
Abstract
Neurotropic viruses target the brain and contribute to neurologic diseases. C-type lectin receptors (CLRs) are pattern recognition receptors that recognize carbohydrate structures on endogenous molecules and pathogens. The myeloid CLR dendritic cell immunoreceptor (DCIR) is expressed by antigen presenting cells and mediates inhibitory intracellular signalling. To investigate the effect of DCIR on neurotropic virus infection, mice were infected experimentally with Theiler’s murine encephalomyelitis virus (TMEV). Brain tissue of TMEV-infected C57BL/6 mice and DCIR−/− mice were analysed by histology, immunohistochemistry and RT-qPCR, and spleen tissue by flow cytometry. To determine the impact of DCIR deficiency on T cell responses upon TMEV infection in vitro, antigen presentation assays were utilised. Genetic DCIR ablation in C57BL/6 mice was associated with an ameliorated hippocampal integrity together with reduced cerebral cytokine responses and reduced TMEV loads in the brain. Additionally, absence of DCIR favoured increased peripheral cytotoxic CD8+ T cell responses following TMEV infection. Co-culture experiments revealed that DCIR deficiency enhances the activation of antigen-specific CD8+ T cells by virus-exposed dendritic cells (DCs), indicated by increased release of interleukin-2 and interferon-γ. Results suggest that DCIR deficiency has a supportive influence on antiviral immune mechanisms, facilitating virus control in the brain and ameliorates neuropathology during acute neurotropic virus infection.
Similar content being viewed by others


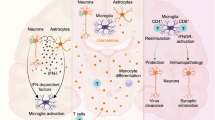
Introduction
Neurotropic viruses target the brain and can cause asymptomatic or acute and fatal diseases1,2. Moreover, cognitive deficits and memory impairment, suggestive of hippocampal dysfunction, as well as an increased risk of developing epilepsy are often observed in patients surviving acute viral encephalitis3,4,5.
Theiler’s murine encephalomyelitis virus (TMEV) is a neurotropic picornavirus that preferentially infects the hippocampus6,7,8. While TMEV persists in the central nervous system (CNS) of SJL mice, C57BL/6 mice eliminate the virus following acute polioencephalitis. C57BL/6 mice mount early robust antiviral immune responses, but are prone to develop hippocampal injury with neuronal loss during the acute infection phase1,9. TMEV infection was shown to increase the susceptibility to develop seizures in C57BL/6 mice6,7. Moreover, neuronal damage is associated with impaired cognition and spatial memory, making TMEV infection a valuable model for brain damage in neurotropic virus infections10,11,12. Innate immune responses during the initial phase significantly contribute to the development of antiviral T cell responses and virus elimination in TMEV-infected mice13,14,15. However, CNS-infiltrating macrophages and activated microglia also account for hippocampal degeneration following TMEV infection by releasing pro-inflammatory factors16,17,18,19,20,21. Surveillance of virus infection and initiation of innate immune responses are mediated by pattern recognition receptors (PRRs) on professional antigen presenting cells (APCs), such as dendritic cells (DCs). C-type lectin receptors (CLRs) are PRRs that recognise a variety of glycan structures present on pathogens, including viruses, damage-associated molecular patterns and self-glycoproteins22,23,24,25,26,27.
The C-type lectin receptor Dendritic cell immunoreceptor (DCIR, human gene: CLEC4A, murine gene: Clec4a2) contains an immunoreceptor tyrosine-based inhibition motif (ITIM), which delivers inhibitory signals predominantly in DCs28,29,30,31. Thus, DCIR is a negative regulator of intracellular signalling of APCs, including microglia, and contributes to immune homeostasis in immune mediated disorders31,32,33,34. However, DCIR signalling seems to play an ambiguous role in infectious diseases30,35,36. For instance, while DCIR signalling limits immunopathology in Chikungunya virus-infected mice, the receptor is thought to trigger brain pathology in cerebral malaria models37,38. The role of DCIR signalling in neurotropic virus infections and its impact on neuropathology have not yet been investigated.
The aim of the present study was to investigate DCIR-mediated effects on the balance of immune responses, virus load and neuropathology in Theiler’s murine encephalomyelitis (TME). Genetic ablation of DCIR indicates that receptor signalling contributes to neuroinflammation and brain injury in C57BL/6 mice following TMEV infection. DCIR−/− mice show an improved hippocampal integrity and are able to control neurotropic virus infection more efficiently. In vitro studies reveal that DCIR deficiency enhanced T cell activation in a dendritic cell/T cell co-culture system.
Results
DCIR−/− mice show preservation of hippocampal integrity and reduced viral load in the brain
The effect of DCIR deficiency on hippocampal and neuronal integrity following TMEV infection was determined (Fig. 1). Two-way ANOVA yielded a significant effect of DCIR deficiency on neuronal integrity determined by histology (HE score; p = 0.0008) and immunohistochemistry (NeuN+ area/mm2, p = 0.0006), as well as on TMEV load (TMEV+ cells/mm2, p = 0.0257). Subsequent Mann–Whitney U tests at different time points post infection revealed a diminished hippocampal damage of infected DCIR−/− mice with a significant difference compared to WT mice at 14 dpi (p = 0.005, Fig. 1a–c). Similarly, a significantly reduced loss of NeuN+ neurons in the hippocampus of DCIR−/− animals compared to WT controls was found at 14 dpi (p = 0.002, Fig. 1d–f). Although increased numbers of β-APP+ axons (damaged axons) were found in hippocampal regions with severe neuronal damage and loss mainly in TMEV-infected WT mice, group differences did not reach the level of significance (Supplementary Fig. S1a). Likewise, an elevation of GFAP+ astrocytes (astrogliosis) was present within the hippocampus of WT animals compared to DCIR−/− mice at 14 dpi, but differences did not reach the level of significance (Supplementary Fig. S1b). No hippocampal inflammation and damage were found in non-infected, age-matched WT and DCIR−/− animals. In addition, non-infected groups showed a similar amount of NeuN+ neurons and GFAP+ astrocytes in the hippocampus (Supplementary Fig. S5).

Neuropathology and neuronal loss within the hippocampus following Theiler’s murine encephalomyelitis virus (TMEV) infection. (a–c) Diminished hippocampal damage in DCIR−/− mice. (a, b) Representative images from 14 dpi show neuronal loss (arrows) and perivascular inflammatory infiltrates (asterisk) in the hippocampus of wild type (WT) and DCIR−/− mice (hematoxylin and eosin staining). (d–f) Decreased neuronal loss in DCIR−/− mice confirmed by NeuN-specific immunohistochemistry. (d, e) Representative images from 14 dpi show neuronal loss of hippocampal pyramidal layer (arrows) in WT and DCIR−/− mice. (c, f) Statistical analysis: Mann–Whitney U test (statistical differences: *p ≤ 0.05, **p ≤ 0.01); data are shown as mean with SEM. n: 7 dpi = 9 WT and 12 DCIR−/− mice; 14 dpi = 10 WT and 9 DCIR−/− mice. (a, b, d, e) 200 × magnification; dpi days post infection.
Viral quantification within the brain was performed by RT-qPCR and TMEV-specific immunohistochemistry. At 7 dpi, TMEV RNA concentration was significantly decreased in DCIR−/− mice compared to WT mice (p = 0.047, Fig. 2a). Immunohistochemistry revealed a preferential infection of hippocampal neurons of infected mice in both groups at 7 dpi. Similar to the diminished viral RNA load, reduced numbers of TMEV-infected cells were observed in the brain of DCIR−/− mice at 7 dpi, but differences did not reach level of significance (p = 0.12, Fig. 2b). Both, WT and DCIR−/− mice, showed reduced viral RNA levels and TMEV antigen at 14 dpi, indicating viral elimination (Fig. 2a,b). TMEV RNA concentration in the brain did not differ significantly between both groups at 14 dpi (p = 0.87, Fig. 2b). However, the number of TMEV+ cells within the hippocampus was significantly reduced at 14 dpi in DCIR−/− mice compared to WT mice, indicating a reduced residual infection following acute infection phase in DCIR deficient animals (p = 0.005, Fig. 2b–d). No TMEV was detected in non-infected WT mice and DCIR−/− mice by immunohistochemistry and RT-qPCR (data not shown). Data show that preserved hippocampal morphology in mice lacking DCIR is associated with an enhanced early virus elimination from the brain, indicating a refined induction of protective responses in DCIR−/− mice following TMEV infection.

Theiler’s murine encephalomyelitis virus (TMEV) infection in the brain. (a) Quantification of TMEV RNA concentrations by reverse transcriptase quantitative polymerase chain reaction. (b–d) Quantification of the TMEV+ area within the hippocampus by immunohistochemistry. Representative images show TMEV antigen (arrows) in the hippocampus of (c) wild type (WT) and (d) DCIR−/− mice at 14 dpi. (a, b) Statistical analysis: Mann–Whitney U test (statistical differences: *p ≤ 0.05, **p ≤ 0.01); data are shown as mean with SEM. n: 7 dpi = 9 WT and 12 DCIR−/− mice; 14 dpi = 10 WT and 9 DCIR−/− mice. (c, d) 600 × magnification; dpi days post infection.
Weekly clinical examination, body weight recordings, Racine score evaluation and RotaRod performance test revealed no symptoms in both groups, indicating a subclinical acute infection (Supplementary Fig. S4)39.
DCIR deficiency leads to diminished brain sequestration of effector immune cells
Immunohistochemistry revealed a reduced infiltration of CD3+ T cells (p = 0.007, Fig. 3a) and CD45R+ B cells (p = 0.005, Fig. 3b) in the hippocampus of DCIR−/− mice compared to WT controls at 14 dpi. At both time points, the hippocampus of DCIR−/− mice contained similar numbers of activated CD107b+ macrophages/microglia in comparison to WT controls (Fig. 3c).

Phenotyping of lymphocyte subsets in the hippocampus following Theiler’s murine encephalomyelitis virus (TMEV) infection. Quantification of (a) CD3+ T cells, (b) CD45R+ B cells, (c) CD107b+ macrophages/microglia, (d) GrB+ cells, (e) CD4+ T cells, and (f) CD8+ T cells in the hippocampus of infected wild type (WT) and DCIR−/− mice, and (g) ratio of CD4+ T cells to CD8+ T cells in the cerebrum determined by immunohistochemistry. (a–g) Statistical analysis: Mann–Whitney U test (statistical differences: *p ≤ 0.05, **p ≤ 0.01); data are shown as mean with SEM (a–f) and box plots display median with 5–95% percentiles (g). n: 7 dpi = 9 WT and 12 DCIR−/− mice; 14 dpi = 10 WT and 9 DCIR−/− mice. dpi days post infection.
Brain-infiltrating GrB+ effector cells decreased in DCIR−/− mice at 14 dpi (p = 0.05, Fig. 3d). Moreover, reduced numbers of CD4+ and CD8+ T cells were found in the hippocampus of DCIR−/− mice at 14 dpi (CD4+ T cells: p = 0.002, Fig. 3e, CD8+ T cells: p = 0.011, Fig. 3f), likely related to decreased virus-trigged immune responses in receptor deficient animals. Comparison of CD4+ and CD8+ T cell proportions revealed a slightly reduced ratio of CD4+ to CD8+ T cells in the brain of DCIR−/−mice at 14 dpi (p = 0.00045, Fig. 3g), showing a relative increase of cytotoxic T cells in animals lacking DCIR. Statistical analyses (Pearson’s correlation coefficient R) revealed negative correlations between neuronal integrity of the hippocampus (NeuN+ area/mm2) and the amount of CD107b+, arginase 1+ and CD45R+ cells at 7 and 14 dpi. Foxp3+ cells and GFAP+ astrocytes were negatively correlated with neuronal integrity at 14 dpi (Supplemental Table S6).
Non-infected control animals of both groups showed no leukocyte infiltrations in the hippocampus.
Collectively, these findings suggest that the reduced viral brain load in DCIR−/− mice leads to an accelerated termination of brain inflammatory responses.
Reduced cerebral cytokine expression in DCIR−/− mice is associated with preserved hippocampal integrity
At 7 dpi, mRNA levels of IL-1α (p = 0.047, Fig. 4a), TNF-α (p = 0.039, Fig. 4b) and IFN-β (p = 0.009, Fig. 4c) were significantly reduced in TMEV-infected DCIR−/− animals compared to the WT group. Data show a reduced pro-inflammatory response to virus infection due to receptor deficiency during the early acute polioencephalitis phase (7 dpi). In addition to mRNA levels of IL-1α (p = 0.018, Fig. 4a), TNF-α (p = 0.022, Fig. 4b), and IFN-β (p = 0.034, Fig. 4c), a significantly lower IFN-γ transcription was detected in DCIR−/− animals compared to WT mice also at 14 dpi (p = 0.027, Fig. 4d). The mRNA levels of the pro-inflammatory cytokines IL-1β, IL-2, IL-6 and IL-23, as well as the anti-inflammatory cytokines IL-4, IL-5 and TGF-β1 did not differ significantly between both groups (Supplementary Fig. S2). Non-infected DCIR−/− mice showed a significantly lower base line mRNA expression of IL-5 compared to WT mice (p = 0.021, Supplementary Fig. S5). Other cytokines mRNA levels of non-infected controls showed no differences between DCIR−/− and WT animals (Supplementary Fig. S5). Statistical analyses (Pearson’s correlation coefficient R) revealed negative correlations between neuronal integrity of the hippocampus (NeuN+ area/mm2) and mRNA expression levels of IFN-β and IL-1β, and a positive correlation with IL-2 at 7 dpi. IFN-γ was negatively correlated with neuronal integrity at 14 dpi (Supplemental Table S6).

Cytokine expression within the cerebrum of mice infected with Theiler’s murine encephalomyelitis virus. Quantification of (a) interleukin (IL)-1α, (b) tumor necrosis factor (TNF)-α, (c) interferon (IFN)-β and (d) IFN-γ mRNA in the cerebrum of wild type (WT) and DCIR−/− mice by reverse transcriptase quantitative polymerase chain reaction. (a–d) Statistical analysis: Mann–Whitney U test (statistical differences: *p ≤ 0.05, **p ≤ 0.01); data are shown as mean with SEM. n: 7 dpi = 9 WT and 12 DCIR−/− mice; 14 dpi = 10 WT and 9 DCIR−/− mice. dpi days post infection.
Reduced pro-inflammatory cytokine expression in the brain at the later stage of acute polioencephalitis (14 dpi) in DCIR−/− mice is likely a direct consequence of reduced viral burden and accelerated termination of neuroinflammation in comparison to WT mice.
Diminished induction of immunomodulatory responses in DCIR−/− mice following neurotropic virus infection
The suppressive cytokine interleukin-10, secreted by regulatory T cells (Treg) and M2-type macrophages/microglia, is thought to exhibit neuroprotective effects in infectious disorders40. On the other hand, Treg and M2-type myeloid cells may dampen effective antiviral responses and thus promote deleterious effects on tissue integrity41,42,43. In order to test whether neuronal preservation in the hippocampus in DCIR−/− mice is accompanied by reduced virus load or attributed to immunomodulatory mechanisms, Foxp3+ Treg, arginase 1+ M2-type macrophages/microglia and the key immunomodulatory cytokine IL-10 were quantified. At 14 dpi, numbers of Foxp3+ regulatory T cells (p = 0.018, Fig. 5a) and Foxp3 mRNA copy numbers (p = 0.009, Fig. 5b), determined by immunohistochemistry and RT-qPCR, respectively, were significantly increased in brain samples of mice with intact DCIR signalling (WT mice) compared to DCIR−/− mice upon infection. Similarly, an increase of arginase 1+ cells was found in the hippocampus of WT mice compared to DCIR−/− mice at both investigated time points with significant differences at 14 dpi (p = 0.006, Fig. 5c). Moreover, significantly increased transcription of IL-10 was detected in WT controls compared to DCIR−/− mice at 14 dpi (p = 0.034, Fig. 5d). In non-infected WT and DCIR−/− mice cerebral IL-10 mRNA expression was not detectable.

Quantification of arginase 1+ macrophages/microglia, Foxp3+ regulatory T cells, and interleukin 10 in the brain of Theiler’s murine encephalomyelitis virus infected mice. Quantification of (a) Foxp3+ regulatory T cells by immunohistochemistry, (b) Foxp3 mRNA transcription by reverse transcriptase quantitative polymerase chain reaction, (c) arginase 1+ M2-type macrophages/microglia by immunohistochemistry, (d) interleukin (IL)-10 mRNA transcription by reverse transcriptase quantitative polymerase chain reaction in wild type (WT) and DCIR−/− mice. (a–d) Statistical analysis: Mann–Whitney U test (statistical differences: *p ≤ 0.05, **p ≤ 0.01); data are shown as mean with SEM. n: 7 dpi = 9 WT and 12 DCIR−/− mice; 14 dpi = 10 WT and 9 DCIR−/− mice. dpi days post infection.
Results suggest that TMEV infection elicits compensatory immune pathways, mediated by Treg and M2-type macrophages/microglia in the brain of DCIR intact C57BL/6 mice. Consequently, the observed neuroprotective effect of DCIR deficiency is likely not mediated by classical immunomodulatory mechanisms at the infection site, but rather due to improved virus elimination and timely onset of peripheral protective immune responses. In addition, diminished induction of immunomodulatory and suppressive mechanisms, including decreased numbers of arginase 1+ M2-type macrophages/microglia and Treg, may promote virus control in DCIR deficient animals.
DCIR deficiency enhances peripheral T cell responses following neurotropic virus infection
The accelerated TMEV elimination observed in the brain of DCIR−/− mice suggests an enhanced antiviral immune response. Priming of naïve T cells in lymphoid organs during the early phase of TME was shown to be crucial for robust antiviral responses44,45. Therefore, splenic cytokine mRNA expression was quantified by RT-qPCR and splenic T cell responses were analysed by flow cytometry.
At 7 dpi, splenic IFN-γ mRNA levels were significantly increased in TMEV-infected DCIR−/− animals compared to the WT group (p = 0.049, Fig. 6a), indicating an enhanced pro-inflammatory immune response. At 14 dpi, IL-1α (p = 0.006, Fig. 6b) and IL-1β (p = 0.001, Fig. 6c) mRNA expression was reduced in TMEV-infected DCIR−/− animals compared to WT mice, likely related to the reduced viral burden and accelerated termination of neuroinflammation in DCIR deficient mice. Splenic IL-2, IL-4, IL-5, IL-6, IL-10, IL-23, IFN-β, TGFβ1, and TNF-α mRNA levels did not differ significantly between both groups (Supplementary Fig. S3).

Splenic cytokine expression (a–c) and flow cytometric analysis of splenic T cells (d–m) in Theiler’s murine encephalomyelitis virus-infected DCIR−/− and wild type (WT) mice. Quantification of (a) interferon (IFN)- γ, (b) interleukin (IL)-1α, (c) IL-1β mRNA in the spleen of wild type (WT) and DCIR−/− mice by reverse transcriptase quantitative polymerase chain reaction. (d) Gating strategy of splenic T cells. Here, the gating for CD4+CD62Llow T helper cells is shown. The same procedure was used for all activation markers. Percentages of (e) CD4+ T helper cells, (f) CD8+ cytotoxic T cells, (g) ratio of CD4+ T helper cells to CD8+ cytotoxic T cells, (h) CD4+CD25+ T helper cells, (i) CD4+CD44+ T helper cells, (j) CD4+CD62Llow T helper cells, (k) CD8+CD25+ cytotoxic T cells, (l) CD8+CD44+ cytotoxic T cells and (m) CD8+CD62Llow cytotoxic T cells in spleen samples. (a–c, e–m) Statistical analysis: Mann–Whitney U test (statistical differences: *p ≤ 0.05, **p ≤ 0.01); data are shown as mean with SEM (a–c, e–f, h–m) and box plots display median with 5–95% percentiles (g). (a–c) n: 7 dpi = 9 WT and 12 DCIR−/− mice; 14 dpi = 10 WT and 9 DCIR−/− mice. (e–m) n: 5 mice per group and time point. dpi days post infection.
Flow cytometric analysis of TMEV-infected groups revealed an increased fraction of CD8+ T cells in the spleen of DCIR−/− mice compared to WT group at 7 dpi (p = 0.016, Fig. 6f), while CD4+ T cell frequency remained unchanged at this time point (Fig. 6e). Accordingly, a significant shift of the CD4+/CD8+ T cell ratio with dominance of cytotoxic T cells was observed at 7 dpi (p = 0.009, Fig. 6g) as well as at 14 dpi (p = 0.028, Fig. 6g). Subsequently, the portion of splenic CD4+ T cells decreased in DCIR−/− mice during the disease course, resulting in a significant difference between the groups at 14 dpi (p = 0.028, Fig. 6e).
Further characterization of splenic T cell subsets revealed a significantly higher proportion of activated CD4+ T cells, displayed by higher fractions of CD4+CD62Llow T cells in DCIR−/− mice compared to WT mice at 7 dpi (p = 0.016) and 14 dpi (p = 0.047, Fig. 6d,j). Moreover, the level of activated CD4+CD44+ T cells was elevated at both time points in DCIR−/− animals compared to WT mice with statistically significant difference between the groups at 7 dpi (p = 0.047, Fig. 6i). Similarly, CD8+CD44+ cell populations were significantly increased in DCIR−/− animals compared to WT mice at 7 dpi (p = 0.009) and 14 dpi (p = 0.016, Fig. 6l). Splenic CD4+- and CD8+ T cell subpopulations expressing CD25 as well as CD8+CD62Llow T lymphocytes did not significantly differ between groups at any time point (Fig. 6h,k,m). Flow cytometry of non-infected control mice showed a slight difference for the portion of CD8+ T cells and the CD4+/CD8+ T cell ratio comparing the spleens of WT and DCIR−/− mice. However, no major differences in surface expression of T cell activation markers were observed between both non-infected control groups (Supplementary Fig. S6). Thus, flow cytometry and cytokine expression analysis revealed an enhanced peripheral T cell activation and an early proportional shift towards cytotoxic CD8+ T cell responses in DCIR−/− mice during viral encephalitis.
Identification of potential influencing factors for hippocampal damage using regression analyses
Regression analyses were performed to identify factors that correlate with hippocampal damage following TMEV infection. Simple regression models confirmed that neuronal integrity of the hippocampus (NeuN+ area/mm2) was significantly associated with the TMEV load. In addition, hippocampal damage was significantly associated with the amount of CD107b+, CD3+, CD45R+ Foxp3+, arginase 1+, GFAP+, and granzyme B+ cells in the hippocampus, as well as with IFN-β mRNA expression in the brain. The amount of TMEV+ cells in the hippocampus remains the only significant parameter in the multiple regression model, pointing at a high collinearity among explanatory variables (Supplementary Table S5).
Reduced neuroinflammation in DCIR−/− mice following neurotropic virus infection correlates with diminished responses of antigen presenting cells
At 14 dpi, significantly reduced CD11c mRNA copy numbers were detected within the brains (p = 0.017, Fig. 7a) and spleens of DCIR−/− mice (p = 0.035, Fig. 7e). Moreover, CD80 mRNA expression levels were significantly reduced within the brains (p = 0.049, Fig. 7b) and spleens (p = 0.034, Fig. 7f) of DCIR−/− mice in comparison to WT animals at 7 dpi. CD86 mRNA transcript levels were significantly diminished within the brains of DCIR−/− mice in comparison to WT mice at both time points (7 dpi: p = 0.049, 14 dpi: p = 0.001, Fig. 7c). Within the spleen, CD86 mRNA transcript levels showed no differences between DCIR−/− and WT animals (Fig. 7g). MHC-I mRNA expression was significantly reduced in spleens of DCIR deficient mice at 7 dpi (p = 0.049, Fig. 7h), whereas cerebral MHC-I mRNA quantities were significantly decreased in DCIR−/− mice compared to WT animals at 14 dpi (p = 0.003, Fig. 7d). Reduced expression of CD11c and co-stimulatory molecules in the brains and spleens of DCIR−/− mice may be linked to the accelerated resolution of TMEV infection and termination of neuroinflammatory response in comparison to WT mice.

Analysis of CD11c, CD80, CD86 and MHC-I mRNA expression within the cerebrum and spleen of Theiler’s murine encephalomyelitis virus-infected DCIR−/− and wild type (WT) mice. Quantification of (a) CD11c, (b) CD80, (c) CD86, and (d) MHC-I mRNA level within the cerebrum and of (e) CD11c, (f) CD80, (g) CD86 and (h) MHC-I mRNA expression within the spleen by reverse transcriptase quantitative polymerase chain reaction. (a–h) Statistical analysis: Mann–Whitney U test (statistical differences: *p ≤ 0.05, **p ≤ 0.01); data are shown as mean with SEM. n: 7 dpi = 9 WT and 12 DCIR−/− mice; 14 dpi = 10 WT and 9 DCIR−/− mice. dpi days post infection.
Lack of DCIR in bone marrow-derived dendritic cells causes an increased CD8+ T cell response against Theiler’s murine encephalomyelitis virus in vitro
MHC-I-restricted CD8+ cytotoxic T cells are important for TMEV elimination in C57BL/6 mice46. To determine the impact of DCIR deficiency on early T cell responses upon TMEV infection in vitro, antigen presentation assays using WT and DCIR−/− MEGs or BMDCs were performed. T cells were isolated from OT-I TCR-transgenic mice, which specifically recognise the OVA-peptide presented via the MHC-I molecule H2-Kb. T cells were co-cultured with MEGs or BMDCs, previously exposed to TMEV-OVA47,48,49. BMDCs were used for the in vitro stimulation of OT-I T cells since BMDCs from WT and DCIR−/− mice had been previously compared in a global and unbiased manner through genome-wide transcriptome analysis and thus represent a well characterized source of APCs36. To analyse CD8+ T cell activation, cytokine release was measured by ELISA, and expression of the early T cell activation marker CD69 was measured by flow cytometry. Microglia, as part of the glial cell mixtures, represent the CNS’ local APC population. Incubating MEGs with TMEV-OVA, however, did not result in a difference between WT or DCIR−/− microglia-mediated CD8+ T cell response (Fig. 8a). Combined with the marginal levels of released pro-inflammatory cytokines (data not shown), these findings suggest that the potential of microglia to process and present antigens is limited. As an additional source of APCs, BMDCs were used in a co-culture system to stimulate antigen-specific CD8+ T cells. To avoid alterations in TMEV antigenicity and modification of (potential) DCIR ligands, live virus was used for incubation with BMDCs. However, to exclude a productive infection of BMDCs leading to classical antigen presentation via MHC-I molecules, viral RNA loads in TMEV DA-exposed BMDCs as well as viral titers in the supernatant were determined (Supplementary Fig. S7). While an initial increase in viral RNA load in BMDCs between 2 and 6 h was observed (Supplementary Fig. S7a), viral titers in the supernatant decreased continuously from 2 to 22 h after TMEV DA incubation (Supplementary Fig. 7b). Thus, the initial increase of TMEV RNA in BMDCs may be mediated by initial TMEV replication, but it most likely does not reflect a productive infection of BMDCs, but rather an increased TMEV internalisation. In addition, incubation with live TMEV did not lead to a significant decrease in MEG and BMDC viability compared to OVA- or mock-stimulated samples and the vast majority of the cells remained viable (Supplementary Fig. S8), further supporting that BMDCs present viral antigens to CD8+ T cells. Upon TMEV DA incubation, BMDCs were activated, but no difference between WT and DCIR−/− BMDCs was detected (Supplementary Fig. S9). Similarly, the activation status of BMDCs did not differ between WT and DCIR−/− BMDCs following co-cultivation with OT-I T cells (Supplementary Fig. S10). However, TMEV-OVA stimulation of DCIR−/− BMDCs led to an increased expression of CD69 by CD8+ T cells compared to WT BMDCs (Fig. 8b). Further, the release of IL-2, IFN-γ and GrB by CD8+ T cells was elevated if co-cultured with DCIR−/− BMDCs (Fig. 8c–e). These results indicate that DCIR deficiency in BMDCs impacts subsequent CD8+ T cell activation and T cell effector functions in this BMDC/T cell co-culture system. Possibly, DCIR in DCs may balance type I and II IFN signaling directly influencing T cell priming36,50. Additionally, cross-talk of DCIR with other immune receptors, such as Toll-like receptors, is conceivable, which can affect the quality of induced T cell responses even without alterations in the expression of co-stimulatory markers CD80 and CD86, as it was shown for human DCIR51,52,53. However, the mechanism by which the differential CD8+ T cell activation by DCIR−/− BMDCs shown here is mediated, remains to be determined in future studies.

CD8+ T cell response induced by pulsing of microglia-enriched glial cell mixtures (MEGs) or bone marrow-derived dendritic cells (BMDCs) with Theiler’s murine encephalomyelitis virus Daniels strain XhoI-OVA8 (TMEV-OVA) in vitro. Flow cytometric assessment of mean fluorescence intensity (MFI) of activation marker CD69 gated on CD8+ cytotoxic T cells derived from OT-I mice after co-cultivation with (a) MEGs and (b) BMDCs. Analysis of the cytokines (c) interleukin 2 (IL-2), (d) interferon γ (IFN-γ) and (e) granzyme B (GrB) by ELISA following co-cultivation with BMDCs. (a–e) Statistical analysis: Mann–Whitney U test (statistical differences: *p ≤ 0.05); data are shown as mean with SEM. n.d. = not detectable. (a–d) Representative for n = 3. (e) Relative amount of granzyme B compared to OVA control, representative for n = 2.
Discussion
This study highlights the role of DCIR in neuropathology of C57BL/6 mice following acute TMEV infection. Genetic ablation of DCIR appears to exert a supporting effect on viral clearance from the CNS and ameliorates hippocampal damage following virus infection.
While susceptible mouse strains (e.g. SJL mice) show an inefficient antiviral immunity and persistent TMEV infection in the CNS, C57BL/6 mice develop vigorous TMEV-specific responses during acute infection54. The ability of C57BL/6 mice to eliminate TMEV is caused by robust MHC class I-restricted antiviral CD8+ T cell responses39,40,46,55,56,57. As shown in the present study, the lack of DCIR contributes to a more effective priming of peripheral T cells with increased CD44 and reduced CD62L expression by CD4+ T cells together with an increased IFN-γ expression in the spleen during the early phase of TMEV infection. In general, DCIR−/− mice show an age-related increase of CD4+CD44high and CD4+CD62low T cells by expanding DC populations in lymphoid organs, demonstrating that DCIR deficiency predisposes to effector-memory T cell development. CD4+ T cells are required for protective immunity in TMEV infection, since CD4 deficiency has been shown to cause virus persistence in C57BL/6 mice. CD4+ helper T cells support antiviral CD8+ T cell responses by cytokine release (e.g. IL-2) and by improving the ability of DCs to prime cytotoxic T cell responses (DC licensing)33,58,59. An increased frequency of splenic CD8+ T cells together with an upregulation of the activation marker CD44 was found in infected DCIR−/− mice, suggesting an enhancement of cytotoxic CD8+ T cell responses. The skewed ratio of CD4+ to CD8+ T cells observed in DCIR−/− mice indicates an early dominance of peripheral cytotoxic responses. Increased frequencies of circulating CD8+ T cells were shown to improve antiviral immunity and account for TMEV elimination in C57BL/6 mice39,60. In agreement with the present findings, enhanced T cell responses in DCIR−/− mice control experimental mycobacteria infection better than WT controls. Of note, DCs of DCIR−/− mice exhibit several transcriptional changes that promote Th1 immunity also under non-infectious conditions36.
Noteworthy, besides protecting from viral infection, T cell immunity has the ability to contribute to acute brain pathology following TMEV infection. Virus-specific CD8+ T cells target infected neurons of the hippocampus in acutely infected C57BL/6 mice. MHC class I-restricted cytotoxicity towards TMEV epitopes contributes to neuronal loss and brain atrophy57,61,62. Moreover, cytotoxicity boosted by TMEV peptides leads to fatal CNS inflammation in infected C57BL/6 mice, demonstrating the difficulty of balancing immune responses in neurotropic virus infection62,63. As observed in experimental autoimmune encephalomyelitis, rheumatoid arthritis models, and experimental colitis, DCIR−/− mice are prone to develop autoimmunity and T cell-mediated immunopathology, respectively33,64,65. Strikingly, despite enhanced peripheral cytotoxic responses in the present study, no exacerbated brain injury was observed in DCIR−/− mice, but on the contrary, a reduced hippocampal damage following TMEV infection. DCIR deficiency seems to fine-tune protective immune responses without evoking additional virus-mediated immunopathology in the TME model. The underlying mechanisms remain speculative, but might be associated with diminished pro-inflammatory cytokine responses found in the brain of DCIR−/− mice. Reduced expression of IFN-β and TNF-α in the brain of DCIR−/− mice during the early phase of polioencephalitis (7 dpi) indicates a diminished cytokine response at the infection site. Particularly, increased IFN-β mRNA levels were significantly associated with hippocampal damage in TMEV-infected mice as determined by correlation analyses. IFN-β (type I interferon) expression in the brain is driven by TMEV infection and involved in the induction of innate and adaptive immune responses66. Robust antiviral immunity trigged by type I interferons accounts for viral elimination in C57BL/6 mice but also elicit neuronal damage following TMEV infection10. Thus, reduced IFN-β expression might have contributed to diminished T cell sequestration in the brain and decreased hippocampal damage in DCIR−/− mice during advanced infection (14 dpi). Similarly, TNF-α is a cytokine produced by activated microglia and macrophages, which initiate protective responses against certain viral infections, including TMEV infection67,68. However, TNF-α also displays cytotoxic effects and contributes to hippocampal damage in C57BL/6 mice following TMEV infection7,10,20. In addition, TNF-α has been shown to cause excitotoxicity and neuronal damage in murine HIV encephalitis models69. Thus, in addition to the accelerated virus elimination, the alleviated brain cytokine response at the infection site might also contribute to the neuroprotective effect observed in DCIR−/− mice. Despite differences of hippocampal integrity and cytokine expression profiles, no obvious clinical changes were observed between DCIR−/− mice and WT mice (subclinical infection). More targeted diagnostic methods such as video/EEG monitoring and behavioral tests (e.g. Morris water maze) are needed to detect subtle clinical changes and fully discover the functional relevance CNS alteration in receptor deficient animals in future studies.
In addition to an enhanced CD8+ T cell activation in the periphery during the early infection phase, an altered immune environment at the site of infection, including reduced infiltrations of Foxp3+ Treg and arginase 1+ M2-type cells in DCIR−/− animals at 14 dpi might have influenced TMEV control. Consistent with this, a decreased expression of genes specific for M2-type cells (including arginase 1) can be found in DCIR−/− mice following mycobacteria infection36. Moreover, arginase 1+ myeloid cells have been shown to exert suppressive effects on antiviral immunity42. For instance, ablation of arginase 1 in macrophages reduces the viral load and ameliorates tissue integrity after experimental Ross River virus infection of mice43. Similarly, Treg are able to dampen antiviral responses during TMEV infection39.
The interplay between innate and adaptive immunity is mediated by APCs, such as macrophages, microglia and DCs, which have the ability to recognize pathogens and induce effector T cell responses51,70,71. Microglia are CNS-resident APCs and play an important role in TMEV-mediated hippocampal damage and seizure development17. However, within the present study, in vitro TMEV exposure of DCIR−/− and WT MEGs did not show differences in CD8+ T cell activation. Although there was a slight OVA- and TMEV-OVA-mediated increase of CD69 observed in the MEG/T cell co-cultivation assay, cytokine levels were not elevated. Thus, in comparison to DCs, the in vitro potential of adult microglia to perform APC function and present specific antigens is apparently limited, as previously shown72,73.
DCIR is expressed on all DC subsets and exerts mainly inhibitory effects on immune responses via its intracellular ITIM28,30,33,50,74,75. The present study shows an enhanced activation of CD8+ T cells when DCIR−/− BMDCs were used to prime CD8+ T cells. In addition, the release of IL-2 by activated T cells was elevated upon co-cultivation with DCIR−/− BMDCs. DCIR deficiency results also in an increased production of IFN-γ by lymphocytes which was also observed in the present study50. Likewise, Chikungunya virus infection of DCIR−/− mice causes an elevation of IFN-γ in vivo37. However, in contrast to the TME model, intact DCIR signalling in experimental Chikungunya virus infection contributes to protection against virus-induced pathology of the joint, demonstrating that the effect of DCIR signalling on disease progression is clearly context dependent and differs between pathogens and the primarily affected organ in infectious disorders37.
Conclusively, DCIR deficiency seems to support antiviral immune responses of C57BL/6 mice during the initial phase of TMEV infection and to reduce virus-induced neuropathology. Previous studies highlight the potential of DCIR for cell specific targeting and immune modulation51,52,76. Thus, this CLR represents a potential target for intervention strategies to selectively enhance protective immunity in neurotropic virus infection.
Material and methods
Animals
DCIR−/− mice (C57BL/6-Clec4a2tm1.1Cfg/Mmucd; RRID:MMRRC_031932-UCD) were obtained from the National Institutes of Health-sponsored Mutant Mouse Resource & Research Center (MMRRC) National System38. The mouse strain was backcrossed on C57BL/6 background over more than ten generations38. DCIR−/− and respective C57BL/6 mice (WT) were used for the infection experiment. All mice were housed in the animal facility of the University of Veterinary Medicine (Hannover, Germany) in individually ventilated cages under controlled conditions (12 h light/12 h dark cycle, 22–24 °C, humidity 50–60%) with permanent access to water and standard rodent feed. Animal experiments were conducted in accordance with the German law for animal protection and the Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for scientific purposes and the ARRIVE guidelines77. The study was approved and authorized by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES), Oldenburg, Germany (permission number 33.19-42502-04-16/2225, date of approval: October 7, 2016).
Virus and cell lines
For intracerebral injection, the Daniels strain of TMEV (TMEV DA) was used78. TMEV DA and live ovalbumin (OVA) peptide-expressing TMEV DA XhoI-OVA8 (TMEV-OVA) were utilized for in vitro bone marrow-derived dendritic cell (BMDC) and adult microglia-enriched glial cell mixtures (MEG)/T cell co-cultivation assays. TMEV-OVA was generated by integrating the coding sequence corresponding to the amino acid sequence OVA(251–267) of chicken egg albumin. Flanking sequences were included to assure natural processing of the immunodominant H-2 Kb restricted epitope OVA(257–264; SIINFEKL). Previous use of this virus has demonstrated viral replication in the CNS of intracranial infected C57BL/6 mice49. Furthermore this live virus vector has demonstrated robust generation of H-2 Kb restricted CD8+ T cell responses to the OVA(257–264) antigen after infection and in tumor models47,48,49. Virus strains were cultivated and passaged in BHK-21 cells and plaque assays were performed using L-cells for virus titration79,80,81.Virus isolation was performed by freezing and thawing. Plaque assays were performed as independent duplicates. Virus titres were determined by calculating the plaque forming units per ml (PFU/ml) as previously described82,83.
Experimental design
Five-week old female DCIR−/− and WT mice were anaesthetised with medetomidine (1 mg/kg, Domitor) and ketamine (100 mg/kg) and inoculated into the right cerebral hemisphere with TMEV DA in a total volume of 20 µl DMEM (Biochrom GmbH, Berlin, Germany) supplemented with 2% FCS (PAA Laboratories GmbH, Pasching, Austria) and 50 µg/kg gentamicin (Sigma Aldrich Chemie GmbH, Taufkirchen, Germany) as described39. Weekly clinical examination included body weight recordings as well as clinical scorings with evaluation of “posture and outer appearance”, “behaviour and activity” and “gait”39. Additionally, a 5 point scale scoring system according to Racine (Racine score) was applied for recording motor seizures84. A RotaRod (TSE Systems GmbH, Bad Homburg, Germany) performance test for motor function and coordination was carried out weekly85.
At 7 and 14 days post infection (dpi) mice were anaesthetised as described above and euthanised with an overdose of medetomidine (1 mg/kg) and ketamine (200 mg/kg). The rostral part of the left cerebrum (contralateral to injection site) was formalin fixed and paraffin embedded (FFPE), and caudal part of the left cerebrum was snap frozen and stored at − 80 °C39,86. Spleens were taken for flow cytometry and parts of splenic tissue were snap frozen and stored at − 80 °C. Serial sections (2–3 µm thickness) of FFPE coronal brain sections at the hippocampal level (Bregma − 1.46 to − 1.82) were used for histology (hematoxylin and eosin staining) and immunohistochemistry, respectively39,87. In addition, non-infected age matched controls were used to determine baseline differences of splenic and hippocampal immune cell compositions as well as cerebral cytokine and transcription factor expression profiles between DCIR−/− and WT mice.
Histologic scoring of hippocampal lesions
Hippocampal damage was evaluated using a semiquantitative scoring system, assessing the integrity of the pyramidal neurons: score 0 = no obvious damage; score 1 = loss involving < 10% of neurons; score 2 = loss involving < 20% of neurons; score 3 = loss involving 20–50% of neurons; score 4 = loss involving > 50% of neurons88.
Immunohistochemistry
Immunohistochemistry was used to detect macrophages/microglia (CD107b, arginase 1), T cells (CD3, CD4, CD8), B cells (CD45R), granzyme B (GrB), regulatory T cells (Foxp3), neurons (neuronal nuclei, NeuN), axons (β-amyloid precursor protein, β-APP), astrocytes (glial fibrillary acidic protein, GFAP), and TMEV capsid protein VP1 as described89,90,91. Used antibodies and staining procedures are listed in Supplementary Table S1. Primary antibodies were diluted in PBS including 1% bovine serum albumin (BSA). In brief, endogenous peroxidase was inhibited by 0.5% H2O2 in ethanol for 30 min. For antigen retrieval (CD107b, arginase 1, CD3, CD45R, Foxp3, NeuN, GrB, β-APP), slides were incubated in citrate buffer within a microwave oven for 20 min. Blocking of unspecific bindings was conducted with either goat serum (TMEV, arginase 1, CD3, NeuN, GrB, β-APP, GFAP) or rabbit serum (CD107b, Foxp3, CD4, CD8). Following, primary antibodies were incubated over night at 4 °C. Biotinylated goat anti-rabbit IgG antibody was used as secondary antibody for TMEV-, arginase 1-, CD3-, and GFAP-specific immunohistochemistry. For CD107b-, CD4-, CD8-, and Foxp3-specific staining, a biotinylated rabbit anti-rat IgG antibody was utilised, and a biotinylated goat anti-mouse IgG antibody was used for NeuN- and β-APP-specific staining. Slides were incubated with the avidin–biotin-peroxidase complex. For visualisation, slides were incubated with 3.3-diaminobenzidine-tetrahydrochloride in PBS containing 0.125% H2O2 and counterstained with Mayer’s hematoxylin.
Hippocampi were digitalised and measured by using the bright field mode of the fluorescence microscope BZ-9000 BIOREVO (HS All-in-one fluorescence microscope, Keyence Corporation, Osaka, Japan) and BZ-II Analyzer software (BZ-H2AE, Keyence Corporation, Osaka, Japan). For CD3-, CD107b-, TMEV-, NeuN- and GFAP-specific immunohistochemistry, the proportion of immunolabelled area within the hippocampus was quantified by densitometric analysis. Moreover, for quantifying TMEV-infected cells, arginase 1+ macrophages/microglia, CD45R+ B cells, Foxp3+ regulatory T cells, CD4+ T helper cells, CD8+ cytotoxic T cells, and GrB+ effector cells, absolute numbers of labelled cells within the hippocampus were counted (cells/mm2). In addition to densitometric analysis, neuronal loss (NeuN) was graded semiquantitavely as described above, too88,92. Evaluation of axonal damage (β-APP) was performed by a semiquantitative scoring system93. Axonal damage in the hippocampus was graded as followed: score 0 = no β-APP+ axons; score 1 = 1–25 β-APP+ axons; score 2 = 26–50 β-APP+ axons; score 3 = 51–75 β-APP+ axons; score 4 = 76–100 β-APP+ axons; score 5 = more than 100 β-APP+ damaged axons93.
Ribonucleic acid isolation and reverse transcription
For RNA isolation, snap frozen tissue of the cerebrum was homogenized in 1 ml QIAzol lysis reagent (Qiagen, Hilden, Germany) with Omni Tip PCR Tissue Homogenizing Kit (Süd-Laborbedarf GmbH, Gauting, Germany). Subsequently, homogenates were treated with RNeasy Lipid tissue Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Likewise, RNA isolation of snap frozen splenic tissue has been performed using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The purity and amount of RNA was measured with a Multiskan GO microplate spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) using a µDrop plate (Thermo Fisher Scientific, Waltham, MA, USA) and SkanIt software (version 3.2.1.4 RE, Thermo Fisher Scientific, Waltham, MA, USA)94. Equal amounts of RNA were translated into cDNA using Omniscript Reverse Transcription Kit (Qiagen, Hilden, Germany), RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) and random primers (Promega Corporation, Madison, WI, USA).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
To determine viral load (TMEV RNA) and mRNA expression levels of CD11c, CD80, CD86, Foxp3, interleukin (IL)-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-23, interferon (IFN)-β, IFN-γ, MHC-I, tumor necrosis factor (TNF)-α, transforming growth factor (TGF)-β1, and the three housekeeping genes, β-actin, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and hypoxanthine–guanine phosphoribosyltransferase (HPRT), RT-qPCR was carried out using the Mx3005P Multiplex Quantitative PCR System (Agilent Technologies Deutschland GmbH, Waldbronn, Germany) and Brilliant III Ultra-Fast SYBR Green QPCR Mastermix (Agilent Technologies Deutschland GmbH, Waldbronn, Germany). Primer details are listed in Supplementary Table S2. Quantification of copy numbers was achieved by parallel, duplicate amplification of tenfold serial dilution of standards ranging from 108 to 102 copies/µl. Melting curve analysis proved specificity of each reaction86. The geNorm software (version 3.4) was utilised for normalisation of qPCR data95,96.
Flow cytometry of murine splenocytes
Spleens were removed and immediately flushed mechanically with a syringe and 1 × PBS to a single cell suspension. Subsequently erythrocytes were lysed using RBC lysis buffer (10% 100 mM Tris–HCl [Tris-(hydroxymethyl)-aminomethanhydrochloride], 90% 160 mM NH4Cl [ammonium chloride], Carl Roth, Karlsruhe, Germany). Afterwards, cells were incubated with rat-anti CD16/32 monoclonal antibody (1:100) to block the Fc gamma receptor and therefore to avoid unspecific binding. Cell solutions were stained with following monoclonal anti-mouse antibodies: CD4-FITC, CD62L-PE-Cy7, CD4-PerCP-Cy5.5, CD25-FITC, CD44-APC, CD8a-PE, CD8a-APC and CD19-FITC. Details for all flow cytometry antibodies are listed in Supplementary Table S3. For fixation, cells were incubated with 1% paraformaldehyde (PFA, Carl Roth, Karlsruhe, Germany). Flow cytometry was performed at the Attune NxT cytometer (Thermo Fisher Scientific, Waltham, MA, USA). Data analysis was conducted with FlowJo software (version 10, FloJo LLC, Ashland, OR, USA)97.
Isolation of an adult microglia-enriched glial cell mixture (MEG)
To isolate MEGs, a previously used method was modified98. Brains of WT and DCIR−/− mice were dissected and stored temporarily in HBSS (Sigma Aldrich, St. Louis, MO, USA) containing 15 mM HEPES (Carl Roth, Karlsruhe, Germany) and 0.5% glucose (Carl Roth, Karlsruhe, Germany). For dissociation, brains were squashed with the top end of a syringe in a 6-well plate containing a digestion cocktail (HBSS, 1 mg/ml collagenase D, 5 U/ml DNase I; Roche, Basel, Switzerland). After 10 min of incubation at 37 °C, brains were gently dissociated manually. Afterwards, a 40% Percoll centrifugation (10 min, 350×g, 18 °C; GE Healthcare, Chicago, IL, USA) and erythrocyte lysis were performed. To check the percentage of microglia within the glial cell mixture, cells were blocked with anti-mouse CD16/32, stained with anti-mouse CD11b-PE and anti-mouse CD45-APC and fixed in 1% PFA. Flow cytometry was performed using an Attune NxT Flow Cytometer. Data analysis was conducted with FlowJo software97. The purity of microglia (CD11b+/CD45low+) within MEG used for co-culture experiments ranged between 40 to 60% for both WT and DCIR−/− cell suspensions.
Microglia-enriched glial cell mixture/T cell co-cultivation
Following MEG isolation, glial cells were seeded with 4 × 105 cells/ml in culture medium (IMDM medium, 10% FCS, 2 mM l-glutamine, 100 U/ml penicillin 100 µg/ml streptomycin; Pan-Biotech, Aidenbach, Germany) in a 96-well U-bottom plate and stimulated with EndoGrade ovalbumin (0.3 mg/ml, LIONEX, Braunschweig, Germany) or TMEV-OVA (MOI 200) at 37 °C for 22 h. T cells were isolated from spleens of 8 to 12 week old OT-I transgenic mice using magnetic activated cell sorting (MACS, Pan T Cell Isolation Kit II mouse, Miltenyi Biotec, Bergisch Gladbach, Germany). Purified T cells were adjusted to 1 × 106 cells/ml, added to the glial cells and co-cultured at 37 °C for 48 h. After incubation, supernatants were harvested and IL-2 and IFN-γ cytokine concentrations were analysed by ELISA (murine IL-2 and IFN-γ Standard ABTS ELISA Development Kit, PeproTech, Rocky Hill, NJ, USA). Co-cultured cells were blocked with anti-mouse CD16/32, stained with anti-mouse CD8a-FITC, CD62L-PE and CD69-APC and fixed in 1% PFA. Flow cytometry was performed using an Attune NxT Flow Cytometer. Data analysis was conducted with FlowJo software97.
Bone marrow-derived dendritic cells/T cell co-cultivation
To generate BMDCs, bone marrow cells were isolated from femurs and tibias of DCIR−/− and C57BL/6 control mice and differentiated into BMDCs by cultivation with differentiation medium (culture medium + 10% X63-GM-CSF supernatant) at 37 °C for 8 to 10 days. Following generation and differentiation, BMDCs were seeded with 2 × 105 cells/ml in culture medium in a 96-well U-bottom plate and co-cultivation was performed as described above.
Statistical analysis
Statistical analyses were performed using SPSS for Windows (version 21, SPSS Inc., IBM Corp.)99 applying multiple Mann–Whitney U tests (Supplementary Table S4) and the statistics software R (version 4.0.4)100 for nonparametric two-way analysis of variance (ANOVA). Moreover, statistics software R was used for applying simple and multiple regression models to study the influence of infiltrating immune cell composition, virus load and cytokine profile on hippocampal neuronal integrity. First independent parameters were preselected by single regression models. Surviving parameters were subjected to multiple regression models, which were further reduced by automatic backwards variable selection. Due to lower sample sizes at individual time points, regression models were avoided at time-specific subgroup analyses. Instead, correlation analyses using Pearson’s correlation coefficient R regarding specific analyses at time points 7 dpi and 14 dpi were performed. Graphs were designed using GraphPad Prism software (version 8, GraphPad Software Inc., San Diego, CA, USA)101. Statistical tests were performed with a significance level of α = 5%.
Data availability
The data presented in this study are available on request from the corresponding author.
References
Bröer, S. et al. Viral mouse models of multiple sclerosis and epilepsy: Marked differences in neuropathogenesis following infection with two naturally occurring variants of Theiler’s virus BeAn strain. Neurobiol. Dis. 99, 121–132. https://doi.org/10.1016/j.nbd.2016.12.020 (2017).
Ludlow, M. et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 131, 159–184. https://doi.org/10.1007/s00401-015-1511-3 (2016).
Betourne, A. et al. Hippocampal expression of a virus-derived protein impairs memory in mice. Proc. Natl. Acad. Sci. USA. 115, 1611–1616. https://doi.org/10.1073/pnas.1711977115 (2018).
De Chiara, G. et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 15, e1007617. https://doi.org/10.1371/journal.ppat.1007617 (2019).
Vezzani, A. et al. Infections, inflammation and epilepsy. Acta Neuropathol. 131, 211–234. https://doi.org/10.1007/s00401-015-1481-5 (2016).
Stewart, K. A., Wilcox, K. S., Fujinami, R. S. & White, H. S. Theiler’s virus infection chronically alters seizure susceptibility. Epilepsia 51, 1418–1428. https://doi.org/10.1111/j.1528-1167.2009.02405.x (2010).
Libbey, J. E. et al. Seizures following picornavirus infection. Epilepsia 49, 1066–1074. https://doi.org/10.1111/j.1528-1167.2008.01535.x (2008).
Theiler, M. Spontaneous encephalomyelitis of mice: A new virus disease. Science 80, 122. https://doi.org/10.1126/science.80.2066.122-a (1934).
Libbey, J. E. & Fujinami, R. S. Neurotropic viral infections leading to epilepsy: Focus on Theiler’s murine encephalomyelitis virus. Future Virol. 6, 1339–1350. https://doi.org/10.2217/fvl.11.107 (2011).
Gerhauser, I., Hansmann, F., Ciurkiewicz, M., Löscher, W. & Beineke, A. Facets of Theiler’s Murine encephalomyelitis virus-induced diseases: An update. Int. J. Mol. Sci. 20, 448. https://doi.org/10.3390/ijms20020448 (2019).
Buenz, E. J., Rodriguez, M. & Howe, C. L. Disrupted spatial memory is a consequence of picornavirus infection. Neurobiol. Dis. 24, 266–273. https://doi.org/10.1016/j.nbd.2006.07.003 (2006).
Umpierre, A. D. et al. Impaired cognitive ability and anxiety-like behavior following acute seizures in the Theiler’s virus model of temporal lobe epilepsy. Neurobiol. Dis. 64, 98–106. https://doi.org/10.1016/j.nbd.2013.12.015 (2014).
Bowen, J. L. & Olson, J. K. IFNgamma influences type I interferon response and susceptibility to Theiler’s virus-induced demyelinating disease. Viral Immunol. 26, 223–238. https://doi.org/10.1089/vim.2013.0004 (2013).
Drappier, M. et al. A novel mechanism of RNase L inhibition: Theiler’s virus L* protein prevents 2–5A from binding to RNase L. PLoS Pathog. 14, e1006989. https://doi.org/10.1371/journal.ppat.1006989 (2018).
Li, L., Ulrich, R., Baumgärtner, W. & Gerhauser, I. Interferon-stimulated genes-essential antiviral effectors implicated in resistance to Theiler’s virus-induced demyelinating disease. J. Neuroinflamm. 12, 242. https://doi.org/10.1186/s12974-015-0462-x (2015).
Libbey, J. E., Kennett, N. J., Wilcox, K. S., White, H. S. & Fujinami, R. S. Interleukin-6, produced by resident cells of the central nervous system and infiltrating cells, contributes to the development of seizures following viral infection. J. Virol. 85, 6913–6922. https://doi.org/10.1128/JVI.00458-11 (2011).
Waltl, I. et al. Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage. Brain. Behav. Immun. 74, 186–204. https://doi.org/10.1016/j.bbi.2018.09.006 (2018).
Howe, C. L., Lafrance-Corey, R. G., Sundsbak, R. S. & Lafrance, S. J. Inflammatory monocytes damage the hippocampus during acute picornavirus infection of the brain. J. Neuroinflamm. 9, 50. https://doi.org/10.1186/1742-2094-9-50 (2012).
Howe, C. L. et al. Hippocampal protection in mice with an attenuated inflammatory monocyte response to acute CNS picornavirus infection. Sci. Rep. 2, 545. https://doi.org/10.1038/srep00545 (2012).
Kirkman, N. J., Libbey, J. E., Wilcox, K. S., White, H. S. & Fujinami, R. S. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia 51, 454–464 (2010).
Cusick, M. F., Libbey, J. E., Patel, D. C., Doty, D. J. & Fujinami, R. S. Infiltrating macrophages are key to the development of seizures following virus infection. J. Virol. 87, 1849–1860 (2013).
Marzi, A. et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 78, 12090–12095. https://doi.org/10.1128/JVI.78.21.12090-12095.2004 (2004).
Curtis, B. M., Scharnowske, S. & Watson, A. J. Sequence and expression of a membrane-associated C-type lectin that exhibits CD4-independent binding of human immunodeficiency virus envelope glycoprotein gp120. Proc. Natl. Acad. Sci. USA. 89, 8356–8360. https://doi.org/10.1073/pnas.89.17.8356 (1992).
Tassaneetrithep, B. et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197, 823–829. https://doi.org/10.1084/jem.20021840 (2003).
Simmons, G. et al. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 305, 115–123. https://doi.org/10.1006/viro.2002.1730 (2003).
Monteiro, J. T. & Lepenies, B. Myeloid C-type lectin receptors in viral recognition and antiviral immunity. Viruses 9, E59. https://doi.org/10.3390/v9030059 (2017).
Mayer, S., Raulf, M. K. & Lepenies, B. C-type lectins: Their network and roles in pathogen recognition and immunity. Histochem. Cell Biol. 147, 223–237. https://doi.org/10.1007/s00418-016-1523-7 (2017).
Billadeau, D. D. & Leibson, P. J. ITAMs versus ITIMs: Striking a balance during cell regulation. J. Clin. Invest. 109, 161–168. https://doi.org/10.1172/JCI14843 (2002).
Burshtyn, D. N., Yang, W., Yi, T. & Long, E. O. A novel phosphotyrosine motif with a critical amino acid at position-2 for the SH2 domain-mediated activation of the tyrosine phosphatase SHP-1. J. Biol. Chem. 272, 13066–13072. https://doi.org/10.1074/jbc.272.20.13066 (1997).
Bates, E. E. et al. APCs express DCIR, a novel C-type lectin surface receptor containing an immunoreceptor tyrosine-based inhibitory motif. J. Immunol. 163, 1973–1983 (1999).
Eklöw, C. et al. Cellular distribution of the C-type II lectin dendritic cell immunoreceptor (DCIR) and its expression in the rheumatic joint: Identification of a subpopulation of DCIR+ T cells. Ann. Rheum. Dis. 67, 1742–1749. https://doi.org/10.1136/ard.2007.076976 (2008).
Veillette, A., Latour, S. & Davidson, D. Negative regulation of immunoreceptor signaling. Annu. Rev. Immunol. 20, 669–707. https://doi.org/10.1146/annurev.immunol.20.081501.130710 (2002).
Fujikado, N. et al. Dcir deficiency causes development of autoimmune diseases in mice due to excess expansion of dendritic cells. Nat. Med. 14, 176–180. https://doi.org/10.1038/nm1697 (2008).
Hickman, S. E. et al. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905 (2013).
Kaifu, T. & Iwakura, Y. in C-Type Lectin Receptors in Immunity (ed S. Yamasaki), 101–113 (Springer, 2016).
Troegeler, A. et al. C-type lectin receptor DCIR modulates immunity to tuberculosis by sustaining type I interferon signaling in dendritic cells. Proc. Natl. Acad. Sci. USA 114, E540–E549. https://doi.org/10.1073/pnas.1613254114 (2017).
Long, K. M. et al. Dendritic cell immunoreceptor regulates Chikungunya virus pathogenesis in mice. J. Virol. 87, 5697–5706. https://doi.org/10.1128/JVI.01611-12 (2013).
Maglinao, M., Klopfleisch, R., Seeberger, P. H. & Lepenies, B. The C-type lectin receptor DCIR is crucial for the development of experimental cerebral malaria. J. Immunol. 191, 2551–2559. https://doi.org/10.4049/jimmunol.1203451 (2013).
Ciurkiewicz, M. et al. Cytotoxic CD8(+) T cell ablation enhances the capacity of regulatory T cells to delay viral elimination in Theiler’s murine encephalomyelitis. Brain Pathol. 28, 349–368. https://doi.org/10.1111/bpa.12518 (2018).
Uhde, A. K. et al. Intact interleukin-10 receptor signaling protects from hippocampal damage elicited by experimental neurotropic virus infection of SJL mice. Sci. Rep. 8, 6106. https://doi.org/10.1038/s41598-018-24378-z (2018).
Ciurkiewicz, M., Herder, V. & Beineke, A. Beneficial and detrimental effects of regulatory T cells in neurotropic virus infections. Int. J. Mol. Sci. 21, 1705. https://doi.org/10.3390/ijms21051705 (2020).
Burrack, K. S. & Morrison, T. E. The role of myeloid cell activation and arginine metabolism in the pathogenesis of virus-induced diseases. Front. Immunol. 5, 428–428. https://doi.org/10.3389/fimmu.2014.00428 (2014).
Stoermer, K. A. et al. Genetic ablation of arginase 1 in macrophages and neutrophils enhances clearance of an arthritogenic alphavirus. J. Immunol. 189, 4047–4059. https://doi.org/10.4049/jimmunol.1201240 (2012).
Oleszak, E. L., Chang, J. R., Friedman, H., Katsetos, C. D. & Platsoucas, C. D. Theiler’s virus infection: A model for multiple sclerosis. Clin. Microbiol. Rev. 17, 174–207. https://doi.org/10.1128/cmr.17.1.174-207.2004 (2004).
Chang, J. R., Zaczynska, E., Katsetos, C. D., Platsoucas, C. D. & Oleszak, E. L. Differential expression of TGF-beta, IL-2, and other cytokines in the CNS of Theiler’s murine encephalomyelitis virus-infected susceptible and resistant strains of mice. Virology 278, 346–360. https://doi.org/10.1006/viro.2000.0646 (2000).
Rodriguez, M. et al. Abrogation of resistance to Theiler’s virus-induced demyelination in H-2b mice deficient in beta 2-microglobulin. J. Immunol. 151, 266–276 (1993).
Renner, D. N. et al. Improved treatment efficacy of antiangiogenic therapy when combined with picornavirus vaccination in the GL261 glioma model. Neurotherapeutics 13, 226–236. https://doi.org/10.1007/s13311-015-0407-1 (2016).
Pavelko, K. D. et al. The epitope integration site for vaccine antigens determines virus control while maintaining efficacy in an engineered cancer vaccine. Mol. Ther. 21, 1087–1095. https://doi.org/10.1038/mt.2013.52 (2013).
Pavelko, K. D. et al. Theiler’s murine encephalomyelitis virus as a vaccine candidate for immunotherapy. PLoS ONE 6, e20217. https://doi.org/10.1371/journal.pone.0020217 (2011).
Maruhashi, T. et al. DCIR maintains bone homeostasis by regulating IFN-gamma production in T cells. J. Immunol. 194, 5681–5691. https://doi.org/10.4049/jimmunol.1500273 (2015).
Klechevsky, E. et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood 116, 1685–1697. https://doi.org/10.1182/blood-2010-01-264960 (2010).
Meyer-Wentrup, F. et al. Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-alpha production. Blood 111, 4245–4253. https://doi.org/10.1182/blood-2007-03-081398 (2008).
Meyer-Wentrup, F. et al. DCIR is endocytosed into human dendritic cells and inhibits TLR8-mediated cytokine production. J. Leukoc. Biol. 85, 518–525. https://doi.org/10.1189/jlb.0608352 (2009).
Brinkmeyer-Langford, C. L. et al. Host genetic background influences diverse neurological responses to viral infection in mice. Sci. Rep. 7, 12194. https://doi.org/10.1038/s41598-017-12477-2 (2017).
Dethlefs, S., Brahic, M. & Larsson-Sciard, E. L. An early, abundant cytotoxic T-lymphocyte response against Theiler’s virus is critical for preventing viral persistence. J. Virol. 71, 8875–8878 (1997).
Getts, M. T., Kim, B. S. & Miller, S. D. Differential outcome of tolerance induction in naive versus activated Theiler’s virus epitope-specific CD8+ cytotoxic T cells. J. Virol. 81, 6584–6593. https://doi.org/10.1128/JVI.00008-07 (2007).
Huseby Kelcher, A. M. et al. Brain atrophy in picornavirus-infected FVB mice is dependent on the H-2Db class I molecule. FASEB J. 31, 2267–2275 (2017).
Murray, P., Pavelko, K., Leibowitz, J., Lin, X. & Rodriguez, M. CD4+ and CD8+ T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J. Virol. 72, 7320–7329 (1998).
Laidlaw, B. J., Craft, J. E. & Kaech, S. M. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat. Rev. Immunol. 16, 102 (2016).
Richards, M. H. et al. Virus expanded regulatory T cells control disease severity in the Theiler’s virus mouse model of MS. J. Autoimmun. 36, 142–154. https://doi.org/10.1016/j.jaut.2010.12.005 (2011).
McDole, J. R. et al. Rapid formation of extended processes and engagement of Theiler’s virus-infected neurons by CNS-infiltrating CD8 T cells. Am. J. Pathol. 177, 1823–1833. https://doi.org/10.2353/ajpath.2010.100231 (2010).
Pirko, I. et al. Contrasting roles for CD4 vs. CD8 T-cells in a murine model of virally induced “T1 black hole” formation. PLoS ONE 7, e31459. https://doi.org/10.1371/journal.pone.0031459 (2012).
Willenbring, R. C. et al. Modulatory effects of perforin gene dosage on pathogen-associated blood-brain barrier (BBB) disruption. J. Neuroinflammation 13, 222. https://doi.org/10.1186/s12974-016-0673-9 (2016).
Seno, A. et al. Exacerbation of experimental autoimmune encephalomyelitis in mice deficient for DCIR, an inhibitory C-type lectin receptor. Exp. Anim. 64, 109–119. https://doi.org/10.1538/expanim.14-0079 (2015).
Hütter, J. et al. Role of the C-type lectin receptors MCL and DCIR in experimental colitis. PLoS ONE 9, e103281. https://doi.org/10.1371/journal.pone.0103281 (2014).
Jin, Y. H. et al. Type I interferon signals control Theiler’s virus infection site, cellular infiltration and T cell stimulation in the CNS. J. Neuroimmunol. 226, 27–37. https://doi.org/10.1016/j.jneuroim.2010.05.028 (2010).
Akdis, M. et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor beta, and TNF-alpha: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 138, 984–1010. https://doi.org/10.1016/j.jaci.2016.06.033 (2016).
Rodriguez, M. et al. Tumor necrosis factor alpha is reparative via TNFR2 [corrected] in the hippocampus and via TNFR1 [corrected] in the striatum after virus-induced encephalitis. Brain Pathol. 19, 12–26 (2009).
Ye, L. et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: A potential role for neuronal glutaminase. J. Neurochem. 125, 897–908 (2013).
Banchereau, J. & Steinman, R. M. Dendritic cells and the control of immunity. Nature 392, 245–252. https://doi.org/10.1038/32588 (1998).
Ueno, H. et al. Dendritic cell subsets in health and disease. Immunol. Rev. 219, 118–142. https://doi.org/10.1111/j.1600-065X.2007.00551.x (2007).
Beauvillain, C. et al. Neonatal and adult microglia cross-present exogenous antigens. Glia 56, 69–77. https://doi.org/10.1002/glia.20565 (2008).
Olson, J. K., Girvin, A. M. & Miller, S. D. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler’s virus. J. Virol. 75, 9780–9789. https://doi.org/10.1128/JVI.75.20.9780-9789.2001 (2001).
Burshtyn, D. N. & Long, E. O. Regulation through inhibitory receptors: lessons from natural killer cells. Trends Cell Biol. 7, 473–479. https://doi.org/10.1016/S0962-8924(97)01167-7 (1997).
Barrow, A. D. & Trowsdale, J. You say ITAM and I say ITIM, let’s call the whole thing off: The ambiguity of immunoreceptor signalling. Eur. J. Immunol. 36, 1646–1653. https://doi.org/10.1002/eji.200636195 (2006).
Klauber, T. C. B. et al. Delivery of TLR7 agonist to monocytes and dendritic cells by DCIR targeted liposomes induces robust production of anti-cancer cytokines. Acta Biomater. 53, 367–377. https://doi.org/10.1016/j.actbio.2017.01.072 (2017).
du Sert, N. P. et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. J. Cereb. Blood Flow Metab. 40, 1769–1777 (2020).
Daniels, J. B., Pappenheimer, A. M. & Richardson, S. Observations on encephalomyelitis of mice (DA strain). J. Exp. Med. 96, 517–530. https://doi.org/10.1084/jem.96.6.517 (1952).
Kumnok, J. et al. Differential transcription of matrix-metalloproteinase genes in primary mouse astrocytes and microglia infected with Theiler’s murine encephalomyelitis virus. J. Neurovirol. 14, 205–217. https://doi.org/10.1080/13550280802008305 (2008).
Falke, D. Uber die zuchtung des theiler-to-virus in der gewebekultur. Z. Hyg. Infektionskr. 143, 645–655. https://doi.org/10.1007/Bf02149971 (1957).
Lipton, H. L. Characterization of the TO strains of Theiler’s mouse encephalomyelitis viruses. Infect. Immun. 20, 869–872 (1978).
Rabinowitz, S. G. & Lipton, H. L. Cellular immunity in chronic Theiler’s virus central nervous system infection. J. Immunol. 117, 357–363 (1976).
Gerhauser, I. In vitro Expressions-Analyse von Transkriptionsfaktoren muriner Astrozyten und Mikrogliazellen nach Infektion mit dem Theiler-Enzephalomyelitis-Virus Dr. med vet. thesis, Hannover, tierärztl. Hochschule (2009).
Racine, R. J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294 (1972).
Uhde, A. K. et al. Viral infection of the central nervous system exacerbates interleukin-10 receptor deficiency-mediated colitis in SJL mice. PLoS ONE 11, e0161883. https://doi.org/10.1371/journal.pone.0161883 (2016).
Gerhauser, I., Alldinger, S., Ulrich, R. & Baumgärtner, W. Spatio-temporal expression of immediate early genes in the central nervous system of SJL/J mice. Int. J. Dev. Neurosci. 23, 637–649. https://doi.org/10.1016/j.ijdevneu.2005.06.004 (2005).
Paxinos, G. & Franklin, K. B. J. The Mouse Brain in Stereotaxic Coordinates 3rd edn. (Elsevier Academic Press, 2007).
Rattka, M., Brandt, C. & Löscher, W. The intrahippocampal kainate model of temporal lobe epilepsy revisited: Epileptogenesis, behavioral and cognitive alterations, pharmacological response, and hippoccampal damage in epileptic rats. Epilepsy Res. 103, 135–152. https://doi.org/10.1016/j.eplepsyres.2012.09.015 (2013).
Kummerfeld, M., Meens, J., Haas, L., Baumgärtner, W. & Beineke, A. Generation and characterization of a polyclonal antibody for the detection of Theiler’s murine encephalomyelitis virus by light and electron microscopy. J. Virol. Methods 160, 185–188. https://doi.org/10.1016/j.jviromet.2009.04.030 (2009).
Ulrich, R. et al. MMP-12, MMP-3, and TIMP-1 are markedly upregulated in chronic demyelinating theiler murine encephalomyelitis. J. Neuropathol. Exp. Neurol. 65, 783–793. https://doi.org/10.1097/01.jnen.0000229990.32795.0d (2006).
Herder, V. et al. Interleukin-10 expression during the acute phase is a putative prerequisite for delayed viral elimination in a murine model for multiple sclerosis. J. Neuroimmunol. 249, 27–39. https://doi.org/10.1016/j.jneuroim.2012.04.010 (2012).
Bröer, S. et al. Brain inflammation, neurodegeneration and seizure development following picornavirus infection markedly differ among virus and mouse strains and substrains. Exp. Neurol. 279, 57–74. https://doi.org/10.1016/j.expneurol.2016.02.011 (2016).
Thornton, E., Vink, R., Blumbergs, P. C. & Van Den Heuvel, C. Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 1094, 38–46. https://doi.org/10.1016/j.brainres.2006.03.107 (2006).
SkanIT Software Version 3.2.1.4 RE. Thermo Fisher Scientific (2018). https://www.thermofisher.com/order/catalog/product/5187139#/5187139.
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, 0034. https://doi.org/10.1186/gb-2002-3-7-research0034 (2002).
geNorm Version 3.4 (2002). https://genorm.cmgg.be/.
FlowJo software Version 10, FloJo LLC (2020). https://www.flowjo.com/.
Prajeeth, C. K. et al. Limited role of regulatory T cells during acute Theiler virus-induced encephalitis in resistant C57BL/6 mice. J. Neuroinflamm. 11, 180. https://doi.org/10.1186/s12974-014-0180-9 (2014).
SPSS for Windows, Version 21, SPSS Inc. IBM Corp. (2020). https://www.ibm.com/de-de/analytics/spss-statistics-software.
The R Project for Statistical Computing Version 4.0.4 (2020). https://www.r-project.org/.
GraphPad Prism Version 8, GraphPad Software Inc. (2020). https://www.graphpad.com/.
Acknowledgements
This research was funded by the German Research Foundation (DFG, BE4200/1-3 and LE 2498/6-1). This publication was supported by Deutsche Forschungsgemeinschaft and University of Veterinary Medicine Hannover, Foundation within the funding programme Open Access Publishing. The authors thank Prof. Robert S. Fujinami, PhD for kindly providing TMEV DA strain, Prof. C. Jane Welsh, PhD for kindly providing L-cells, Prof. Dr. Ludwig Haas for kindly providing BHK-21 cells and Prof. Dr. Reinhold Förster for kindly providing OT-I mice. We thank Petra Grünig, Claudia Hermann, Caroline Schütz and Danuta Waschke for their excellent technical assistance.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: B.L. and A.B.; Data curation: A.B.; Formal analysis: M.S., T.E., S.M.-L., K.J. and A.B.; Funding acquisition: B.L. and A.B.; Investigation: M.S., T.E., M.C., S.P., K.J., S.M.-L., T.S. and A.B.; Methodology: K.D.P., W.B., B.L. and A.B.; Project administration: B.L. and A.B.; Resources: K.D.P., W.B., B.L. and A.B.; Supervision: B.L. and A.B.; Validation: B.L. and A.B.; Visualization: M.S. and T.E.; Writing—original draft: M.S., T.E., K.J., B.L. and A.B.; Writing—review & editing: M.S., T.E., M.C., S.M.-L., T.S., K.J., K.D.P., W.B., B.L. and A.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stoff, M., Ebbecke, T., Ciurkiewicz, M. et al. C-type lectin receptor DCIR contributes to hippocampal injury in acute neurotropic virus infection. Sci Rep 11, 23819 (2021). https://doi.org/10.1038/s41598-021-03201-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03201-2
This article is cited by
-
The C-type lectin DCIR contributes to the immune response and pathogenesis of colorectal cancer
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
