quantity, namely color of leaf surface (QL1), color of reverse side leaf (QL2), stem color (QL3), leaf shape (QL4), degree of pubescence (QL5), flowering time (QL6), seed color (QL7), and seed hardness (QL8) at the appropriate growth stages. These morphological traits, as shown in Supplementary Table 2, were selected based on a previous report by Lee and Ohnishi (2001). In our study, 28 accessions were not used for measurement because of failure of plant growth in the field. Therefore, in this study, only 372 accessions out of the 400 accessions of cultivated P. frutescens var. frutescens were evaluated for morphological characteristics (Supplementary Table 1).
SSR analysis and DNA electrophoresis
SSR amplification was performed in a total volume of 20 μL containing 20 ng genomic DNA, 1 × polymerase chain reaction (PCR) buffer, 0.2 mM dNTPs, 0.5 μM forward and reverse primers, and 1 unit of Taq polymerase (Bio Rad, Hercules, CA). The PCR profile consisted of initial denaturation at 95 °C for 3 min, followed by 36 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min 30 s, with a final extension step of 5 min at 72 °C. After PCR, DNA electrophoresis analysis was performed with a QIAxcel advanced system (QIAGEN Co., Hilden, Germany) according to the protocol described in the QIAxcel DNA Handbook. The samples were run in the QIAxcel advanced electrophoresis system, and sample separation was performed over 15 min. Gel images were obtained as the results, and the quantification analysis was performed with QIAxcel software. The results were displayed as gel images and electropherograms acquired from the QIAxcel advanced system software.
Data analysis
The fragments amplified for each SSR primer set were scored as present (1) or absent (0). Power Marker version 3.2557 was used to confirm information on the number of alleles, allele frequency, and genetic diversity index (GDI), such as major allele frequency (MAF), gene diversity (GD), and polymorphic information content (PIC). Genetic similarities (GS) were calculated for each pair of accessions using the Dice similarity index58. To illustrate the genetic relationships of the total accessions, a similarity matrix was used to construct an unweighted pair group method with arithmetic mean (UPGMA) dendrogram by the application of SAHN-Clustering from NTSYS-pc V2.159. Principal component analysis (PCA) was performed to detect differences between and within accessions of P. frutescens var. frutescens. The PCA analysis was performed using NTSYS-pc V2.159. Population structure was investigated for the 400 accessions of cultivated P. frutescens var. frutescens using STRUCTURE 2.2 software60. Five independent runs with K values ranging from one to ten were performed with 100,000 cycles for both burn-in and the run length. The delta K statistic, based on the rate of change in the log probability of data between K values32, was calculated with STRUCTURE HARVESTER. For extraction of a core collection, this study used PowerCore software, which uses the advanced M strategy implemented through the modified heuristic algorithm for the core collection as described in the user’s manual20.
References
Frankel, O. H. & Soule, M. E. Conservation and Evolution. Cambridge University Press (Cambridge, 1981).
Mongkolporn, O., Hanyong, S., Chunwongse, J. & Wasee, S. Establishment of a core collection of chilli germplasm using microsatellite analysis. Plant Genet. Resour. 13, 104–110 (2015).
van Hintum, T. J. L., Brown, A. H. D., Spillane, C. & Hodgkin, T. Core Collections of Plant Genetic Resources. IPGRI Technical Bulletin No. 3 (2000).
Belaj, A. et al. Developing a core collection of olive (Olea europaea L.) based on molecular markers (DArTs, SSRs, SNPs) and agronomic traits. Tree Genet. Genom. 8, 365–378 (2012).
Frankel, O. H. Genetic perspectives of germplasm conservation. In Genetic Manipulation: Impact on Man and Society (eds Arber, W. K. et al.) 161–170 (Cambridge University Press, 1984).
Charmet, G. & Balfourier, F. The use of geostatistics for sampling a core collection of perennial ryegrass populations. Genet. Resour. Crop Evol. 42, 303–309 (1995).
Yonezawa, K., Nomura, T. & Morishima, H. Sampling strategies for use in stratified germplasm collections. In: Hodgkin T, Brown AHD, van Hintum ThJL, Morales EAV (eds) Core collections of plant genetic resources. IPGRI, Wiley, Baffins Lane, 35–54 (1995).
Noirot, M., Hamon, S. & Anthony, F. The principal component scoring: a new method of constituting a core collection using quantitative data. Genet. Resour. Crop Evol. 43, 1–6 (1996).
Wada, T. et al. Development of a core collection of strawberry cultivars based on SSR and CAPS marker polymorphism. Hort. J. 86, 365–378 (2017).
Ebana, K., Kojima, Y., Fukuoka, S., Nagamine, T. & Kawase, M. Development of mini core collection of Japanese rice landrace. Breed. Sci. 58, 281–291 (2008).
Kaga, A. et al. Evaluation of soybean germplasm conserved in NIAS genebank and development of mini core collections. Breed. Sci. 61, 566–592 (2012).
Balfourier, F. et al. A worldwide bread wheat core collection arrayed in a 384-well plate. Theor. Appl. Genet. 114, 1265–1275 (2007).
Upadhyaya, H. D., Bramiel, P. J. & Sube, S. Development of a chickpea core subset using geographic distribution and qualitative traits. Crop Sci. 41, 206–210 (2001).
Amalraj, V. A., Balakrishnan, R., Jebadhas, A. W. & Balasundaram, N. Constituting a core collection of Saccharum spontaneum L. and comparison of three stratified random sampling procedures. Genet. Resour. Crop Evol. 53, 1563–1572 (2006).
Quenouille, J., Saint-Felix, L., Moury, B. & Palloix, A. Diversity of genetic backgrounds modulating the durability of a major resistance gene. Analysis of a core collection of pepper landraces resistant to Potato virus Y. Mol. Plant Pathol. 17, 296–302 (2016).
Zhang, C. Y. et al. A method for constructing core collection of Malus sieversii using molecular markers. Sci. Agric. Sin. 42, 597–604 (2009).
Liu, W. et al. Evaluation of genetic diversity and development of a core collection of wild rice (Oryza rufipogon Griff.) populations in China. PLoS ONE 10, e0145990 (2015).
Escribano, P., Viruel, M. A. & Hormaza, J. I. Comparison of different methods to sequence repeat markers. A case study in cherimoya (Annona cherimola, Annonaceae), an underutilised subtropical fruit tree species. Ann. Appl. Biol. 153, 25–32 (2008).
Richards, C. M. et al. Selection of stratified core sets representing wild apple (Malus sieversii). J. Am. Soc. Hort. Sci. 134, 228–235 (2009).
Kim, K. W. et al. PowerCore: a program applying the advanced M strategy with a heuristic search for establishing core sets. Bioinformatics 23, 2155–2162 (2007).
Agrama, H. A. et al. Genetic assessment of a mini-core subset developed from the USDA Rice Genebank. Crop Sci. 49, 1336–1346 (2009).
Jayarame, G., Bharathi, S., Somu, G., Krishnappa, M. & Rekha, D. Formation of core set in barnyard millet [Echinochloa frumentacea (Roxb.) Link] germplasm using data on twenty four morpho-agronomic traits. Inter. J. Plant Sci. 4, 1–5 (2009).
Chandrashekhar, H., Gowda, J. & Ugalat, J. Formation of core set in Indian and African finger millet [Eleusine coracana (L.) Gaertn] germplasm accessions. Indian J. Genet. Plant Breed. 72, 358–363 (2012).
Oliveira, E. J., Ferreira, C. F., Santos, V. S. & Oliveira, G. A. Development of a cassava core collection based on single nucleotide polymorphism markers. Genet. Mol. Res. 13, 6472–6485 (2014).
Lee, J. K. & Ohnishi, O. Geographic differentiation of morphological characters among Perilla crops and their weedy types in East Asia. Breed. Sci. 51, 247–255 (2001).
Nitta, M., Lee, J. K. & Ohnishi, O. Asian Perilla crops and their weedy forms: their cultivation, utilization and genetic relationships. Econ. Bot. 57, 245–253 (2003).
Lee, J., Lee, M. H., Cho E. J. & Lee, S. High-yield methods for purification of a-linolenic acid from Perilla frutescens var. Japonica oil. Appl. Biol. Chem. 59, 89–94 (2016).
Park, H., Sa, K. J., Hyun, D. Y., Lee, S. & Lee, J. K. Identifying SSR Markers related to seed fatty acid content in Perilla crop (Perilla frutescens L.). Plants 10, 1404 (2021).
Lee, J. K. et al. Genetic diversity of Perilla and related weedy types in Korea determined by AFLP analyses. Crop Sci. 42, 2161–2166 (2002).
Raom, R. & Hodgkin, T. Genetic diversity and conservation and utilization of plant genetic resources. Plant Cell Tissue Organ Cult. 68, 1–19 (2002).
Palmer, J. D., Jansen, R. K., Michaels, H. J., Chase, M. W. & Manhart, J. R. Chloroplast DNA variation and plant phylogeny. Ann. Missouri Bot. Gard. 75, 1180–1206 (1988).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Wang, R. et al. Population structure and linkage disequilibrium of a mini core set of maize inbred lines in China. Theor. Appl. Genet. 117, 1141–1153 (2008).
D’hoop, B. B. et al. Population structure and linkage disequilibrium unravelled in tetraploid potato. Theor. Appl. Genet. 121, 1151–1170 (2010).
Odong, T. L., van Heerwaarden, J., Jansen, J., van Hintum, T. J. L. & van Eeuwijk, F. A. Determination of genetic structure of germplasm collections: Are traditional hierarchical clustering methods appropriate for molecular marker data. Theor. Appl. Genet. 123, 195–205 (2011).
Nitta, M. & Ohnishi, O. Genetic relationships among two Perilla crops, shiso and egoma, and the weedy type revealed by 536 RAPD markers. Jpn. J. Genet. 74, 43–48 (1999).
Park, Y. J. et al. Evaluation of genetic diversity and relationships within an on-farm collection of Perilla frutescens (L.) Britt. Using microsatellite markers. Genet. Resour. Crop Evol. 55, 523–535 (2008).
Sa, K. J. et al. Identification of genetic variations of cultivated and weedy types of Perilla species in Korea and Japan using morphological and SSR markers. Genes Genom. 35, 649–659 (2013).
Park, D. H., Sa, K. J., Lim, S. E., Ma, S. J. & Lee, J. K. Genetic diversity and population structure of Perilla frutescens collected from Korea and China based on simple sequence repeats (SSRs). Genes Genom. 41, 1329–1340 (2019).
Ma, S. J., Sa, K. J., Hong, T. K. & Lee, J. K. Genetic diversity and population structure analysis in Perilla crop and their weedy types from northern and southern areas of China based on simple sequence repeat (SSRs). Genes Genom. 41, 267–281 (2019).
Powell, W. et al. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 2, 225–238 (1996).
Park, Y. J., Lee, J. K. & Kim, N. S. Simple sequence repeat polymorphisms (SSRPs) for evaluation of molecular diversity and germplasm classification of minor crops. Molecules 14, 4546–4569 (2009).
Sa, K. J., Choi, I. Y., Park, K. C. & Lee, J. K. Genetic diversity and population structure among accessions of Perilla frutescens (L.) Britton in East Asia using new developed microsatellite markers. Genes Genom. 40, 1319–1329 (2018).
Kim, J. Y., Sa, K. J., Ha, Y. J. & Lee, J. K. Genetic variation and association mapping in the F2 population of the Perilla crop (Perilla frutescens L.) using new developed Perilla SSR markers. Euphytica 217, 135 (2021).
Kwon, S. J. et al. Isolation and characterization of SSR markers in Perilla frutescens Britt. Mol. Eco. Notes 5, 454–456 (2005).
Sa, K. J., Lim, S. E., Choi, I. Y., Park, K. C. & Lee, J. K. Development and characterization of new microsatellite markers for Perilla frutescens (L.) Britton. Am. J. Plant Sci. 10, 1623–1630 (2019).
Oh, J. K., Sa, K. J., Hyun, D. Y., Cho, G. T. & Lee, J. K. Assessment of genetic diversity and population structure among a collection of Korean Perilla germplasms based on SSR markers. Genes Genom. 42, 1419–1430 (2020).
Ha, Y. J., Sa, K. J. & Lee, J. K. Identification of SSR markers associated with seed characteristics in Perilla (Perilla frutescens L.). Physiol. Mol. Biol. Plants 27, 93–105 (2021).
Lee, J. K. & Ohnishi, O. Genetic relationships among cultivated types of Perilla frutescens and their weedy types in East Asia revealed by AFLP markers. Genet. Resour. Crop Evol. 50, 65–74 (2003).
Nitta, M., Lee, J. K., Kobayashi, H., Liu, D. & Nagamine, T. Diversification of multipurpose plant, Perilla frutescens. Genet. Resour. Crop Evol. 52, 663–670 (2005).
Vaiman, D., Mercier, D. & Moazai, G. A set of 99 cattle microsatellite, characterization, synteny mapping and polymorphism. Mamm. Genome 5, 288–297 (1994).
Tong, W. et al. Gene set by de novo assembly of Perilla species and expression profiling between P. frutescens (L.) var. frutescens and var. crispa. Gene 559, 155–163 (2015).
Becelaere, G. V., Edward, L. L., Paterson, A. H. & Chee, P. W. Pedigree-vs. DNA marker-based genetic similarity estimates in Cotton. Crop Sci. 45, 2281–2287 (2005).
Xie, C. X. et al. An analysis of population structure and linkage disequilibrium using multilocus data in 187 maize inbred lines. Mol. Breed. 21, 407–418 (2008).
Roy Choudhury, D. et al. Analysis of genetic diversity and population structure of rice germplasm from north-eastern region of India and development of a core germplasm set. PLOS ONE 9, e113094 (2014).
Brown, A. H. D. Core collections: a practical approach to genetic resources management. Genome 31, 818–824 (1989).
Liu, K. & Muse, S. V. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129 (2005).
Dice, L. R. Measures of the amount of ecologic association between species. Ecology 26, 297–302 (1945).
Rohlf, F. NTSYS-pc: numerical taxonomy and multivariate analysis system. version 21. Exter Software Setauket, NY (2000)
Pritchard, J. K. & Wen, W. Documentation for STRUCTURE software: Version 2 (2003).
Acknowledgements
This study was supported by the Cooperative Research Program for Agriculture Science & Technology Development (project no. PJ014227032021 and PJ0142272021), Rural Development Administration, and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (NRF-2021R1A6A1A03044242, #2016R1D1A1B01006461), Republic of Korea.
Author information
Authors and Affiliations
Contributions
D.Y.H., S.L., and J.H. Rhee provided the genetic material. K.J.S., D.M.K., J.S.O., and H.P. conducted morphological characteristic evaluation and molecular biology experiments. D.M.K., J.S.O., and K.J.S. performed statistical analysis. K.J.S. and J.K.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sa, K.J., Kim, D.M., Oh, J.S. et al. Construction of a core collection of native Perilla germplasm collected from South Korea based on SSR markers and morphological characteristics. Sci Rep 11, 23891 (2021). https://doi.org/10.1038/s41598-021-03362-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03362-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
