
Abstract
Amphiregulin (AREG) is an epidermal growth factor receptor (EGFR) ligand. The aim of this study was to investigate the effects of baseline plasma AREG levels in KRAS, NRAS, and BRAF wild-type metastatic colorectal cancer (CRC) on treatment outcome with palliative first-line cetuximab + FOLFIRI chemotherapy. Chemotherapy outcomes were analyzed based on baseline plasma AREG levels. The clinical findings were further validated using an in vitro model of CRC. Among 35 patients, the progression-free survival (PFS) was significantly inferior in patients with high AREG than in those with low AREG levels: 10.9 vs. 24.2 months, respectively (p = 0.008). However, after failure of first-line chemotherapy, AREG levels were associated with neither PFS (4.8 vs. 11.6 months; p = 0.215) nor overall survival (8.4 vs. 13.3 months; p = 0.975). In SNU-C4 and Caco-2 cells which were relatively sensitive to cetuximab among the seven CRC cell lines tested, AREG significantly decreased the anti-proliferative effect of cetuximab (p < 0.05) via AKT and ERK activation. However, after acquiring cetuximab resistance with gradual exposure for more than 6 months, AREG neither increased colony formation nor activated AKT and ERK after cetuximab treatment. Our results suggest that plasma AREG is a potential biomarker to predict clinical outcomes after cetuximab-based chemotherapy.
Similar content being viewed by others
Introduction
Palliative first-line chemotherapy with anti-EGFR (epidermal growth factor receptor) monoclonal antibody, cetuximab, or panitumumab, combined with conventional cytotoxic chemotherapy, has shown preferable outcomes in phase III trials1,2,3,4. Of note, the antitumor effect was conspicuous in the patients with extended RAS wild-type cancers5. However, although overall survival (OS) reached over 2 years with this treatment strategy, most of the patients are deemed to encounter treatment resistance. KRAS, NRAS, and BRAF (RAS/BRAF) mutation status is currently the only factor that predicts the response to cetuximab treatment. However, in clinical practice, the treatment response is varied even among RAS/BRAF wild-type patients.
Amphiregulin (AREG) is an EGFR ligand, which is associated with normal tissue development and proliferation and with a wide variety of human cancers including colorectal cancer6,7,8,9. In colorectal cancer, AREG is known to act competitively with anti-EGFR monoclonal antibodies and activate downstream signaling, and thus may be involved with cetuximab resistance10. It has been speculated that patients with high AREG levels may have worse treatment outcomes with anti-EGFR therapy. However, in some studies, conflicting results have been reported on the effects of AREG on anti-EGFR treatment outcomes11,12,13.
Therefore, in this study, we aimed to investigate the effects of baseline plasma AREG levels in patients with colorectal cancer treated with palliative first-line cetuximab plus FOLFIRI regimen and subsequent second-line chemotherapy. In addition, we conducted an in vitro study to further define the molecular mechanisms of AREG in cetuximab-naïve and cetuximab-resistant colorectal cancer cell lines.
Results
High baseline plasma AREG levels are associated with inferior progression-free survival
From May 2015 to September 2019, a total of 35 patients were consecutively included in this study. All patients were treated with palliative first-line chemotherapy consisting of cetuximab plus FOLFIRI. Among them, 21 patients (60.0%) had left-sided colorectal cancer and most of the cancers (n = 29, 82.9%) showed moderate differentiation. All patients were evaluated for RAS mutation; no one had RAS mutation at the time of diagnosis. BRAF mutation was evaluated in 34 patients; 2 patients (5.9%) had BRAF mutant cancer (Table 1).
The median follow-up time was 19.1 months (range 1.2–56.0 months). Among patients whose disease had target lesions defined by RECIST version 1.1 (n = 34), 21 patients (60.0%) showed partial response (Table 2). In the entire patient cohort, the median PFS and OS were 17.0 months (95% CI 15.0–51.1 months) and 33.0 months (95% confidence interval [CI] 8.7–25.3 months), respectively (Fig. 1A,B).

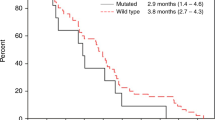
Kaplan–Meier survival analysis of patients who were treated with palliative first-line cetuximab + FOLFIRI chemotherapy. (A) Progression-free survival (PFS), (B) overall survival (OS) of the entire patient population, (C) PFS, (D) OS according to plasma AREG levels.
The median plasma AREG level was 33.6 pg/mL (range 0–1570.54 pg/mL). Using a maximal chi-square test, the cut-off value for AREG level was defined as 12.00 pg/mL. Comparing patients with high (n = 26) vs low plasma AREG levels, PFS was significantly inferior (10.9 months [95% CI 8.0–13.8 months] vs. 24.2 months [95% CI 14.1–34.2]; p = 0.008, respectively) (Fig. 1C). Patients with high plasma AREG levels also showed a trend toward inferior OS (33.0 months [95% CI 0.0–66.1 months] vs. not reached, respectively; p = 0.154) (Fig. 1D).
Baseline plasma AREG levels are correlated with neither PFS nor OS after progression of palliative first-line chemotherapy
PFS2 and OS2 were evaluated among the patients who showed disease progression with cetuximab + FOLFIRI (n = 24). Most of the patients (n = 20) were treated with bevacizumab + FOLFOX after progression, while the others (n = 3) received cetuximab + FOLFIRI retreatment (n = 1), regorafenib (n = 1), or radiation (n = 1). One patient did not receive any subsequent treatment. The median PFS2 and OS2 were 7.2 months (95% CI 1.5–12.9 months) and 13.3 months (95% CI 5.0–21.6 months), respectively (Fig. 2A,B). Baseline plasma AREG levels were associated with neither PFS2 (4.8 months [95% CI 3.4–6.3 months] vs. 11.6 months [95% CI 2.1–21.1 months]; p = 0.215) (Fig. 2C) nor OS2 (8.4 months [95% CI 7.3–9.5 months] vs. 13.3 months [95% CI 2.1–24.5 months]; p = 0.975) (Fig. 2D).

Kaplan–Meier survival analysis of patients who were treated with palliative 2nd-line chemotherapy after progression of 1st-line cetuximab + FOLFIRI treatment. (A) Progression-free survival (PFS), (B) overall survival (OS) among the patients who showed progression with first-line palliative chemotherapy and (C) PFS, (D) OS according to plasma AREG levels.
AREG decreases cellular response to cetuximab
The anti-proliferative effects of cetuximab were evaluated in seven human colorectal cancer cells, SNU-C5, SW480, DLD-1, HCT-15, Caco-2, SNU-C4, and SW48 (Fig. 3A). Cell lines with KRAS or BRAF hotspot mutations were resistant to cetuximab (GI50 ≥ 440.4 μg/mL), while, among those without KRAS and BRAF hotspot mutations, Caco-2 and SNU-C4 were relatively sensitive to cetuximab with GI50 values of 44.0 μg/mL and 198.7 μg/mL, respectively. Based on cetuximab sensitivity, Caco-2 and SNU-C4 cell lines were selected for further study.

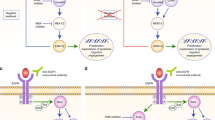
Effect of AREG on cetuximab-induced anti-proliferative effects and EGFR signaling pathways in colorectal cancer cells. (A) Cetuximab cytotoxicity assay in various colorectal cancer cell lines. Serially diluted cetuximab was added for 5 days and cell viability was measured using CellTiter-Glo. (B) western blot of EGFR signaling molecules after treatment with cetuximab and 50 ng/mL AREG. Cetuximab was added at GI50 concentration for 15 min. AREG was added to SNU-C4 and Caco-2 cells 15 min and 24 h prior to cetuximab treatment, respectively. Serum starvation for 24 h was conducted before the addition of cetuximab and AREG.
To determine the effect of cetuximab combined with AREG on the EGFR signaling pathways, Western blot analyses were conducted (Fig. 3B). AREG increased the phosphorylation of EGFR Y1068 (p-EGFR), which is the most sensitive site to EGFR ligand stimulation among p-EGFR residues, in Caco-2 and SNU-C4 cells14,15. In AREG-treated Caco-2 cells, the changes of EGFR downstream molecules involving phosphorylated AKT (p-AKT) and phosphorylated ERK1/2 (p-ERK1/2) were not apparent. In contrast, the phosphorylation of AKT and ERK1/2 strongly increased by adding AREG to SNU-C4 cells, indicating a more responsive phenotype against AREG in terms of signaling pathway activation compared with Caco-2 cells. Further, cetuximab monotherapy decreased the phosphorylation of EGFR, AKT, and ERK1/2 in both cell lines. To examine the effects of AREG on cetuximab response, AREG was administered prior to cetuximab treatment in Caco-2 and SNU-C4 cells. In both Caco-2 and SNU-C4 cells, AREG partially restored the cetuximab-induced inhibition of EGFR downstream molecules, AKT and ERK1/2.
To assess the effect of cetuximab in the presence or absence of AREG on the cell cycle in Caco-2 and SNU-C4 cells, we conducted flow cytometry (Fig. 4A,B). We sought that cetuximab monotherapy significantly increased the G0/G1 phase but decreased the S and G2/M phase in Caco-2 and SNU-C4 cells. However, in the presence of AREG, cetuximab-mediated cell cycle alteration was partially reversed in Caco-2 and SNU-C4 cells. Particularly, in the presence of AREG, subG1 phase was significantly repressed with the cetuximab treatment in both cells, indicating that AREG might reduce the activity of cetuximab in Caco-2 and SNU-C4 cells.

Effect of AREG on cetuximab-mediated cell cycle, apoptosis, and colony forming activities in colorectal cancer cells. In (A) Caco-2 and (B) SNU-C4, cell cycle analysis was conducted using flow cytometry after propidium iodide (PI) staining. The cells were seeded in 60-mm plates, and then treated with or without cetuximab or AREG for 72 h. (C) Caco-2 and (D) SNU-C4 cells were treated with the indicated concentrations of cetuximab. The cells were also exposed to the indicated concentrations of AREG for 96 h. Apoptosis was measured using the Caspase-Glo® 3/7 assay kit. Results indicate mean ± standard deviation. The values were compared using the Student’s t-test. *p < 0.05; **p < 0.01; ***p < 0.001. (E) Colony-forming assays were conducted to evaluate the long-term effect of AREG. AREG 50 ng/mL and cetuximab (10 μg/mL for Caco-2 and 20 μg/mL for SNU-C4) were added to the cells for 3 to 4 weeks. The colonies were quantified for optical density at 595 nm.
To verify whether AREG disturbs cetuximab-mediated apoptosis, the caspase-3/7-positive apoptotic cells were measured in Caco-2 and SNU-C4 cells with AREG in the presence or absence of cetuximab (Fig. 4C,D). AREG alone did not alter the number of apoptotic rates. Notably, in the presence of AREG, the cetuximab-mediated apoptotic cells were significantly suppressed in both Caco-2 and SNU-C4 cells compared with AREG-free condition.
In addition, the colony-forming assays were carried out to analyze the prolonged effect of cetuximab in the presence or absence of AREG on the proliferation of Caco-2 and SNU-C4 cells (Fig. 4E). We observed that cetuximab monotherapy significantly decreased the growth of colonies compared with the control in Caco-2 and SNU-C4 cells. In the absence of cetuximab treatment, high AREG did not significantly increase cell proliferation in Caco-2 cells, whereas in SNU-C4 cells, the proliferation was significantly increased in the presence of AREG relative to that in the control. The colony-forming assay results appeared to be correlated with the western blot data (Fig. 3B): SNU-C4 cells showed higher susceptibility to the effect of AREG on EGFR downstream molecules, AKT and ERK1/2, possibly resulting in higher proliferation, compared with Caco-2 cells. Notably, in the presence of AREG, the anti-proliferative effects of cetuximab were significantly decreased in both Caco-2 and SNU-C4 cells compared to AREG-free environment.
AREG does not confer additional cetuximab resistance in cetuximab acquired resistance cell lines
We established cetuximab-resistant cell lines (Caco-2_R and SNU-C4_R) and their corresponding parental cells (Caco-2_P and SNU-C4_P) to evaluate the effects of AREG in cetuximab-resistant conditions (Fig. 5A). Cetuximab resistance was acquired by gradual dose escalation for 3 months. Caco-2_R and SNU-C4_R cells represented approximately eightfold (R1: 495.4 and R2: 440.7 μg/mL) and fivefold (R1: 442.2 and R2: 511.6 μg/mL) higher GI50 values to cetuximab, respectively, compared with their corresponding parental cells (Fig. 5B).

Effect of AREG on cell proliferation and EGFR signaling pathways in cetuximab-resistant colorectal cancer cells. (A) Scheme of the establishment of the cetuximab acquired resistance model (CET-R). Cetuximab was treated in a dose-escalating manner for 3 months. After acquiring resistance, cells were maintained with 400 μg/mL cetuximab. (B) Cytotoxicity assay was conducted to evaluate the resistance to cetuximab between parental and CET-R. Cetuximab was administered for 5 days, and cell viability was confirmed using CellTiter-Glo. (C) Western blotting of signaling molecules for an examination of the response to AREG in CET-R. 50 ng/mL AREG was added 15 min after 24 h serum starvation. Screening of EGFR down signaling molecules by Western blotting (D) and colony-forming assay (E) were carried out to check effects of AREG on cetuximab in CET-R. The conditions of treatment with cetuximab and AREG were equal in Figs. 3B and 4E.
We examined the influence of AREG on the EGFR signaling pathways by western blot analysis in Caco-2_R and SNU-C4_R cells (Fig. 5C). In line with our western blot data in the previous section (Fig. 3B), AREG increased the phosphorylation of EGFR, AKT, and ERK1/2 in SNU-C4_P cells, whereas SNU-C4_R cells did not respond to AREG. In contrast, in Caco-2_P cells, high AREG did not cause AKT phosphorylation, but led to ERK1/2 phosphorylation. High AREG slightly increased the expression of p-ERK1/2 in Caco-2_R1 cells but had no such effect in Caco-2_R2. In Caco-2_R1 and Caco-2_R2 cells, p-AKT slightly decreased in the presence of AREG (Supplementary Fig. S1).
Further, in cetuximab resistant cells, EGFR and its downstream signaling molecules were examined after cetuximab treatment in the presence or absence of AREG (Fig. 5D). Based on the anti-proliferation assay data (Fig. 5B), relatively more resistant clones, Caco-2_R1 and SNU-C4_R2, were selected for the study. In these cetuximab-resistant cells, additional western blot analysis showed that, after cetuximab treatment, the presence or absence of AREG did not elicit apparent differences in terms of EGFR downstream signaling (Fig. 5D).
Next, we carried out colony-forming assays to determine the prolonged effect of AREG on proliferation in cetuximab-resistant cells (Fig. 5E). Colony-forming assays of Caco-2_P and SNU-C4_P cells showed similar results as in cetuximab-naïve cells (Fig. 4E). However, in Caco-2_R1 and SNU-C4_R2 cells, the anti-proliferative effects of cetuximab were not significant both in the presence and absence of AREG.
Discussion
This study was conducted to investigate the effects of AREG on cetuximab response in patients with metastatic colorectal cancer and determine the mode of action via the EGFR signaling pathway using in vitro experiments with both cetuximab-naïve and resistant colorectal cancer cell lines. Our results indicated that high baseline plasma AREG levels could predict PFS after first-line cetuximab plus FOLFIRI chemotherapy in patients with RAS/BRAF wild-type colorectal cancer. Our in vitro study also indicated that AREG significantly enhanced the growth of cetuximab-sensitive RAS/BRAF wild-type colorectal cancer cell lines with increased phosphorylation of AKT and ERK1/2. However, after progression of first-line chemotherapy, the plasma AREG levels could not predict treatment outcomes of second-line non-EGFR-directed treatment, suggesting that AREG may not be a prognostic biomarker in these patients. In line with this clinical finding, in cetuximab-resistant cell lines, AREG neither impeded the signal transduction inhibition by cetuximab nor was associated with the cetuximab-induced anti-proliferative effects.
It is still controversial whether AREG protein or mRNA expression levels are prognostic factors in patients with colorectal cancer who did not undergo cetuximab- or panitumumab-based chemotherapy. For example, Ohchi et al. showed that higher AREG expression by IHC was significantly associated with worse relapse-free survival and OS16. In contrast, Stahler et al. reported that AREG mRNA expression in tumor tissue was not prognostic17. Besides, several studies demonstrated that higher AREG expression levels by tissue immunohistochemistry (IHC) and serum ELISA were associated with worse pathologic parameters18,19,20.
Other studies evaluated the predictive role of mRNA expression of AREG in the patients receiving cetuximab- or panitumumab-based treatment. In most studies, mRNA expression of AREG predicted better response to anti-EGFR treatment. However, mRNA-based analyses are less likely to be reproducible and thus difficult to adopt in clinical practice. Yonesaka et al. evaluated plasma AREG levels by ELISA in colorectal cancer patients treated with cetuximab in combination with cytotoxic chemotherapy21. However, treatment in the patients were heterogeneous in terms of combined cytotoxic chemotherapy and prior lines of treatment. In addition, this study evaluated the role of plasma AREG levels in combination with heregulin, therefore, further validation is required to determine whether plasma AREG levels by ELISA could be a predictive biomarker for cetuximab-based treatment.
Furthermore, our study group previously conducted a biomarker study of serum AREG in patients with HER2-positive breast cancer treated with trastuzumab plus taxane and found that high AREG levels were associated with poor clinical outcomes22. EGFR and HER2 belong to the epidermal growth factor family of receptor tyrosine kinases (ErbBs), which includes EGFR, HER2, HER3, and HER423. HER2 is a receptor which has no identified ligand but shares the same downstream pathway with EGFR. AREG is a ligand of EGFR, which can form a heterodimer with HER2, leading to down-stream signaling activation24. Therefore, our interpretation in our previous study was that AREG indirectly confers trastuzumab resistance via EGFR activation in HER2-positive breast cancer. In contrast, in the present study, AREG-induced cetuximab resistance may be attributed to direct EGFR activation in RAS/BRAF wild-type colorectal cancer.
We further analyzed the biological mechanisms of AREG using in vitro experiments. In our clinical data, plasma AREG was a negative predictive biomarker of cetuximab-based treatment, which was validated in our in vitro study as well. However, it was no longer predictive after patients acquired cetuximab resistance. Thus, it is speculated that AREG interferes with the inhibitory effect of cetuximab on EGFR. To prove the hypothesis, we generated cells with acquired cetuximab resistance using cetuximab-sensitive Caco-2 and SNU-C4 cell lines. As a result, AREG could not activate the EGFR down-stream signaling in cetuximab-resistant Caco-2 and SNU-C4 cells.
Based on these findings, it is hypothesized that AREG inhibition might have a therapeutic potential in patients receiving anti-EGFR treatment. Several preclinical studies have investigated the effect of AREG inhibition using anti-AREG antibodies or AREG shRNA in various human cancer cell lines25,26,27. However, there is still no clinical data on AREG inhibition in patients with high plasma AREG levels receiving anti-EGFR treatment. Our results may provide evidence of an AREG-targeting strategy in patients with RAS/BRAF wild-type colorectal cancer receiving cetuximab-based chemotherapy.
One of the limitations of our study is a small patient population and lack of validation cohort. Hence, it should be further validated in a larger patient cohort whether amphiregulin is an independent biomarker in this patient population. In addition, our experiment did not include various mechanisms of action of cetuximab including the antibody-dependent cell mediated cytotoxicity28. In this regard, further research is warranted.
In conclusion, high baseline plasma AREG levels predicted worse PFS in patients with RAS/BRAF wild-type metastatic colorectal cancer treated with palliative first-line cetuximab plus FOLFIRI chemotherapy, but not with non-EGFR-directed second-line treatment. AREG reversed the inhibitory effects of cetuximab in RAS/BRAF wild-type cetuximab-naïve colorectal cancer cells, but not in cetuximab-resistance cells in vitro. Our results suggest that plasma AREG is a potential biomarker to predict clinical outcomes after cetuximab-based chemotherapy.
Patients and methods
Patient population
In total, 35 consecutive patients were enrolled in this study between May 2015 and September 2019. The patients were treated with palliative first-line cetuximab plus FOLFIRI chemotherapy at Seoul National University Bundang Hospital, Seongnam, Korea. The chemotherapy regimen consisted of intravenous cetuximab 500 mg/m2 combined with FOLFIRI. The FOLFIRI regimen consisted of an intravenous infusion of irinotecan 180 mg/m2 on day 1, followed by leucovorin 400 mg/m2 infusion and 5-fluorouracil 400 mg/m2 bolus, and then 5-fluorouracil 2400 mg/m2 infusion over 46 h every 2 weeks. Before the first dose of cetuximab + FOLFIRI treatment, blood samples were collected after obtaining informed consent during two preceding prospective studies: pharmacogenomic study in patients with advanced solid tumors receiving palliative chemotherapy (IRB registration No. B-1603/340-305) and biomarker study in patients with metastatic colorectal cancer (IRB registration No. B-1211/180-007). The present study was conducted using these consecutively collected blood samples. Retrospective medical record reviews were conducted for these patients. The study was approved by the institutional review board of Seoul National University Bundang Hospital (IRB registration No. B-2102-667-301) and performed in accordance with the ethical principles of the Declaration of Helsinki.
Plasma sample collection and AREG quantification
Plasma was separated from the peripheral blood samples of patients by centrifugation at 3000×g for 10 min. Human recombinant AREG levels in the plasma samples were quantified using a Quantikine Human AREG ELISA (enzyme-linked immunosorbent assay) kit (DAR00, R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocol.
Cell culture
The human colorectal cancer cell lines, SNU-C5, SW480, DLD-1, HCT-15, Caco-2, and SNU-C4, were obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea). The cells were cultured with RPMI 1640 medium (LM011-51, WELGENE, Gyeongsan, Korea) supplemented with 10% fetal bovine serum (FBS) (26140079, Gibco, Waltham, MA, USA) and 1% penicillin–streptomycin (LS202-02, WELGENE) solution. SW48 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured with DMEM medium (11995065, Gibco) supplemented with 10% FBS and 1% penicillin–streptomycin. Cetuximab-resistant cells were maintained in a medium with 400 μg/mL cetuximab. All the cells were maintained every 3–4 days and cultured at 37 ℃ in a humidified 5% CO2 incubator.
Cell viability assay
Cells were seeded in 384-well plates (781080, Greiner Bio-One, Kremsmünster, Austria) at 1,000 cells/well 1 day prior to cetuximab treatment. At day 0 and 5, cell viability was measured by CellTiter-Glo luminescent cell viability assay (G7573, Promega, Madison, WI, USA) according to the manufacturer’s protocol. Luminescence was measured with the Synergy H1 microplate reader (BioTek, Winooski, VT, USA). The half-maximal growth inhibition (GI50) values were determined using CalcuSyn 2.0 software (Biosoft, Cambridge, UK).
Establishment of acquired cetuximab-resistant colorectal cancer cell lines
Cetuximab-resistant Caco-2 and SNU-C4 cell lines were generated by continuous exposure to increasing concentrations of cetuximab up to 400 μg/mL for 3 months, and then maintained in cetuximab 400 μg/mL-containing media for more than 6 months (Fig. 3A). Parental Caco-2 or SNU-C4 cells were simultaneously maintained by continuous exposure to cetuximab-free media. Parental (P) or cetuximab resistant (R) cells were designated as Caco-2_P, Caco-2_R (clone 1 and 2), SNU-C4_P, and SNU-C4_R (clone 1 and 2). Caco-2_R (clone 1 and 2) and SNU-C4_R (clone 1 and 2) cell lines showed approximately five to ninefold higher GI50 values than their corresponding parental cells.
Western blot analysis
The cells were washed with cold Dulbecco’s phosphate-buffered saline (PBS) (LB 001-02, WELGENE) and then lysed using radio immunoprecipitation assay (RIPA) buffer with protease and phosphatase inhibitors. The bicinchoninic acid (BCA) protein assay kit (23227, Thermo Fisher Scientific, Waltham, MA, USA) was used for protein quantification. Protein samples were separated on 8–10% sodium dodecyl sulfate (SDS)-polyacrylamide gel and then transferred onto 0.45 μM pore polyvinylidene fluoride (PVDF) membrane (IPVH00010, Millipore, Burlington, MA, USA). The membrane was blocked with 5% skim milk (70166, Sigma-Aldrich, St. Louis, MO, USA) solution and was probed with primary antibody at 4 ℃ overnight. After incubation with horseradish peroxidase (HRP)-conjugated secondary antibody for 2 h at room temperature, the blot was detected using ChemiDoc Touch Imaging System (BioRad, Hercules, CA, USA). Protein expression was analyzed by ImageJ software (NIH, Baltimore, MD, USA).
Antibodies and reagents
Phospho-EGFR (Y1068) (#3777), EGFR (#2646), phospho-AKT (S473) (#4058), AKT (#4685), phospho-ERK1/2 (T202/Y204) (#4376), ERK1/2 (#4695), and vinculin (#13901) primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Vinculin was used as loading control. HRP-conjugated anti-rabbit (#111-035-003) and mouse (#115-035-003) secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA, US). Cetuximab (A2000) was purchased from Selleckchem (Houston, TX, USA). The recombinant human AREG (262-AR-100) was purchased from R&D Systems.
Cell cycle analysis
The Caco-2 and SNU-C4 cells were plated in 60-mm culture dishes at a density of 1 × 106 cells per dish for 72 h, and then treated with AREG and/or cetuximab. Next, the cells were harvested at 72 h by trypsinization, centrifugation, and fixation with 70% ethanol. A staining solution containing 0.05 μg/mL of propidium iodide (PI) and 0.2 mg/mL of RNase was used for DNA staining. Attune® NxT cytometer (Thermo Fisher Scientific) was used for cell cycle analysis. The flow cytometry data was processed using FlowJo software (BD Biosciences, San Jose, CA, USA).
Apoptosis assay
Apoptosis assays were performed according to manufacturer’s recommendations using the Caspase-Glo® 3/7 assay kit (Promega). Significant differences between values obtained in DMSO control groups and different treatment groups were determined using the Student’s t-test.
Colony-forming assay
Cells were seeded in 6-well plates at 1000 cells/well (30006, SPL Life Science, Pocheon, Korea). The treatments, 10 and 20 μg/mL of cetuximab and 50 ng/mL of AREG, were added three times weekly for 3 or 4 weeks to SNU-C4 or Caco-2, respectively. The cell colonies were stained with Coomassie brilliant blue R-250 solution (1610436, BioRad) for 2 h at room temperature. The representative colony images were photographed by ChemiDoc Touch Imaging System. The procedure of colony de-staining was performed using a 1% SDS solution to analyze the relative colony area. The relative colony area was determined at 595 nm using a Synergy H1 microplate reader. Protein expression was analyzed by ImageJ software (NIH, Baltimore, MD, USA).
Statistical analysis
The Eastern Cooperative Oncology Group (ECOG) scale was used to evaluate performance status (PS) of patients. KRAS, NRAS, and BRAF mutation status was evaluated using polymerase chain reaction (PCR) or next generation sequencing as a part of clinical practice. Microsatellite instability was determined according to the revised Bethesda guidelines (14). Disease status was evaluated using computed tomography (CT) scans or magnetic resonance imaging (MRI). Treatment response was assessed in accordance with the response evaluation criteria in solid tumor (RECIST) version 1.129.
To identify the optimal cut-off value for plasma AREG levels, Maxstat R package (a maximal chi-square method) was used in R 4.0.3 (R Development Core Team, Vienna, Austria). PFS was defined from the start of systemic chemotherapy to the date of documented disease progression, relapse, or death of any cause. The second PFS (PFS2) was defined from the start of second-line anticancer treatment to the date of documented disease progression, relapse, or death of any cause. OS was measured from the start day of systemic chemotherapy to the date of death of any cause. The second OS (OS2) was defined from the start of the second-line anticancer treatment to the date of death of any cause. The survival outcomes were calculated using the Kaplan–Meier method and compared with the log-rank test.
All in vitro experiments were conducted at least three times independently. The differences between groups were evaluated by Student’s t-test using GraphPad Prism 5 software (San Diego, CA, USA). Error bars represent mean ± standard deviation. Two-sided p-values < 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, and ***p < 0.001).
Data availability
The datasets generated during and/or analysis during the current study are available from the corresponding author on reasonable request.
References
Douillard, J. Y. et al. Final results from PRIME: Randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann. Oncol. 25, 1346–1355. https://doi.org/10.1093/annonc/mdu141 (2014).
Douillard, J. Y. et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME study. J. Clin. Oncol. 28, 4697–4705. https://doi.org/10.1200/JCO.2009.27.4860 (2010).
Qin, S. et al. Efficacy and tolerability of first-line cetuximab plus leucovorin, fluorouracil, and oxaliplatin (FOLFOX-4) versus FOLFOX-4 in patients with RAS wild-type metastatic colorectal cancer: The open-label, randomized, phase III Tailor trial. J. Clin. Oncol. 36, 3031–3039. https://doi.org/10.1200/JCO.2018.78.3183 (2018).
Van Cutsem, E. et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 360, 1408–1417. https://doi.org/10.1056/NEJMoa0805019 (2009).
Van Cutsem, E. et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 33, 692–700. https://doi.org/10.1200/JCO.2014.59.4812 (2015).
Berasain, C. & Avila, M. A. Amphiregulin. Semin. Cell Dev. Biol. 28, 31–41. https://doi.org/10.1016/j.semcdb.2014.01.005 (2014).
Fontanini, G. et al. Evaluation of epidermal growth factor-related growth factors and receptors and of neoangiogenesis in completely resected stage I-IIIA non-small-cell lung cancer: Amphiregulin and microvessel count are independent prognostic indicators of survival. Clin. Cancer Res. 4, 241–249 (1998).
Normanno, N. et al. Expression of messenger RNA for amphiregulin, heregulin, and cripto-1, three new members of the epidermal growth factor family, in human breast carcinomas. Breast Cancer Res. Treat. 35, 293–297. https://doi.org/10.1007/BF00665981 (1995).
Saeki, T. et al. Differential immunohistochemical detection of amphiregulin and cripto in human normal colon and colorectal tumors. Cancer Res. 52, 3467–3473 (1992).
Martini, G. et al. Resistance to anti-epidermal growth factor receptor in metastatic colorectal cancer: What does still need to be addressed? Cancer Treat Rev. 86, 102023. https://doi.org/10.1016/j.ctrv.2020.102023 (2020).
Jacobs, B. et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J. Clin. Oncol. 27, 5068–5074. https://doi.org/10.1200/JCO.2008.21.3744 (2009).
Khambata-Ford, S. et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 25, 3230–3237. https://doi.org/10.1200/JCO.2006.10.5437 (2007).
Stahler, A. et al. Amphiregulin expression is a predictive biomarker for EGFR inhibition in metastatic colorectal cancer: Combined analysis of three randomized trials. Clin. Cancer Res. 26, 6559–6567. https://doi.org/10.1158/1078-0432.CCR-20-2748 (2020).
Yen, H. Y. et al. Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc. Natl. Acad. Sci. U.S.A. 112, 6955–6960. https://doi.org/10.1073/pnas.1507329112 (2015).
Tanaka, T. et al. Ligand-activated epidermal growth factor receptor (EGFR) signaling governs endocytic trafficking of unliganded receptor monomers by non-canonical phosphorylation. J. Biol. Chem. 293, 2288–2301. https://doi.org/10.1074/jbc.M117.811299 (2018).
Ohchi, T. et al. Amphiregulin is a prognostic factor in colorectal cancer. Anticancer Res. 32, 2315–2321 (2012).
Stahler, A. et al. Influence of mRNA expression of epiregulin and amphiregulin on outcome of patients with metastatic colorectal cancer treated with 5-FU/LV plus irinotecan or irinotecan plus oxaliplatin as first-line treatment (FIRE 1-trial). Int. J. Cancer 138, 739–746. https://doi.org/10.1002/ijc.29807 (2016).
Yamada, M. et al. Amphiregulin is a promising prognostic marker for liver metastases of colorectal cancer. Clin. Cancer Res. 14, 2351–2356. https://doi.org/10.1158/1078-0432.CCR-07-4499 (2008).
Li, X. D. et al. Amphiregulin and epiregulin expression in colorectal carcinoma and the correlation with clinicopathological characteristics. Onkologie 33, 353–358. https://doi.org/10.1159/000315380 (2010).
Chayangsu, C., Khunsri, S., Sriuranpong, V. & Tanasanvimon, S. The correlations between serum amphiregulin and other clinicopathological factors in colorectal cancer. J. Gastrointest. Oncol. 8, 980–984. https://doi.org/10.21037/jgo.2017.08.15 (2017).
Yonesaka, K. et al. Combined analysis of plasma amphiregulin and heregulin predicts response to cetuximab in metastatic colorectal cancer. PLoS ONE 10, e0143132. https://doi.org/10.1371/journal.pone.0143132 (2015).
Kim, J. W. et al. Amphiregulin confers trastuzumab resistance via AKT and ERK activation in HER2-positive breast cancer. J. Cancer Res. Clin. Oncol. 142, 157–165. https://doi.org/10.1007/s00432-015-2012-4 (2016).
Wieduwilt, M. J. & Moasser, M. M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol. Life Sci. 65, 1566–1584. https://doi.org/10.1007/s00018-008-7440-8 (2008).
Scaltriti, M. & Baselga, J. The epidermal growth factor receptor pathway: A model for targeted therapy. Clin. Cancer Res. 12, 5268–5272. https://doi.org/10.1158/1078-0432.CCR-05-1554 (2006).
Carvalho, S. et al. An antibody to amphiregulin, an abundant growth factor in patients’ fluids, inhibits ovarian tumors. Oncogene 35, 438–447. https://doi.org/10.1038/onc.2015.93 (2016).
Lindzen, M., Lavi, S., Leitner, O. & Yarden, Y. Tailored cancer immunotherapy using combinations of chemotherapy and a mixture of antibodies against EGF-receptor ligands. Proc. Natl. Acad. Sci. U.S.A. 107, 12559–12563. https://doi.org/10.1073/pnas.1006218107 (2010).
Baillo, A., Giroux, C. & Ethier, S. P. Knock-down of amphiregulin inhibits cellular invasion in inflammatory breast cancer. J. Cell Physiol. 226, 2691–2701. https://doi.org/10.1002/jcp.22620 (2011).
Kimura, H. et al. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 98, 1275–1280. https://doi.org/10.1111/j.1349-7006.2007.00510.x (2007).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247. https://doi.org/10.1016/j.ejca.2008.10.026 (2009).
Acknowledgements
This research was funded by Seoul National University Bundang Hospital Research Fund (No. 14-2016-001) and the National Research Foundation of Korea (NRF) Grant funded by the Korea government (MIST) (No. 2018R1D1A1A02086240). The funders had no role in the study design, data collection and analysis, the decision to publish, or the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.A.K., H.J.P., K.J.K. and J.-W.K. drafted the article; Prof. J.-W.K. and K.W.L. revised it critically for important intellectual content; S.A.K., H.J.P., K.J.K., J.-W.K., J.H.S., M.L.N., K.J.S., J.Y.L., S.H.K., J.-O.L., J.W.K., Y.J.K., J.H.K., S.-M.B., J.S.L. and K.-W.L. contributed to acquisition of data, analysis and interpretation of data, and approved the final version to be published.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, SA., Park, H., Kim, KJ. et al. Amphiregulin can predict treatment resistance to palliative first-line cetuximab plus FOLFIRI chemotherapy in patients with RAS wild-type metastatic colorectal cancer. Sci Rep 11, 23803 (2021). https://doi.org/10.1038/s41598-021-03197-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03197-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
