
Abstract
Human induced pluripotent stem cells (hiPSCs) can differentiate into cells of the three germ layers and are promising cell sources for regenerative medicine therapies. However, current protocols generate hiPSCs with low efficiency, and the generated iPSCs have variable differentiation capacity among different clones. Our previous study reported that MYC proteins (c-MYC and MYCL) are essential for reprogramming and germline transmission but that MYCL can generate hiPSC colonies more efficiently than c-MYC. The molecular underpinnings for the different reprogramming efficiencies between c-MYC and MYCL, however, are unknown. In this study, we found that MYC Box 0 (MB0) and MB2, two functional domains conserved in the MYC protein family, contribute to the phenotypic differences and promote hiPSC generation in MYCL-induced reprogramming. Proteome analyses suggested that in MYCL-induced reprogramming, cell adhesion-related cytoskeletal proteins are regulated by the MB0 domain, while the MB2 domain regulates RNA processes. These findings provide a molecular explanation for why MYCL has higher reprogramming efficiency than c-MYC.
Similar content being viewed by others


Introduction
Human induced pluripotent stem cells (hiPSCs) are generated from somatic cells and can differentiate into cells of all three germ layers1,2. They are functionally identical to human embryonic stem cells (hESCs) but do not require the destruction of the embryo, which has made them attractive sources for regenerative medicine3. The original reprogramming was induced by four factors, OCT3/4, SOX2, KLF4, and c-MYC (OSKM). Since then, several new methods have been developed to improve the yield and quality of iPSCs, but the cost remains high and the production remains technically difficult4,5. Further complicating the application of hiPSCs is the wide variability in the differentiation capacity of different hiPSC clones6.
We have shown that excluding c-MYC from the reprogramming factors significantly lowers the reprogramming and differentiation efficiencies of the resulting iPSCs7. The MYC family consists of the oncogenes c-MYC, MYCN, and MYCL in humans8. c-MYC was the first MYC gene discovered in human and has been a topic of cancer research ever since9. Tumorigenesis depends on high transformation activity derived from the N-terminus region of c-MYC protein10. Consequently, OSKM-based reprogramming may not be appropriate for the clinical application of iPSCs. Many groups have reported reprogramming methods that exclude c-MYC overexpression but at the cost of lower reprogramming efficiency5,7. MYCL is about 30 amino acids shorter in the N-terminus region than c-MYC and has lower transformation activity10. We found that substituting c-MYC for MYCL in reprogramming can increase the number of iPSC colonies and maintain the ability to differentiate into the cells of three germ layers7. Furthermore, fewer chimeric mice died by tumorigenesis after the transplantation of MYCL-iPSCs, whereas the transplantation of c-MYC-iPSCs caused lethal tumorigenesis in more than 50% of mice during two years of observation. Despite these observations, little is known about the molecular function of MYCL and the different mechanisms between c-MYC and MYCL to promote reprogramming.
MYC proteins have six MYC Box (MB) domains: MB0, 1, 2, 3a, 3b, and 4 in the N-terminus and a basic helix-loop-helix leucine zipper (bHLHLZ) in the C-terminus11, but MYCL does not have MB3a. The C-terminus of c-MYC and MYCL is essential in reprogramming due to its binding with MAX protein, allowing MYC to access the DNA7,12. The N-terminus is mainly known as a transactivation domain (TAD), which regulates the target gene, but its function in reprogramming is less clear13. We found that a mutant of c-MYC lacking the N-terminal showed low transformation activity and promoted reprogramming7. However, which domain on the N-terminal side is essential for reprogramming and what function it performs were not resolved. In addition, MYC proteins act as transcription factors upon interacting with several binding proteins14. Although MYCL-binding proteins are important for MYCL function, there are no reports about MYCL-binding proteins during reprogramming.
In this study, using domain deletion mutants of MYC proteins, we found that the MB0 and MB2 domains promote iPSC-like colonies and that the MB0 domain is functionally different between c-MYC and MYCL. In c-MYC, it induced non-iPSC-like colonies by increasing nucleic proteins related to transcription, but in MYCL, the MB0 domain induced iPSC-like colonies by increasing the expression of cell adhesion-related proteins. We also found that deletion of the MB2 domain in MYC proteins prevented colony formation and that MYCL could interact with RNA-binding proteins (RBPs) via this domain. These results suggested that MYCL promotes reprogramming by regulating RNA processing.
Results
MYCL promotes reprogramming more efficiently than c-MYC
To compare the reprogramming phenotypes of MYCL and c-MYC, we used Sendai virus (SeV)-based reprogramming (CytoTune-iPS) and StemFit AK03N medium without bFGF (Fig. 1A). The SeV method has high reprogramming efficiency without genome integration, and c-MYC and MYCL SeV kits are already available15. The bFGF exclusion is based on the data in Supplementary Fig. S1. DMEM supplemented with 10% FBS (DMEM + 10%FBS) is the standard medium to induce reprogramming. We used DMEM + 10%FBS when introducing the reprogramming factors, but after 7 days of reprogramming, we replated the cells and used StemFit AK03N without bFGF (03N (-)) from that point on. The MOI (multiplicity of infection) of each SeV was 20. To improve the reprogramming efficiency, we compared three media combinations (Supplementary Fig. S1A). The highest number of colonies was obtained using 03N (-) during reprogramming and 03N ( +) after replating (Supplementary Fig. S1B). These results indicated that the 03N (-) reprogramming condition in the first 7 days enhances the reprogramming efficiency compared to 03N ( +). We then examined the optimal MOI of SeV for the reprogramming (Supplementary Fig. S1C). A lower MOI induced more colonies (Supplementary Fig. S1D), indicating a higher reprogramming efficiency. Following these results, we applied SeV for the transduction at an MOI of 4.3 using 03N (-) during reprogramming.

MYCL promotes reprogramming more efficiently than c-MYC. (A) Schematic representation of HDF reprogramming with Sendai virus (SeV). HDFs were transduced with SeV carrying KLF4-OCT3/4-SOX2 (KOS), KLF4 (K), and c-MYC or MYCL on day 0. We used an MOI (multiplicity of infection) of 4.3 for each virus. StemFit AK03N without bFGF was used during the transduction and subsequent induction of iPSC-like colonies. We performed immunostaining of the reprogramming HDFs 1 to 7 days after the transduction and analyzed the results using ArrayScan. (B) Representative immunostaining images of reprogramming HDFs stained by anti-TRA-1-60 antibody (green) and Hoechst (blue) 7 days after the transduction. Scale bar, 300 μm. Ph, phase contrast. (C) Proliferation and expression of TRA-1-60 ( +) cells during reprogramming. HDFs were transduced with SeV, including c-MYC or MYCL, and immunostaining was performed from days 1 to 7. The number of total cells was counted as Hoechst-positive cells. Mean ± SD values are shown. n = 3, *p < 0.05 by paired t-test. (D) Schematic representation of HDF reprogramming with episomal plasmid vector (EpiP). HDFs were transduced with EpiP carrying SOX2, KLF4, OCT3/4-shp53, LIN28A, EBNA1, and c-MYC or MYCL. StemFit AK03N without bFGF was used during the transfection and subsequent induction of iPSC-like colonies. We performed flow cytometry of the reprogramming HDFs every three days from 1 to 19 days plus day 21 after the transduction. (E) Representative immunostaining images of reprogramming HDFs stained by anti-TRA-1-60 antibody (green) and Hoechst (blue) 21 days after the transduction. Scale bar, 300 μm. Ph, phase contrast. (F) Proliferation and expression of TRA-1-60 ( +) cells during reprogramming were analyzed by flow cytometry. HDFs were transduced with EpiP, including c-MYC or MYCL. Flow cytometry was performed every three days from days 1 to 19 days plus day 21. Mean ± SD for n = 3, *p < 0.05 and **p < 0.01 by paired t-test. (G) The number of iPSC-like and non-iPSC-like colonies derived from 1 × 105 HDFs transduced with EpiP including c-MYC or MYCL on day 21. Mean ± SD values are shown. n = 3, **p < 0.01 by unpaired t-test. (H) Percentage of CD13 ( +) cells during EpiP reprogramming determined by flow cytometry. Mean ± SD values are shown. n = 3, *p < 0.05 and **p < 0.01 by paired t-test.
Next, we conducted immunostaining to analyze the expression of TRA-1-60 from days 1 to 7 after the transduction (Fig. 1A). TRA-1-60 is a glycoprotein and major cell surface marker of hiPSCs and hESCs16. We quantified the results using a high-content imaging system, ArrayScan, because the cell number was small during SeV reprogramming for the first seven days, making flow cytometry challenging. On day 7, we observed that c-MYC and MYCL induced a small cell mass to form colonies, but only the colonies induced by MYCL expressed TRA-1-60, while those induced by c-MYC looked like cell aggregations (Fig. 1B and Supplementary Fig. S2). Cell proliferation was highly increased in human dermal fibroblasts (HDFs) transduced with c-MYC compared to MYCL. On the other hand, the percentage of TRA-1-60 ( +) cells increased more in MYCL-transduced HDFs on day 3 after the transduction (Fig. 1C). This difference may be because c-MYC has higher transformation activity than MYCL, which causes different phenotypes, especially cell proliferation10.
We confirmed these reprogramming phenotypes using episomal plasmid vector (EpiP)17 (Fig. 1D). SeV systems have a higher gene transfer efficiency, leading to more efficient reprogramming. However, we could modify the reprogramming vectors, which is useful for evaluating the molecular mechanism of c-MYC and MYCL, only when using the EpiP system.
Similar to the results with the SeV method, few colonies expressed TRA-1-60 in c-MYC-transfected HDFs (Fig. 1E and Supplementary Fig. S3A). However, the transfection of MYCL resulted in a higher percentage of TRA-1-60 ( +) cells and lower cell proliferation than the transfection of c-MYC (Fig. 1F and Supplementary Fig. S3B). These differences between MYCL and c-MYC were more obvious with EpiP reprogramming than SeV reprogramming (Fig. 1C, F), probably because of differences in the gene transfer efficiency15,17, the expression of the transfected factors, cell toxicity, and the time required for the iPSC-like colonies to appear: the SeV system requires about 7 days, but the EpiP system needs about 21 days based on our observations.
We found two types of colonies: “iPSC-like” and “non-iPSC-like” colonies. The iPSC-like colonies produced by MYCL were more flattened and showed a monolayered colony morphology, with each cell tightly packed and expressing TRA-1-60. The non-iPSC-like colonies produced by c-MYC showed a cell aggregation-like morphology, in which individual cells were irregularly aggregated and did not express TRA-1-60. We counted the number of iPSC-like and non-iPSC-like colonies on day 21 and found that c-MYC induced iPSC-like colonies as well as many non-iPSC-like colonies, but MYCL induced almost only iPSC-like colonies and more of them than c-MYC (Fig. 1G and Supplementary Fig. S3).
It has been reported that before the increase in the expression of TRA-1-60, a decrease in the expression of CD13, a marker of fibroblasts18, is observed in somatic cell reprogramming. Therefore, we confirmed the expression of CD13 during reprogramming. The percentage of CD13 ( +) cells decreased daily in HDFs transduced with c-MYC or MYCL, but the number of CD13 (-) cells rapidly increased in c-MYC compared to MYCL (Fig. 1H and Supplementary Fig. S4). In particular, the CD13 (-) TRA-1-60 (-) population was larger on day 10 with c-MYC reprogramming than MYCL reprogramming, but the CD13 (-) TRA-1-60 ( +) population from days 16 to 21 was larger with MYCL reprogramming (Supplementary Fig. S4). These results suggested that MYCL promotes TRA-1-60 ( +) cells more than c-MYC, but c-MYC suppresses CD13 expression more than MYCL.
MYC Box 0 and 2 domains are crucial for colony formation during reprogramming
Next, we prepared domain deletion mutants to identify which domains in the N-terminus of MYC proteins influence reprogramming (Fig. 2A and Supplementary Fig. S5). We previously showed that a c-MYC mutant lacking transformation activity enhances the formation of iPSC-like colonies. This mutant has a point mutation in the transactivation domain of the N-terminal region, W135E (Fig. 2A and Supplementary Fig. S5B), but can bind to genomic DNA7. On the other hand, the bHLHLZ domain in the C-terminus region is a well-known binding domain of MAX19. Mutants in the C-terminus region prevent MYC proteins from binding to DNA and thus reprogramming7. Finally, we tested the reprogramming activities of these mutants using the EpiP reprogramming system because, as explained above, this method provided a clearer phenotype and was easier to manipulate than the SeV method.

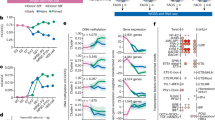
MYC Box 0 and 2 domains are crucial for colony formation during reprogramming. (A) Schematic representation of WT c-MYC and MYCL protein. Black boxes show important domains for MYC function, including MB0, 1, 2, 3a (c-MYC only), 3b, 4, and basic-helix-loop-helix leucine zipper motif (bHLHLZ). The percentage of common amino acids in each MYC box domain between MYCL and c-MYC is shown (Identity%). The numbers on the right indicate amino acid lengths. (B) Number of iPSC-like and non-iPSC-like colonies transduced with EpiP including c-MYC-WT/mutants (left) or MYCL-WT/mutants (right) on day 21. Mean ± SD values are shown. n = 3, **p < 0.01, ***p < 0.001 and ****p < 0.0001 by ordinary one-way ANOVA and Dunnett’s test vs. WT. (C) Expression of TRA-1-60 ( +) HDFs and CD13 ( +) HDFs transduced with EpiP including c-MYC-WT/mutants (left) or MYCL-WT/mutants (right) on day 16. Mean ± SD values are shown. n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 by ordinary one-way ANOVA and Dunnett’s test vs. WT. (D) Representative flow cytometry images for TRA-1-60 and CD13 for HDFs transduced with EpiP including c-MYC-WT/ΔMB0 or MYCL-WT/ΔMB0 10 and 21 days after the transduction. Numbers indicate the expression percentage of each quadrant. (E) Proliferation of HDFs transduced with EpiP including c-MYC-WT/ΔMB0 or MYCL-WT/ΔMB0 10 and 21 days later. Mean ± SD values are shown. n = 3, ***p < 0.001 by ordinary one-way ANOVA and Dunnett’s test vs. c-MYC-WT. The number of cells was counted using a Cell Counter model R1 (OLYMPUS).
The EpiP mutants were transfected into HDFs with other reprogramming factors, and the number of iPSC-like and non-iPSC-like colonies was counted (Fig. 2B and Supplementary Fig. S6). c-MYC-ΔMB0 promoted the formation of iPSC-like colonies and inhibited the formation of non-iPSC-like colonies compared to c-MYC-WT. In contrast, MYCL-ΔMB0 showed almost no ability to form iPSC-like colonies (Fig. 2B). We confirmed that the protein expression of each domain deletion mutant by western blotting showed no difference compared with c-MYC- or MYCL-WT (Supplementary Fig. S7 and S8). These results demonstrate that the MB0 domain has different functions in c-MYC and MYCL for reprogramming and that c-MYC-ΔMB0 has a similar function as MYCL-WT.
Figure 2B shows that c-MYC-ΔMB1 promoted iPSC-like colony formation like c-MYC-ΔMB0, but it also led to the formation of non-iPSC-like colonies. The formation of iPSC-like colonies by MYCL-ΔMB1 was about a quarter that by MYCL-WT. Unlike c-MYC-WT, c-MYC-ΔMB2 did not induce non-iPSC-like colonies, but it did induce a rate of iPSC-like colonies similar to c-MYC-WT. MYCL-ΔMB2 showed little ability to form iPSC-like colonies, resembling MYCL-ΔMB0. c-MYC-ΔMB3a, -ΔMB3b, and -ΔMB4 had similar colony-forming activities as c-MYC-WT. MYCL-ΔMB3b showed the same reprogramming efficiency as MYCL-WT, but MYCL-ΔMB4 formed about the same small number of iPSC-like colonies as MYCL-ΔMB1. The ΔbHLHLZ mutants of both c-MYC and MYCL failed to induce colonies and were therefore considered to have lost MYC function completely. Thus, the results indicate that in c-MYC, the MB0 and MB2 domains are repressive for iPSC-like colony formation, but in MYCL, they are promotive. Other domains also influenced the colony formation efficiency, but the effect was small.
Next, we analyzed the effect of the MYC-deletion mutants on the expression of TRA-1-60 and CD13 by flow cytometry 16 days after the start of reprogramming (Fig. 2C). Mutants that increased the number of iPSC-like colonies also increased the expression of TRA-1-60, while those that reduced the number of iPSC-like colonies lowered the TRA-1-60 expression (Fig. 2C and Supplementary Fig. S9). c-MYC-WT showed little TRA-1-60 expression, whereas c-MYC-ΔMB0 upregulated the expression. MYCL-ΔMB0, unlike MYCL-WT, failed to upregulate the expression of TRA-1-60. The CD13 expression was also correlated with colony formation. In c-MYC, a significant decrease in CD13 expression was observed for mutants that promoted non-iPSC-like colony formation. As for MYCL, only a slight decrease in CD13 expression was observed for mutants that promoted iPSC-like colony formation. From these results, we concluded that the MB0 domain is essential for the function of MYC in reprogramming but functions differently between c-MYC and MYCL.
To analyze the function of the MB0 domain in more detail, we analyzed the expression of TRA-1-60 and CD13 10 and 21 days after the start of reprogramming by flow cytometry (Fig. 2D). In the case of c-MYC-WT, there was a strong decrease in CD13 expression on day 10, and most cells were CD13 negative on day 21. In the cases of c-MYC-ΔMB0 and MYCL-WT, there was a slight decrease in CD13 expression on day 10, and more than half of cells were expressing TRA-1-60 on day 21. Finally, in the case of MYCL-ΔMB0, there was no change in CD13 or TRA-1-60 expression. More study is needed to determine how CD13 is regulated by c-MYC and MYCL.
Additionally, c-MYC-WT showed higher cell proliferation on day 10, but c-MYC-ΔMB0 resulted in a lower cell proliferation comparable more with MYCL-WT than with c-MYC-WT on day 10 (Fig. 2E). We attributed this effect to the lost transformation activity of c-MYC-ΔMB0. From days 10 to 21, the cell proliferation increased significantly in c-MYC-ΔMB0 and MYCL-WT, and a concomitant increase in the CD13 (-) TRA-1-60 ( +) population was observed (Fig. 2D, E). These observations suggest that the number of cells that were reprogrammed increased rapidly with c-MYC-ΔMB0 and MYCL-WT. With c-MYC-WT, the cell proliferation continued until day 21. However, the CD13 (-) TRA-1-60 ( +) population hardly increased (Fig. 2D), indicating that these cells were not reprogramming but changing to other highly proliferative cell types. From these results, we concluded that the MB0 domain functions negatively in c-MYC and positively in MYCL for reprogramming.
MYCL regulates cytoskeleton- and cell adhesion-related proteins during reprogramming via the MB0 domain
To confirm which genes are regulated by the MYCL MB0 domain in reprogramming, we analyzed protein expressions during reprogramming because it was reported that gene expressions do not correlate well with protein expressions20. We performed a comprehensive analysis of expressed proteins during reprogramming induced by c-MYC and MYCL WT and ΔMB0 mutants. We used SeV-reprogramming HDFs on days 3, 5, and 7 days and EpiP-reprogramming HDFs on day 10 as samples for mass spectrometry (MS) (Fig. 3A) because the percentage of TRA-1-60 ( +) cells was much higher with SeV than with EpiP for observations up to day 7 (Fig. 1C, F). There was more than a two-fold increase in the expression of 520 (SeV) and 128 (EpiP) proteins with MYCL-WT reprogramming compared to c-MYC-WT reprogramming (Fig. 3B, groups (i) and (ii), respectively) and 183 (EpiP) proteins with c-MYC-ΔMB0 reprogramming compared to c-MYC-WT reprogramming (Fig. 3B, group (iii)). Overall, we identified 18 proteins common to the three groups (Fig. 3B, group (iv)). Then, we applied a Gene Ontology (GO) analysis using DAVID and detected enriched terms during reprogramming21,22 (Fig. 3C, D, and Table 1), finding cytoskeleton- and cell adhesion-related proteins are involved in the promotion of reprogramming by MYCL-WT. The same analysis was performed to identify proteins whose expression was upregulated by c-MYC-WT compared with MYCL-WT and c-MYC-ΔMB0 (Supplementary Fig. S10A and Table 2). These proteins were associated with the proliferation of non-iPSC-like colonies. We found that c-MYC-WT regulates proteins involved in cell proliferation, such as the cell cycle and DNA replication. To understand the function of the MB0 domain in reprogramming, MS analysis was applied to HDF samples transfected with MYCL-WT, MYCL-ΔMB0, or c-MYC-ΔMB0 (Supplementary Fig. S10B and Table 3). GO analysis indicated that these proteins were associated with cell adhesion and RNA processing.

MYCL regulates cytoskeleton- and cell adhesion-related proteins during reprogramming via the MB0 domain. (A) Schematic of the mass spectrometry (MS) and GO analysis (DAVID). (B) Venn diagram of upregulated proteins during iPSC-like colony formation. (C) Molecular functions from the GO analysis of the four groups in (B). (D) KEGG pathways from the GO analysis of the four groups in (B).
We also compared phosphorylated proteins during SeV reprogramming with MYCL and c-MYC. In total, there was more than a two-fold relative increase of 17 phosphorylated proteins with MYCL-WT and 132 phosphorylated proteins with c-MYC-WT. The GO analysis indicated that the phosphorylated proteins increased by MYCL included cytoskeleton-related proteins and those increased by c-MYC included transcription-related proteins (Supplementary Fig. S11).
MYCL regulates RNA processing-related proteins during reprogramming via the MB2 domain
Our analysis also revealed that, along with the MYCL MB0 domain, the MYCL MB2 domain is important for reprogramming (Fig. 2B). It has been reported that the c-MYC MB2 domain is involved in transformation activity, and tryptophan 135 within the MB2 domain is necessary for this activity10. MYCL also has a tryptophan residue within its MB2 domain but little transformation activity23. We hypothesized that this domain in MYCL has reprogramming function. We therefore produced a mutant in which tryptophan 96 was substituted with glutamate (W96E). This tryptophan is equivalent to tryptophan 135 in c-MYC (Fig. 4A and Supplementary Fig. S5B). We confirmed the expression of MYCL-W96E by western blotting (Supplementary Fig. S12). Next, we examined the effect of MYCL-W96E for reprogramming. HDFs were transfected with reprogramming factors including MYCL-WT or -W96E. MYCL-W96E could not induce iPSC-like colonies, suggesting tryptophan 96 is crucial for reprogramming (Fig. 4B, C). We thus hypothesized that the residue might be important for MYCL to bind to other proteins. To identify the binding proteins, we produced GST-fusion recombinant proteins of the MYCL MB2 domain (Fig. 4A). GST-MYCL-MB2-WT or -W96E proteins were immobilized on glutathione Sepharose, and affinity columns were prepared. Cell lysates were applied to the column, and, after washing, the bound proteins were eluted. We used the cell lysates from reprogramming HDFs, but since it was difficult to collect a large amount, we also used cell lysates from hiPSCs. The reason for using the hiPSC lysates is that many of the proteins expressed in reprogramming HDFs are highly expressed in hiPSCs as well16,24,25,26,27.

MYCL regulates RNA processing-related proteins during reprogramming via the MB2 domain. (A) W96 and W135 in the MB2 domain of MYCL and c-MYC, respectively. The structure with the recombinant protein of the MB2 domain of MYCL-WT/W96E is shown below. The numbers on the right indicate amino acid lengths. (B) The number of iPSC-like and non-iPSC-like colonies derived from 1 × 105 HDFs transduced with EpiP including MYCL-WT or MYCL-W96E on day 21. Mean ± SD values are shown. n = 3, *p < 0.05 by unpaired t-test. (C) Representative images of reprogramming HDFs 21 days after the transduction of EpiP, including MYCL-WT or MYCL-W96E. Scale bars, 100 μm. (D) Venn diagram of enriched proteins between reprogramming HDFs and hiPSCs by AP-MS. A list of the 25 commonly enriched proteins is shown below. Blue indicates RBP (23 in total). (E) Molecular function from the GO analysis of the 25 commonly identified proteins in (D).
We identified 31 candidate proteins that bind to the MB2 domain of MYCL-WT but not of MYCL-W96E during reprogramming in the HDF lysates (Fig. 4D and Table 4). Of those 31 proteins, 25 proteins were also identified using hiPSC lysates, and 23 were RNA-binding proteins (RBPs; Fig. 4D, genes written in blue). Six proteins were identified only in the reprogramming HDFs lysates: HNRNPK, DDX17, C1QBP, KBTBD3, COPG2, and SIKE1, of which HNRNPK, DDX17, and C1QBP are RBPs. From these results, there were 26 RBPs identified in the HDF lysates in total. We confirmed the function of the 31 proteins using a public database (https://www.nextprot.org/)28 and found 16 of them are involved in RNA processing. A GO analysis using DAVID also showed that the 31 proteins are related to controlling pre-mRNA splicing, capping, and polyadenylation, suggesting functions in mRNA export, turnover, localization, and translation (Fig. 4E). These results suggested that MYCL interacts with RBPs via its MB2 domain and promotes reprogramming by post-transcriptional regulation.
Discussion
Here we described the molecular function of MYCL during reprogramming and compared it to the c-MYC function by focusing on MYC Box domains. We found that the MB0 and MB2 domains are important for reprogramming, and deleting either region compromised the reprogramming ability of MYCL. Proteomic analysis revealed that MYCL regulates the expression of cell adhesion-related proteins during reprogramming via the MB0 domain (Fig. 3C, D). We also found the possibility that the same domain is regulated by post-translational modifications (PTM), as discussed below. It is known that cell-substrate adhesion is closely related to the mesenchymal-epithelial transition (MET)29 and that MET occurs during the reprogramming process30,31,32. We speculate that MYCL promotes iPSC-like colony formation via the MET process by upregulating cell adhesion-related genes. Furthermore, we identified that the MB2 domain is required for MYCL to promote reprogramming by binding to RBPs, especially RNA processing-related proteins (Fig. 4D, E). It has been reported that RBPs regulate MET through post-transcriptional regulation. For example, heterogeneous nuclear ribonucleoprotein (hnRNP) A1 regulates the alternative splicing of Rac1 to control MET33. These findings suggest that MYCL regulates the RNA processing of cell adhesion-related genes transcribed by MYCL itself or other genes. Therefore, we hypothesize that transcriptional and post-transcriptional regulation by MYCL promotes MET, which increases the efficiency of reprogramming and leads to higher quality iPSCs.
Western blotting revealed that MYCL protein has a unique expression pattern (Supplementary Fig. S8 and S12). The calculated molecular weight of MYCL is about 40 kDa (364 aa), but we detected three strong bands at around 60 kDa, which we verified with second antibody (Supplementary Fig. S13). Since the expression of MYCL-ΔMB0 showed a strong single band, we speculate that the MYCL MB0 domain is the PTM site (Supplementary Fig. S8). Such a phenomenon was not observed in c-MYC (Supplementary Fig. S7). One possible type of relevant PTM is phosphorylation. Phosphorylation is crucial for protein function. For example, RNA polymerase II (Pol II) is required for transcription pauses in a promoter-proximal position during transcription initiation. In order to initiate transcription, the C-terminal domain of Pol II must be phosphorylated by P-TEFb34. In addition, the phosphorylation of c-MYC on threonine 58 in the MB1 domain promotes c-MYC binding to F-box and WD repeat domain containing 7 (FBXW7), causing the ubiquitination of c-MYC, which triggers c-MYC degradation35. Similarly, MYCL might undergo phosphorylation to change its activity and interaction with binding proteins. However, this hypothesis requires further study.
Comprehensive proteomic analysis suggested that the MYCL MB0 domain influences the expression of cell adhesion-related proteins, and MYCL shows an up-regulation of phosphorylated cytoskeletal proteins (Fig. 3C, D, and Supplementary Fig. S11A). Cell adhesion is mediated by adhesion molecules, such as integrins and cadherins, which function in the extracellular matrix (ECM) and cell–cell adhesion and are important for cell communication and the regulation of fundamental physiological processes such as tissue development and maintenance36,37. Human iPSCs and hESCs have unique focal adhesion localization, and appropriate adhesion to the ECM is required to regulate reprogramming via MET and maintain pluripotency38,39,40. Accordingly, our study supports MYCL regulating cell-substrate adhesion through its MB0 domain to promote reprogramming. In other words, MYCL might regulate proteins involved in cell adhesion and the cytoskeleton directly or indirectly to cause MET and promote reprogramming. In c-MYC, loss of the MB0 domain positively affects iPSC-like colony formation, suggesting that this domain has a different function compared to MYCL. This functional difference is somewhat surprising since the domain is well conserved (Supplementary Fig. S5B). We would like to clarify this point in the future.
We also found that the MB2 domain has an important function in MYCL-reprogramming (Fig. 2B,C). Deleting the MB2 domain completely compromised the reprogramming ability of MYCL. In c-MYC, the MB2 domain has an important function in transformation activity14, and tryptophan 135 in the MB2 domain is essential for this activity. The equivalent tryptophan residue in MYCL is tryptophan 96. MYCL has little transformation activity, but we showed that the mutation of tryptophan 96 completely lost the reprogramming ability of MYCL. To further investigate the function, we sought interacting proteins by affinity column chromatography. We found 31 proteins, including 26 RBPs, that interact with the MYCL MB2 domain (Table 4, genes written in blue). A GO analysis suggested that some of the 31 proteins are involved in RNA processing (Table 4). It has been reported that altered RNA processing affects somatic cell reprogramming41. Therefore, we hypothesize that MYCL also promotes MET in reprogramming by regulating RNA processing via interactions with RBPs at its MB2 domain. An illustrative summary of how MYCL regulates cell reprogramming through these two domains is shown in Fig. 5.

Model of the reprogramming process by MYCL. MYCL promotes iPSC-like colonies via its MB0 and MB2 domains. The MB0 domain regulates the expression of cell-adhesion proteins, possibly via post-translational modifications (PTM). The MB2 domain regulates RNA processing by interacting with RNA-binding proteins (RBP). We speculate that MYCL promotes reprogramming through the synergistic effects of these two mechanisms.
To conclude, we have demonstrated that MYCL promotes more efficient reprogramming than c-MYC, regulates the expression of cell adhesion and cytoskeletal proteins, and is involved in RNA processing via a single tryptophan residue in the MB2 domain. Following these findings, we propose that MYCL causes MET by regulating the expression of proteins involved in the promotion of reprogramming from the RNA-processing stage. Further elucidation of the function of MYCL in reprogramming will improve the quality and efficiency of iPSC generation.
Material and methods
Cell culture
HDFs (106-05f.) were purchased from Cell Applications, Inc. HDFs were cultured in DMEM (08459-64, Nacalai Tesque) supplemented with 10% FBS (10439-024, gibco) and 1% penicillin and streptomycin (15140-122, Pen/Strep, gibco). The hiPSC clone 201B7 was used in this study2. iPSCs were cultivated on iMatrix-511 (NP892-012, Nippi)-coated (0.5 μg/cm2) cell culture plates with StemFit (AK03N, Ajinomoto) supplemented with bFGF and passaged via dissociation into single cells using TrypLE Select (A12859-01, Life Technologies) on day 7 following a previously reported protocol42.
Generation of iPSCs
A frozen stock of HDFs was thawed and cultured for four days, and then 1 × 105 cells were collected by trypsinization. With SeV, HDFs were transduced with the CytoTune-iPS 2.0 (c-MYC) or CytoTune-iPS 2.0L (MYCL) Sendai Reprogramming Kit (DV-0304, DV-0305, ID Pharma). With EpiP, HDFs were electroporated with 1.2 μg of plasmid mixtures with the Neon Transfection System (MPK1096 and MPK10096, Invitrogen). The plasmid mixtures included pCXLE-SOX2, -KLF4, -OCT3/4-shp53, -LIN28A, and pCXWB-EBNA1 with wild-type or mutant pCXLE-c-MYC or -MYCL17. The mixing ratio of SOX2, KLF4, OCT3/4-shp53, LIN28A, EBNA1, and c-MYC/MYCL was 1:1:2:1:0.5:2. After that, the cells were plated in a 6-well plate and cultured in StemFit AK03N without bFGF with iMatrix-511 at 0.25 μg/cm2 in SeV or 0.125 μg/cm2 in EpiP. The culture medium was changed the next day and every three days after that. The colonies were counted 21 days after plating.
Episomal plasmid vector construction for deletion mutants of c-MYC and MYCL
We previously generated pCXLE-c-MYC and -MYCL from human cDNAs encoding c-MYC and MYCL amplified by PCR and cloned into pENTR1A17. Primers for the deletion mutants were designed using the Primer Design tool for the In-Fusion HD Cloning Kit (639650, Takara) and inserted into pENTR1A. The switch from pENTR1A to pCXLE was done using the Gateway system (11791020, Invitrogen). The primers used are listed in Table S1.
Immunostaining
The cells were fixed with 4% formaldehyde (163-20145, Wako) for 20 min at room temperature. Then, the fixed cells were treated with PBS (14249-24, Nacalai Tesque) containing 0.5% Triton X-100 (35501-15, Nacalai Tesque) and 3% bovine serum albumin (01281-84, BSA, Nacalai Tesque) for 20 min at room temperature for permeabilization. The cells were incubated with primary antibodies diluted in PBS containing 3% BSA at 4℃ overnight. After washing with PBS, the cells were incubated with fluorescence-conjugated secondary antibodies for 1 h at room temperature. Nuclei were visualized with Hoechst 33342 (346-07951, DOJINDO). Anti-TRA-1-60 (1:500, 560071, BD Pharmingen, and 1:500, 09-0068, Stemgent) and Alexa 488-conjugated goat anti-mouse IgG, IgM (H + L) (1:250, A10680, Invitrogen) were used as the antibodies.
Imaging and quantification
Stained cells were imaged using a BZ-9000 imaging system (KEYENCE) or ArrayScan High-Content Systems (Thermo Fisher Scientific). HCS Studio 2.0 Cell Analysis Software (Thermo Fisher Scientific) was used to quantify cell counts and signal intensities. The Cellomics BioApplication system (Thermo Fisher Scientific) was programmed to capture and analyze 25 images per well. The total cell number was detected by Hoechst 33342 staining. The number of TRA-1-60 ( +) cells was calculated as the number of TRA-1-60 ( +) cells among Hoechst ( +) cells. TRA-1-60 ( +) cells were calculated by dividing this number by the total cell number.
Flow cytometry
Transduced cells were harvested with 0.25% trypsin/1 mM EDTA (25200-056, gibco) each day after the transduction for the analysis. At least 5 × 104 cells were stained with the following antibodies in FACS buffer (2% FBS, 0.36% glucose (16806-25, Nacalai Tesque), 50 μg/μL Pen/Strep in PBS) for 30 min at room temperature: BV510-conjugated anti-TRA-1-60 (1:40, 563188, BD Biosciences) and PE-Cy7-conjugated anti-CD13 (1:40, 561599, BD Biosciences) antibodies. The analysis was performed using MACSQuant Analyzers (Miltenyi Biotec). Negative controls used a mixture of HDFs without any EpiP transduction and reprogramming HDFs electroporated with EpiP including c-MYC or MYCL. “Isotype” means mixed HDFs stained with the isotype control of anti-TRA-1-60 (1:40, 563082, BD Biosciences) and -CD13 (1:40, 557646, BD Biosciences) antibodies.
SDS-PAGE
Cells were lysed with SDS sample buffer (0.125 M Tris-base (35434-21, Nacalai Tesque), 0.96 M glycine (17109-35, Nacalai Tesque), and 17.3 mM SDS (31606-75, Nacalai Tesque)) containing 3-mercaptoethanol (139-16452, Wako). Samples were applied and separated in an 8% polyacrylamide gel composed of 30% (w/v)-Acrylamide/Bis Mixed Solution (29:1) (06141-35, Nacalai Tesque), Separating Gel Buffer Solution (4x) (30651-05, Nacalai Tesque) and Stacking Gel Buffer Solution (4x) (32158-25, Nacalai Tesque) for SDS-PAGE.
Western blotting
Proteins on an SDS-PAGE gel were transferred to a PVDF membrane (IPVH00010, Immobilon-P, Millipore) and probed with the following antibodies using an iBind Flex system (SLF2000, SLF2010 and SLF2020, Invitrogen): anti-human MYCL (1:250, AF4050, R&D) (1:250, C-20, sc-790, Santa Cruz), anti-human c-MYC (1:500, 9E10, sc-40, Santa Cruz, and 1:500, D84C12, CST), anti-β-actin (1:1000, A5441, SIGMA), anti-Goat (1:3000, ab6741-1, abcam), anti-mouse (1:3000, 7076S, CST), and anti-rabbit (1:3000, 7074S, CST) antibodies.
Preparation of recombinant proteins and affinity purification (AP)
The MB2 region of MYCL-WT or -W96E was cloned into pGEX-6P-1. The plasmids were transformed into BL21 E. coli (DE3) (L1198, Promega) competent cells. The fusion proteins, GST-MYCL-WT-MB2 and GST-MYCL-W96E-MB2, were induced by treatment with 0.5 mM IPTG (19742-94, Nacalai Tesque) for 4 h at 37 °C. The proteins were purified using glutathione Sepharose beads (17-0756-01, GE Healthcare). Human iPSCs or reprogramming HDFs were lysed in RIPA buffer (20 mM Tris/HCl (pH 7.6) (35436-01, Nacalai Tesque), 1% NP-40 (25223-75, Nacalai Tesque), 0.1% SDS, 150 mM NaCl (31320-05, Nacalai Tesque), and protease inhibitor (25955-11, Nacalai Tesque)) and then centrifuged. Cell lysates (supernatant) were transferred into a column (29922, Thermo Fisher Scientific) packed with beads conjugated with GST- MYCL-WT or -W96E proteins. After washing, binding proteins were eluted in lysis buffer (12 mM sodium deoxycholate (190-08313, Wako), 12 mM sodium lauroyl sarcosinate (192-10382, Wako), and 100 mM Tris-HCl (pH9.0) (314-90381, NIPPON GENE)) for the MS analysis. The iPSC lysates were prepared 6 days after passaging in two 10-cm dishes (n = 1), and reprogramming HDF lysates were prepared 3 days after SeV transduction in five 10-cm dishes (n = 1).
GO analysis by DAVID
The Database for Annotation, Visualization, and Integrated Discovery (DAVID Bioinformatics Resources 6.8) was used to identify enriched biological GO terms and KEGG pathway43,44,45. For more information, please visit the DAVID website (https://david.ncifcrf.gov/home.jsp) and KEGG Database website (https://www.kegg.jp/kegg/kegg1.html).
The methods for MS are described in the Supplementary methods.
References
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Azuma, K. & Yamanaka, S. Recent policies that support clinical application of induced pluripotent stem cell-based regenerative therapies. Regen. Ther. 4, 36–47 (2016).
Huang, C.-Y. et al. Human iPSC banking: barriers and opportunities. J. Biomed. Sci. 26, 87 (2019).
Moradi, S. et al. Research and therapy with induced pluripotent stem cells (iPSCs): social, legal, and ethical considerations. Stem Cell Res. Ther. 10, 341 (2019).
Koyanagi-Aoi, M. et al. Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells. Proc. Natl. Acad. Sci. 110, 20569–20574 (2013).
Nakagawa, M., Takizawa, N., Narita, M., Ichisaka, T. & Yamanaka, S. Promotion of direct reprogramming by transformation-deficient Myc. Proc. Natl. Acad. Sci. 107, 14152–14157 (2010).
Baluapuri, A., Wolf, E. & Eilers, M. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol. 21, 255–267 (2020).
Varmus, H. E. The molecular genetics of cellular oncogenes. Annu. Rev. Genet. 18, 553–612 (1984).
Oster, S. K., Mao, D. Y. L., Kennedy, J. & Penn, L. Z. Functional analysis of the N-terminal domain of the Myc oncoprotein. Oncogene 22, 1998–2010 (2003).
Beaulieu, M.-E., Castillo, F. & Soucek, L. Structural and biophysical insights into the function of the intrinsically disordered myc oncoprotein. Cells 9, 1038 (2020).
Amati, B. et al. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 359, 423–426 (1992).
Zhang, Q. et al. MB0 and MBI are independent and distinct transactivation domains in MYC that are essential for transformation. Genes 8, 134 (2017).
Kalkat, M. et al. MYC protein interactome profiling reveals functionally distinct regions that cooperate to drive tumorigenesis. Mol. Cell 72, 836-848.e7 (2018).
Fusaki, N., Ban, H., Nishiyama, A., Saeki, K. & Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 85, 348–362 (2009).
Tanabe, K., Nakamura, M., Narita, M., Takahashi, K. & Yamanaka, S. Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proc. Natl. Acad. Sci. 110, 12172–12179 (2013).
Okita, K. et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 8, 409–412 (2011).
Xing, Q. R. et al. Diversification of reprogramming trajectories revealed by parallel single-cell transcriptome and chromatin accessibility sequencing. Sci. Adv. 6, eaba1190 (2020)
Wolf, E. & Eilers, M. Targeting MYC proteins for tumor therapy. Annu. Rev. Cancer Biol. 4, 61–75 (2020).
Schwanhäusser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl. Acids Res. 37, 1–13 (2009).
Wasylishen, A. R. et al. New model systems provide insights into Myc-induced transformation. Oncogene 30, 3727–3734 (2011).
Okita, K., Ichisaka, T. & Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 448, 313–317 (2007).
Yu, J. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007).
Sone, M. et al. Hybrid cellular metabolism coordinated by Zic3 and Esrrb synergistically enhances induction of naive pluripotency. Cell Metab. 25, 1103-1117.e6 (2017).
Maekawa, M. et al. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 474, 225–229 (2011).
Zahn-Zabal, M. et al. The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucl. Acids Res. 48, D328–D334 (2020).
Rodriguez-Boulan, E. & Macara, I. G. Organization and execution of the epithelial polarity programme. Nat. Rev. Mol. Cell Biol. 15, 225–242 (2014).
Li, R. et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63 (2010).
Sakurai, K. et al. Kinome-wide functional analysis highlights the role of cytoskeletal remodeling in somatic cell reprogramming. Cell Stem Cell 14, 523–534 (2014).
Pei, D., Shu, X., Gassama-Diagne, A. & Thiery, J. P. Mesenchymal–epithelial transition in development and reprogramming. Nat. Cell Biol. 21, 44–53 (2019).
Bonomi, S. et al. HnRNP A1 controls a splicing regulatory circuit promoting mesenchymal-to-epithelial transition. Nucl. Acids Res. 41, 8665–8679 (2013).
Price, D. H. Regulation of RNA polymerase II elongation by c-Myc. Cell 141, 399–400 (2010).
Welcker, M. et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. 101, 9085–9090 (2004).
Tamkun, J. W. et al. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 46, 271–282 (1986).
Hynes, R. Integrins: A family of cell surface receptors. Cell 48, 549–554 (1987).
Närvä, E. et al. A strong contractile actin fence and large adhesions direct human pluripotent colony morphology and adhesion. Stem Cell Rep. 9, 67–76 (2017).
Santoro, R., Perrucci, G. L., Gowran, A. & Pompilio, G. Unchain My heart: integrins at the basis of iPSC cardiomyocyte differentiation. Stem Cells Int. 2019, 1–20 (2019).
Hansson, J. et al. Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell Rep. 2, 1579–1592 (2012).
Ohta, S., Nishida, E., Yamanaka, S. & Yamamoto, T. Global splicing pattern reversion during somatic cell reprogramming. Cell Rep. 5, 357–366 (2013).
Nakagawa, M. et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci. Rep. 4, 3594 (2014).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: integrating viruses and cellular organisms. Nucl. Acids Res. 49, D545–D551 (2021).
Acknowledgements
This research was supported by AMED under Grant Number JP21bm0104001 and a grant from the Fujiwara Memorial Incorporated Foundation. We thank Dr. C. Okubo, Dr. H. Kagawa, Dr. T. Yamakawa, Dr. K. Okita, and Dr. K. Takahashi for scientific discussions; Dr. A. Ohta and Dr. Y. Nishi for technical assistance with the ArrayScan analysis; and Dr. P. Karagiannis for reading the manuscript.
Author information
Authors and Affiliations
Contributions
C.A. and M.N. wrote the main manuscript text. C.A. mainly prepared all figures (supported by C.S., Y.C., T.I., and M.N.). Proteome analysis was done by Y.K. and M.I.. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.I. is a scientific adviser (without salary) of xFOREST therapeutics. Other authors do not provide a competing interest statement.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Akifuji, C., Iwasaki, M., Kawahara, Y. et al. MYCL promotes iPSC-like colony formation via MYC Box 0 and 2 domains. Sci Rep 11, 24254 (2021). https://doi.org/10.1038/s41598-021-03260-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03260-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
