
Abstract
Chronic diabetic complications affect multiple organs causing widespread organ damage. Although there are some commonalities, the phenotype of such changes show tissue specific variation. Given this, we examined whether differences in circular RNA (circRNA) mediated gene regulatory mechanisms contribute to changes in gene expression at the basal level and in diabetes. CircRNAs are single-stranded RNA with covalently closed loop structures and act as miRNA sponges, factors of RNA splicing, scaffolding for proteins, regulators of transcription, and modulators of the expression of parental genes, among other roles. We examined heart and retinal tissue from Streptozotocin-induced diabetic mice with established diabetes related tissue damage and tissue from non-diabetic controls. A custom array analysis was performed and the data were analysed. Two major circRNA mediated processes were uniquely upregulated in diabetic heart tissue, namely, positive regulation of endothelial cell migration and regulation of mitochondria: mitochondrial electron transport. In the retina, circRNAs regulating extracellular matrix protein production and endothelial to mesenchymal transition (EndMT) were found to be upregulated. The current study identified regulatory and potential pathogenetic roles of specific circRNA in diabetic retinopathy and cardiomyopathy. Understanding such novel mechanisms, may in the future, be useful to develop RNA based treatment strategies.
Similar content being viewed by others

Introduction
In diabetes, hyperglycemia causes changes in cellular transcription. Endothelial cells (ECs) alter their synthetic phenotype when in a high glucose environment, and subsequently affect other cells in the target organs of diabetic complications including the heart and retina1,2. Glucose-induced biochemical alterations converge on the EC nucleus and change gene transcription3,4.
Alterations of inflammatory cytokines, extracellular matrix (ECM) proteins and aberrant angiogenesis are some of the key pathologic processes that cause impaired cellular and organ functions1,2,3,4. Although some of the pathologic processes are similar in the retina and heart, such as increased ECM protein production, others, such as angiogenesis, vary between these organs5,6. Angiogenesis is a main feature in proliferative diabetic retinopathy, however this process becomes impaired in the diabetic heart7. Hence, transcriptional and post-transcriptional regulatory mechanisms likely vary between different diabetic organs.. Previously, we have identified that a concerted effort of multiple transcription factors, transcription co-activators and microRNAs/long non-coding RNAs regulate specific vasoactive molecules and ECM proteins in the context of diabetic complications8,9,10,11. However, other recently identified non-coding RNAs such as circular RNAs (circRNAs) also play a significant role in this concert.
CircRNAs are single-stranded RNA molecules. In contrast to linear RNAs, they demonstrate closed loop structures generated as a result of back-splicing12. Due to circularisation, these molecules are devoid of 5-prime caps or poly-A tails. Originally discovered more than fourdecades ago, in recent yearsnew sequencing technologies have been developed allowing for the identificationof circular RNA isoforms from a large number of genes, and demonstratingthat these circRNAs are transcribed in both humans and mice13,14. In addition, expression of circRNAS is cell type dependent. It has been estimated that in human, circRNA expression may account for 1% of mRNAs14. The most common mechanism of circularization involves the spliceosome machinery and occurs in pre-messenger RNAs conventionally transcribed by RNA polymerase II (RNAP II) from nuclear-encoded genes. Physiological RNA circularization by the spliceosome can proceed by three principle mechanisms15,16. CircRNAs exhibit higher stability and act as miRNA sponges, factors of RNA splicing, scaffolding for proteins, regulators of transcription, and modulators of the expression of parental genes. circRNAs can also serve as biomarkers for numerous diseases17,18,19. To date, several circRNAs have been functionally studied in the context of cardiometabolic disease20, The ubiquity of circRNA and their specific regulation could significantly alter our perspective on post-transcriptional regulation and the roles that RNA can play in the cell, making circRNAs a promising candidate for diagnostic modalities and therapies.
Overall, non-coding RNAs play a significant role in all biological processes and diseases. The role of circRNAs has become increasingly important among non-coding RNAs. The availability of RNA deep sequencing and bioinformatics has started to reveal the importance of circRNAs as regulators of gene expression in chronic diabetic compliations. Specifically, in the retina, circHIPK3 acts as an endogenous miR-30a-3p sponge to sequester and inhibit miR-30a-3p activity, which leads to increased vascular endothelial growth factor-C expression21. Studies have demonstrated that circRNAs can be methylated by N6-methyladenosine (m6A), which is the most abundant base modification of RNA, leading to promotion of efficient initiation of protein translation from circRNAs in human cells16,22. m6A translation is enhanced by METTL3 (methyltransferase-like 3) and METTL14 (methyltransferase-like 14), and inhibited by demethylase FTO (obesity-associated protein)22. CircRNA Col1A2 was also found to promote angiogenesis through miR29b/VEGF23. Other circular RNAs, altered in diabetic retinopathy include circular DNMT3B and circRNA_0084043, each working through various pathways24,25. In the heart, 58 significantly differentially expressed circRNAs were identified in db/db mice, a model of type 2 diabetes26. Also identified are alterations of circRNA_010567 and circRNA_000203 working through various pathways to regulate specific transcript altering fibrosis related genes27,28.
The identification of differential expression of various circular RNAs in the heart and retina is important to understanding disease etiologyas diabetes affects these organs differently. For example, abnormal angiogenesis is seen in the retina in diabetes, whereas lack of angiogenesis is observed in the heart6,7. Hence, it is conceptually possible that in diabetes, gene transcription and the regulatory mechanisms thereof, will also be different in these two organs. The aim of the current study if to have a better understanding of the differential expression of circRNA expression in both the basal and diabetic state. This approach will lead to better understanding of the pathogenetic mechanisms of transcription and subsequent tissue damage in diabetic patients.
Methods
Animal models
The Western University Council for Animal Care Committee approved all animal experiments, which were performed in accordance with The Guide for the Care and Use of Laboratory Animals (NIH Publication 85–23, revised in 1996). Western’s Animal Care Committee is responsible for overseeing all aspects of animal ethics, care and use. Mice (C57/BL6 background; 22–24 g, 8 weeks old) were obtained (Charles River, Wilmington, USA) and randomly divided into control and diabetic groups. As our previous data were obtained from male mice and for cost containment, we used only male mice for this initial study. Streptozotocin (STZ) (50 mg/kg IP, 5 injection on consecutive days) was used to generate a type 1 diabetic animal model. Age- and sex-matched littermate controls received identical volumes of citrate buffer. Diabetes was confirmed by measuring blood glucose (> 16.7 mmol/L) from a tail vein using a glucometer. Animals were monitored for changes in body weight and blood glucose. After 8 weeks of diabetes, mice (n = 6/group) were euthanized. Retinal and left ventricular tissues were collected and immediately frozen for further analysis. A small portion of the cardiac tissue from each mouse was formalin fixed, paraffin embedded and stained with hematoxylin/eosin and trichome stain for morphologic analysis. Animal monitoring and tissue collection have been previously described29,30. The microarray study (please see below) included 3 control mice and 3 diabetic mice, with both retina and heart samples collected from each of the 6 mice. This animal study is reported in accordance with ARRIVE guidelines.
Echocardiography
Echocardiography was used to measure possible cardiac functional alterations in diabetes using previously described methodology29,30. Animals were anesthetized (1.5% inhaled isoflurane) and examined on a warm handling platform. A 40-MHz linear array transducer (MS-550D) and Vevo 2100 preclinical ultrasound system (VisualSonics) was used. Left ventricular fractional shortening (FS) was used as the cardiac contractile function index. Pulse-waved color flow-guided Doppler recordings of maximal early (E) and late (A) diastolic transmittal flow velocities and Doppler tissue imaging recordings of peak E = velocity and peak A = velocity were collected. Mitral inflow patterns (E/A ratio) was used to assess diastolic dysfunction as described29,30.
Histological analysis
Tissues collected in formalin were embedded in paraffin and 5 µm sections were cut. The tissues were stained with hematoxylin and eosin and trichrome stain following standard procedure29,30.
RNA analysis
TRIzol™(Invitrogen) was used to extract total RNA. The quality of the extracted RNA was checked spectrophotometrically and via gel analyses. From a portion of the extracted RNA, cDNA for PCR was synthesized using high-capacity cDNA reverse-transcription kit (Applied Biosystems, Burlington, ON). To examine transcriptional alterations in diabetes, mRNA expression of specific transcripts (Collagen, fibronectin) were performed using real-time RT-PCR using a LightCycler (Roche Diagnostics). The housekeeping gene β-actin was used to normalize the data10,29,30.
The remaining RNA samples were then shipped to Arraystar for circular RNA array analysis using the Arraystar Mouse circRNA Array V2 (8 × 15 K) panel.
circRNA microarray
The purity and concentration of total RNA from each sample was quantified using the NanoDrop ND-1000. The integrity of RNA was assessed by electrophoresis on a denaturing agarose gel. The sample preparation and microarray hybridization were performed based on Arraystar’s in-house protocols (Rockville, MD).
Briefly, total RNAs were digested with Rnase R (Epicentre, Inc.) to remove linear RNAs and enrich circular RNAs. Then, the enriched circular RNAs were amplified and transcribed into fluorescent cRNA utilizing a random priming method (Arraystar Super RNA Labeling Kit; Arraystar). The labeled cRNAs were purified by RNeasy Mini Kit (Qiagen) and hybridized onto the Arraystar Mouse circRNA Array V2(8 × 15 K, Arraystar). The concentration and specific activity of the labeled cRNAs (pmol Cy3/μg cRNA) were measured by NanoDrop ND-1000. 1 μg of each labeled cRNA was fragmented by adding 5 μl 10 × Blocking Agent and 1 μl of 25 × Fragmentation Buffer, then heated at 60 °C for 30 min, finally 25 μl 2 × Hybridization buffer was added to dilute the labeled cRNA. 50 μl of hybridization solution was dispensed into the gasket slide and assembled to the circRNA expression microarray slide. The slides were incubated for 17 h at 65 °C in an Agilent Hybridization Oven. The hybridized arrays were washed, fixed and scanned using the Agilent Scanner G2505C (Protocol adapted from in-house protocols developed by Arraystar (Rockville, MD).
Hierarchical clustering
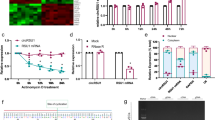
Hierarchical clustering of circRNAs in all samples was conducted using euclidean clustering for computing dissimilarity between rows and between columns. The expression levels of circRNAs were represented by a color scale where blue represents low expression levels and red represents high expression levels. Each column represents a unique sample type and each row represents a distinct circRNA (Supplementary Fig. 1).

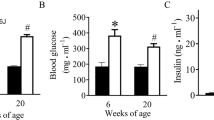
Diabetic (DIA) mice showed animals showed (A) reduced body weight and (B) hyperglycemia following 2 mo of diabetes compared with non-diabetic age- and sex-matched controls (CON). Echocardiographically diabetic animals also showed (C) increased fractional shortening (FS), (D) reduced mitral inflow pattern (E/A ratio). Analysis of (E, F) cardiac and (G, H) retinal tissues showed increased mRNA expression of fibronectin (FN) and collagen 1α1 (Col1a1) (*P = 0.05 or less vs CON, n = 6/group). (I) Trichrome stains showed focal scarring and collagen deposition (green stain in the myocardium of the diabetic animals. Such collagen deposition was not seen in the heart of non-diabetic animals. Such collagen deposition was not seen in the heart of (I) non-diabetic animals (magnification same for I and J).
Differentially expressed circRNAs
Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images. Quantile normalization and subsequent data processing were performed using the R software limma package31. circRNAs that had flags of ‘present’ or ‘marginally present’ for at least 3 out of 12 samples (as defined by GeneSpring software) were retained for further differential analyses. R (version 4.0.4) was used for all downstream data analysis. To mitigate batch effect, harman correction32 was implemented with a confidence limit of 0.875. Reducing the confidence limit further was found to cause a loss of biological information. Correction of batch effect was confirmed through analysis of pre-correction and post-correction principal component analysis. A total of eight comparisons were made as described in Table 1. Equal variance two-sided paired t-tests were conducted for the retina vs heart comparisons and equal variance two-sided unpaired t-tests were conducted for the control vs diabetic comparisons. Differentially expressed circRNAs were defined using a p-value threshold of 0.005 and an absolute fold change value threshold of 1.25.
GO term & KEGG pathway analysis
For each of the 8 directional comparisons, the 100 most significant genes were inputted into GO term and KEGG pathway analysis. The universe consisted of all unique genes corresponding to circRNA probes on the microarray. The genes corresponding to the differentially expressed circRNAs for each GO term and KEGG pathway were extracted. GO term and KEGG pathway analysis was conducted using goana and kegga functions in the limma R package.
circRNA/microRNA interactions
circRNA/microRNA interactions were predicted with Arraystar's home-made miRNA target prediction software based on TargetScan33 and miRanda34. The top 5 circRNA/microRNA interactions were prioritized by using the miRanda structure score.
miRNA to circRNA matchup
miRNA 1, miRNA 133a, miRNA-320, miRNA-195, miRNA-200b, miRNA-146a, and miRNA-9 have been previously established as differentially expressed miRNAs in diabetic tissue. We searched the tissue-specific circular RNAs database35 for these miRNAs and their associated circRNAs. From these identified circRNA's, we determined the circRNAs that were also differentially expressed as determined by our analysis. Specifically, miRNAs associated with circRNAs from the TSCD database from any tissue other than testis, were compared with our differentially expressed circRNAs. Only complete overlaps were considered valid for this analysis.
Results
Diabetic animals showed features of diabetic dysmetabolism
We initially established weather the mice demonstrate features characteristic of diabetic dysmetabolism. Following STZ induction, the mice and age and sex-matched controls were monitored for a period of 2 mo. Hyperglycemia was evident in the diabetic animals along with reduced body weight (Fig. 1) and with polyuria, glycosuria (not shown), distinctive of poorly controlled diabetes. No such changes were seen in the non-diabetic control mice.
Diabetic animals showed characteristic transcriptional and cardiac functional alterations
We performed functional analysis in the heartandechocardiographic assessment prior to sacrific. We have previously demonstrated that cardiac dysfunction, manifested as abnormalities of cardiac contractility is a characteristic feature of diabetic cardiomyopathy36. Hence, we examined whether these animals show similar functional defects. As expected, increased FS and reduced E/A ratio was present in the diabetic mice compared to non-diabetic control mice (Fig. 1).
To confirm whether these animals developed diabetes induced alterations of specific transcripts, we measured extracellular matrix (ECM) protein transcripts. Increased ECM protein production is a characteristic feature of all chronic diabetic complications including those involving the retina and heart1,4,10,11. In the current experiments, we also demonstrated increased ECM protein transcript production both in the heart and in the retina of diabetic animals compared to non-diabetic controls (Fig. 1). At the structural level, such changes were reflected in the trichrome stain where increased collagen deposition was noted in the heart of diabetic mice (Fig. 1).
circRNA differential expression
All of the contrasts between heart and retinal tissue in both diabetic and control mice showed differential expression in a number of circRNAs. Details of all differentially expressed circRNAs can be found in Supplementary Table 1a-h).
Tissue specific differences in diabetic circRNA expression shows upregulation of synaptic activity in retinal tissue and cardiac contractile pathways in heart tissue
At the basal level there were tissue specific variation of circRNA expression suggesting tissue specific differences of circRNA mediated regulatory mechanisms on gene expression (Fig. 2). In non-diabetic mice, cardiac tissue had significantly upregulated expression of 455 circRNAs (FC > 1.25, p < 5e-3) and retinal tissue had significantly upregulated expression of 236 circRNAs (FC > 1.25, p < 5e-3) (Supplemental Table 1).

Heatmap hierarchial clustering of all circRNAs across all samples shows global clustering of the retinal (R) and cardiac (H) tissues of diabetic (D) and non-diabetic control (C) mice.
The circRNA profiles of cardiac tissue and retinal tissues in mice who were diabetic or non-diabetic showed significant differences. In diabetic mice, cardiac tissue had significantly upregulated expression of 660 circRNAs (FC > 1.25, p < 5e-3) and retinal tissue had significantly upregulated expression of 776 circRNAs (FC > 1.25, p < 5e-3).
KEGG pathway analysis of differential circRNAs suggested pathways relating to synaptic activity were significantly upregulated in diabetic retinal tissue in comparison to diabetic cardiac tissue and included glutamatergic synapse (path:mmu04724, P.DE = 4.49e−4), D-gluatmine and D-glutamate metabolism (pat:mmu00471, P.DE = 3.01e−2) (Table 2). This was supported by the upregulated GO term analysis including structural constituent of synapse (GO:0,098,918, P.DE = 1.83e−4), AP-2 adaptor complex (GO:0,030,122, P.DE = 2.48e−4), and clathrin coat of endocytic vesicle (GO:0,030,128, P.DE = 3.51e−4) (Fig. 3A). Similarly, non-diabetic retinal tissue revealed upregulation in dopaminergic synapse (path:mmu04728, P.DE = 1.32e−2) and spontaneous neurotransmitter secretion (GO:0,061,669, P.DE = 8.92e−05), positive regulation of nervous system process (GO:0,031,646, P.DE = 1.67e−4) and regulation of neurotransmitter secretion (GO:0,046,928, P.DE = 2.94e−4) (Fig. 3B, Table 2).


Top 15 (P.DE < 0.05), differentially expressed GO terms by contrast. GO terms are represented with number of DE genes in term/number of genes shown both in brackets after each GO term and controlling dot colour (corresponding heatmap legend). Size of dot represents total N in term. (A) HD_RD_down (B) HC_RC_down (C) HD_RD_up (D) HC_RC_up.
Diabetic cardiac tissue revealed upregulation in cardiac contractile metabolic pathways including calcium signaling pathway (path:mmu04020, P.DE = 4.73e−4), oxytocin signaling pathway (path:mmu04921, P.DE = 6.61e−4), and adrenergic signaling in cardiomyocytes (path:mmu04261, P.DE = 4.77e−3) (Table 2). This was supported by the observed upregulation in GO terms including sarcolemma (GO:0,042,383, P.DE = 9.95e−08), Z-disc (GO:0,030,018 P.DE = 8.00e−05), and contractile fiber (GO:0,043,292, P.DE = 1.18e−4) (Fig. 3C). Similarly, in non-diabetic cardiac tissue we observed upregulation in cardiac contractile pathways including ubiquinone and other terpenoid-quinone biosynthesis (path:mmu00130, P.DE = 2.52e−4), citrate cycle (path:mmu00020, P.DE = 1.00e−3), and metabolic pathways (path:mmu0110, P.DE = 8.13e−3) (Table 2). This was supported by the observed upregulation in GO terms including oxidation–reduction process (GO:0,055,114, P.DE = 2.36e−05), mitochondrion (GO:0.0005739, P.DE = 3.33e−05), and tricarboxylic acid cycle enzyme complex (GO:0,045,239, P.DE = 7.39e−05) (Fig. 3D). Interestingly, circadian entrainment (path:mmu04713) was upregulated in diabetic cardiac tissue (P.DE = 1.51e−4) and upregulated significantly in control retinal tissue (P.DE = 3.69e−2).
Disease specific differences in diabetic circRNA expression shows upregulation of diabetic cardiomyopathy pathways
Non-diabetic cardiac tissue had 105 circRNAs whose expression was significantly upregulated (FC > 1.25, p < 5e−3) and diabetic cardiac tissue had 67 circRNAs whose expression was significantly upregulated (FC > 1.25, p < 5e−3). Non-diabetic retinal tissue had 6 circRNAs whose expression was significantly upregulated (FC > 1.25, p < 5e−3) and diabetic retinal tissue had 3 circRNAs whose expression was significantly upregulated (FC > 1.25, p < 5e−3).
Cardiac tissue in diabetic mice revealed upregulation in diabetic cardiomyopathy and cardiac contractility pathways including calcium signaling pathway (path:mmu04020, P.DE = 2.83e−3), oxidative phosphorylation (path:mmu00190, P.DE = 7.85e−3), cardiac muscle contraction (path:mmu04260, P.DE = 7.85e−3), diabetic cardiomyopathy (path:mmu05415, P.DE = 1.68e−2), and insulin secretion (path:mmu04911, P.DE = 2.89e−3) (Table 2). This was supported by the observed upregulation in GO terms including inner mitochondrial membrane protein complex (GO:0,098,800, P.DE = 1.34e−4), regulation of muscle system process (GO:0,090,257, P.DE = 6.16e−4), and mitochondrial protein complex (GO:0,098,798, P.DE = 3.87e−3) (Supplementary Fig. 1D). Similarly, in diabetic retinal tissue we observed upregulation in calcium signaling pathway (path:mmu04020, P.DE = 4.39e−2) and insulin secretion (path:mmu04911, P.DE = 2.45e−2) pathways (Table 2).
Cardiac tissue in non-diabetic mice revealed upregulation in carbohydrate digestion and absorption (path:mmu04973, P.DE = 2.82e−2) (Table 2). Non-diabetic retinal tissue showed upregulation of three circRNAs which were all associated with the Rmst gene (Table 3).
Given the previous identification by our group of miRNA’s that are known to be associated with these outcomes4,8,36,37,38, we queried miRNA match-ups between those candidates (miR 1, 133a, 320, 195, 200b, 146a and 9) for all differentially expressed circRNAs in this study and identified 30 circRNA-miRNA pairs (Table 4). Of particular interest are mmu_circRNA_36350 and mmu_circRNA_33461 which are upregulated in control heart as compared to diabetic heart and are known to act as sponges for miR-1 and miR-9, respectively.
Discussion
In this research, we have demonstrated qualitative and quantitative differences in circRNA expression in two tissues affected in chronic diabetic complications. We used a microarray based approach for analysis due to the fact that circular RNA identification requires high junction read counts which traditional RNA sequencing only provides at prohibitive costs. The Arraystar circRNA used in this study provides unambiguous and reliable circular junction-specific array probes of high sensitivity and specificity. Although both retina and heart were affected by diabetes and show some similarities (eg. ECM protein expression), there is significant structural, functional, and biochemical differences. Hence it is expected that such differences in the regulatory mechanisms exists. Further, the results show the presence of disease specific variations in circRNA expression and discordant GO pathway enrichments which are of particular interest. The comparison of control heart and diabetic heart implicates two major processes to be uniquely upregulated in diabetic heart tissue: (1) endothelial cells: positive regulation of blood vessel endothelial cell migration (P = 7.03E−03) and blood vessel endothelial cell migration (P = 4.28E−02); (2) mitochondria: mitochondrial electron transport, ubiqunol to cytochrome c (P = 1.6E−02) and mitochondrial protein complex (P = 3.87E−03). It has been well established that mitochondrial dysfunction is a characteristic abnormality in all chronic diabetic complications including diabetic cardiomyopathy6,38. The current data further support this notion and indicate that the critical mediator of such pathogenetic process are regulated by specific circRNAs.
Similarly, in the comparison of control retina to diabetic retina, extracellular matrix is uniquely upregulated in diabetic retinal tissue as evidenced by the enriched GO terms extracellular matrix (P = 1.09E−02), collagen-containing extracellular matrix (P = 1.10E−02). Further, endothelial to mesenchymal transition (EndMT) was uniquely overrepresented (P = 1.47E−02) along with regulation of mesenchymal cell proliferation (P = 3.09E−02), suggesting a possible unique mechanism for mesenchymal cells in diabetic retina. We have previously demonstrated this association in the retina and heart in diabetes. It has been hypothesized that EndMT may be a key mechanism causing tissue damage in all chronic diseases including diabetic cardiomyopathy and retinopathy30,38,39. This research further establishes such changes and identifies novel circRNA mediated regulation of such changes. Also, as previously mentioned, increased ECM protein production is a ubiquitous characteristic feature of chronic diabetic complications4,8,30,38. Our data indicate that these processes are further regulated by circRNA expression. It is however interesting to note that endMT related transcripts based on the GO term search were only significantly altered in the retina in diabetes in comparison with non-diabetic controls. However, other endothelial-related terms were upregulated in the diabetic heart (as compared to control heart) such as ‘blood vessel endothelial cell migration’ and ‘positive regulation of blood vessel endothelial cell migration’ and ‘regulation of blood vessel endothelial cell migration’. Failure to be picked up by the GO term in the heart may be related to analysis limitations or may reflect true biological differences. Similarly, GO terms related to mitochondrial processes were not upregulated in diabetic retina, however, several related terms are downregulated in the diabetic retina such as, negative regulation of mitochondrial RNA catabolic process, mitochondrial inner membrane peptidase complex, negative regulation of mitochondrial calcium ion concentration, mitochondrial RNA catabolic process, and regulation of mitochondrial RNA catabolic process. Further experiments and analyses of individual circRNA are needed to delineate these findings.
The pathological processes in chronic diabetic complications are indeed complex. Multiple pathogenetic mechanisms play roles in this concert. Epigenetic mechanisms in the form of acetylation, methylation and alterations of non-coding RNA likely all play a part in this symphony. We have previously demonstrated roles of specific lncRNAs and microRNAs in these processes8,9,36,37,38. Of specific relevance to this project, one of the mechanisms through which circRNA works is by sponging specific miRs. We have previously demonstrated alterations of specific miRs in chronic diabetic complications4,8,36,37,38. Hence, we specifically explored whether some of these miRs are regulated by the altered circRNA identified in this study. As predicted we found some of the altered circRNAs indeed regulate miRNAs known to play significant regulatory roles in diabetic cardiomyopathy and retinopathy.
There are few studies performed in chronic diabetic complications which interrogate circRNA expression levels. Interestingly, one of the differentially expressed circRNAs in the comparison of heart and retina in the diabetic mouse, mmu_circ_000203 has been previously identified to be upregulated in diabetic mouse myocardium40, and is downregulated in diabetic heart tissue in this study.
Interestingly, there are significant differences compared to other studies40,41,42. Such differences may result from variation in species (human vs rodent), type of diabetes (Type 1 vs Type 2), or duration of diabetes. Our study was also limited due to the inclusion of small number of animals. Although we monitored 6 animals per group we used 3 animals per group for were used for array analyses. Due to resource-related challenges, we had to focus on the male animals from which we were able to obtain high quality RNA. Nevertheless, the current study demonstrates alterations of circRNAs in two target organs of diabetic complications. However, further studies are required to characterize these changes to establish their role and potential clinical utilities.
In summary, we have demonstrated tissue- and diabetes-specific alterations of several circRNAs in the heart and retina. The current study also indicated regulatory and pathogenetic roles of these molecules in the context of diabetic retinopathy and cardiomyopathy. Understanding these novel pathogenetic mechanisms, may in the future, be useful to develop RNA based treatment strategies.
References
Kolluru, G. K., Bir, S. C. & Kevil, C. G. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int. J. Vasc. Med. 2012, 918267 (2012).
Bakker, W., Eringa, E. C., Sipkema, P. & van Hinsbergh, V. W. Endothelial dysfunction and diabetes: roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res. 335, 165–189 (2009).
Farhangkhoee, H. et al. Vascular endothelial dysfunction in diabetic cardiomyopathy: pathogenesis and potential treatment targets. Pharmacol. Ther. 111, 384–399 (2006).
Biswas, S. & Chakrabarti, S. Pathogenetic mechanisms in diabetic retinopathy: From molecules to cells to tissues. In Mechanisms of vascular defects in diabetes mellitus Vol. 17 (eds Kartha, C. C. et al.) 209–247 (Springer, 2017).
Giacco, F. & Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 107, 1058–1070 (2010).
Geraldes, P. & King, G. L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 106, 1319–1331 (2010).
Tahergorabi, Z. & Khazaei, M. Imbalance of angiogenesis in diabetic complications: The mechanisms. Int. J. Prev. Med. 3, 827–833 (2012).
Puthanveetil, P., Thomas, A. A. & Chakrabarti, S. microRNAs in diabetes- are they perpetrators in disguise or just epiphenomena?. ncRNA Endocrinol. 1, 31–43 (2014).
Biswas, S. & Chakrabarti, S. The multifaceted roles of lncRNAs in diabetic complications: A promising, yet perplexing paradigm. In RNA Technologies 11: The Chemical Biology of Long Non-coding RNA (eds Jurga, S. & Barciszewski, J.) 492–514 (Springer Nature, 2020). https://doi.org/10.1007/978-3-030-44743-4.
Chen, S. et al. Transcriptional co-activator p300 regulates glucose induced gene expression in the endothelial cells. Am. J. Physiol. Endocrinol. Metab. 298, E127–E137 (2010) (Epub Nov 10, 2009).
Chen, S., Khan, Z. A., Cukiernik, M. & Chakrabarti, S. Differential activation of NF-κB and AP-1 in mediating endothelin-dependent increased fibronectin synthesis in target organs of diabetic complications. Am. J. Physiol (Endocr. Physiol.) 284, 1089–1097 (2003).
Bolisetty, M. T. & Graveley, B. R. Circuitous route to transcription regulation. Mol. Cell 51, 705–706 (2013).
Salzman, J., Gawad, C., Wang, P. L., Lacayo, N. & Brown, P. O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 7, e30733 (2012).
Salzman, J., Chen, R. E., Olsen, M. N., Wang, P. L. & Brown, P. O. Cell-type specific features of circular RNA expression. PLoS Genet. 9, e1003777 (2013).
Holdt, L. M., Kohlmaier, A. & Teupser, D. Molecular roles and function of circular RNAs in eukaryotic cells. Cell Mol. Life Sci. 75, 1071–1098 (2018).
Barrett, S. P. & Salzman, J. Circular RNAs: Analysis, expression and potential functions. Development 143, 1838–1847 (2016).
Hansen, T. B. et al. Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388 (2013).
Memczak, S. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338 (2013).
Ashwal-Fluss, R. et al. CircRNA Biogenesis competes with Pre-mRNA splicing. Mol. Cell. 56, 55–66 (2014).
Meng, S. et al. CircRNA: Functions and properties of a novel potential biomarker for cancer. Mol. Cancer 16, 94 (2017).
Shan, K. et al. Circular noncoding RNA HIPK3 mediates retinal vascular dysfunction in diabetes mellitus. Circulation 136(17), 1629–1642. https://doi.org/10.1161/CIRCULATIONAHA.117.029004 (2017).
Yang, Y. et al. Extensive translation of circular RNAs driven by N 6-methyladenosine. Cell Res. 27, 626–641 (2017).
Zou, J., Liu, K.-C., Wang, W.-P. & Xu, Y. Circular RNA COL1A2 promotes angiogenesis via regulating miR-29b/VEGF axis in diabetic retinopathy. Life Sci. 256, 117888. https://doi.org/10.1016/j.lfs.2020.117888 (2020).
Zhu, K. et al. Downregulation of circRNA DMNT3B contributes to diabetic retinal vascular dysfunction through targeting miR-20b-5p and BAMBI. EBioMedicine 49, 341–353. https://doi.org/10.1016/j.ebiom.2019.10.004 (2019).
Li, Y., Cheng, T., Wan, C. & Cang, Y. CircRNA_0084043 contributes to the progression of diabetic retinopathy via sponging miR-140-3p and inducing TGFA gene expression in retinal pigment epithelial cells. Gene 747, 1446. https://doi.org/10.1016/j.gene.2020.144653 (2020).
Dong, S. et al. Expression profiling of circular RNAs and their potential role in early-stage diabetic cardiomyopathy. Mol. Med. Rep. 22(3), 1958–1968. https://doi.org/10.3892/mmr.2020.11248 (2020).
Zhou, B. & Yu, J. W. A novel identified circular RNA, circRNA_010567, promotes myocardial fibrosis via suppressing miR-141 by targeting TGF-beta1. Biochem. Biophys. Res. Commun. 487(4), 769–775. https://doi.org/10.1016/j.bbrc.2017.04.044 (2017).
Tang, C. M. et al. CircRNA_000203 enhances the expression of fibrosis-associated genes by derepressing targets of miR-26b-5p, Col1a2 and CTGF, in cardiac fibroblasts. Sci. Rep. 7, 40342. https://doi.org/10.1038/srep40342 (2017).
Biswas, S. et al. The long non-coding RNA HOTAIR is a critical epigenetic mediator of angiogenesis in diabetic retinopathy. Invest. Oph. Vis. Sci. 62, 20. https://doi.org/10.1167/iovs.62.3.20 (2021).
Thomas, A., Feng, B. & Chakrabarti, S. ANRIL regulates production of extracellular matrix proteins and vasoactive factors in Diabetic Complications. Am. J. Physiol. Endocrinol. Metab. 314(3), E191–E200. https://doi.org/10.1152/ajpendo.00268.2017 (2018).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47–e47 (2015).
Oytam, Y. et al. Risk-conscious correction of batch effects: maximising information extraction from high-throughput genomic datasets. BMC Bioinform. 17(1), 1–17 (2016).
Enright, A. et al. MicroRNA targets in Drosophila. Genome Biol. 5, R1 (2003).
Pasquinelli, A. E. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 13, 271–282 (2012).
Xia, S. et al. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief Bioinform. (2016) [Epub ahead of print].
Feng, B., Gordon, A. D. & Chen, S. Chakrabarti S miR-146a mediates inflammatory changes and fibrosis in the heart in diabetes. J. Mol. Cell. Cardiol. 105, 70–76. https://doi.org/10.1016/j.yjmcc.2017.03.00 (2017).
Mortuza, R., Feng, B. & Chakrabarti, S. MiR-195 Regulates SIRT1 mediated changes in diabetic retinopathy. Diabetologia 57(5), 1037–1046 (2014).
Cao, Y., Feng, B., Chen, S., Chu, Y. & Chakrabarti, S. Mechanisms of endothelial to mesenchymal transition in the retina in diabetes. Invest. Ophthalmol. Vis. Sci. 55, 7321–7331. https://doi.org/10.1167/iovs.14 (2014).
Schwartz, M. A., Vestweber, D. & Simons, M. A unifying concept in vascular health and disease. Science 360, 270–271. https://doi.org/10.1126/science.aat3470 (2018).
Tang, C.-M. et al. CircRNA_000203 enhances the expression of fibrosis-associated genes by derepressing targets of miR-26b-5p, Col1a2 and CTGF, in cardiac fibroblasts. Sci. Rep. 7, 40342. https://doi.org/10.1038/srep40342 (2017).
Shan, K. et al. Circular noncoding RNA HIPK3 mediates retinal vascular dysfunction in diabetes mellitus. Circulation 136, 1629–1642 (2017).
Gu, Y. et al. Altered expression profile of circular RNAs in the serum of patients with diabetic retinopathy revealed by microarray. Ophthal. Res. 58, 176–184 (2017).
Acknowledgements
Supported by the Canadian Institutes of Health Research (Funding Reference Number: 169650) (SC), Jiangsu Province 100 talent International collaborative research program (BX2019100) (SC and ZS) and The Department of Pathology and Laboratory Medicine Start-up Funds (CC).
Author information
Authors and Affiliations
Contributions
Experimental conception and design: C.C., S.C. Performed the experiments: B.F., N.P. Reagents/materials/analysis tools contribution: all; Writing of manuscript: S.C., C.C., N.P., B.F.; Manuscript review: all.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patil, N.S., Feng, B., Su, Z. et al. Circular RNA mediated gene regulation in chronic diabetic complications. Sci Rep 11, 23766 (2021). https://doi.org/10.1038/s41598-021-02980-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-02980-y
This article is cited by
-
Hsa_circ_0021727 (circ-CD44) promotes ESCC progression by targeting miR-23b-5p to activate the TAB1/NFκB pathway
Cell Death & Disease (2023)
-
Non-coding RNAs regulating mitochondrial function in cardiovascular diseases
Journal of Molecular Medicine (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
