
Abstract
Neolamarckia cadamba is an important tropical and subtropical tree for timber industry in southern China and is also a medicinal plant because of the secondary product cadambine. N. cadamba belongs to Rubiaceae family and its taxonomic relationships with other species are not fully evaluated based on genome sequences. Here, we report the complete sequences of mitochondrial genome of N. cadamba, which is 414,980 bp in length and successfully assembled in two genome circles (109,836 bp and 305,144 bp). The mtDNA harbors 83 genes in total, including 40 protein-coding genes (PCGs), 31 transfer RNA genes, 6 ribosomal RNA genes, and 6 other genes. The base composition of the whole genome is estimated as 27.26% for base A, 22.63% for C, 22.53% for G, and 27.56% for T, with the A + T content of 54.82% (54.45% in the small circle and 54.79% in the large circle). Repetitive sequences account for ~ 0.14% of the whole genome. A maximum likelihood (ML) tree based on DNA sequences of 24 PCGs supports that N. cadamba belongs to order Gentianales. A ML tree based on rps3 gene of 60 species in family Rubiaceae shows that N. cadamba is more related to Cephalanthus accidentalis and Hymenodictyon parvifolium and belongs to the Cinchonoideae subfamily. The result indicates that N. cadamba is genetically distant from the species and genera of Rubiaceae in systematic position. As the first sequence of mitochondrial genome of N. cadamba, it will provide a useful resource to investigate genetic variation and develop molecular markers for genetic breeding in the future.
Similar content being viewed by others


Introduction
Neolamarckia cadamba is one of two species (N. macrophylla for the other species) in genus Neolamarckia of Rubiaceae1, one of the largest families in flowering plants. The species is naturally distributed in Vietnam, Malaysia, Myanmar, India and Sri Lanka, and mainly grows in Guangdong, Guangxi and Yunnan Provinces in China. It grows in the habitat of high temperature and humidity, with the average temperature of 20–24 °C and the annual precipitation of 1200–2400 mm, and also in the fertile, loose and humid soil or in the humid sandy soil. N. cadamba, aka a miraculous tree, is a fast-growing species2 and commercially important materials. Its wood is good for building construction, wood board making, furniture, pulp and paper production3. In addition, the tree fruits can be used for nutraceutical enriched beverage4. Leaves are used as woody forage to feed livestock5 and have effects of antibacterial and anti-inflammatory to animals6. One particular value is that the species has enormous pharmacological implications due to its rich secondary metabolites (e.g., phenols and alkaloids)6,7,8,9. The monoterpenoids, alkaloids and triterpenoids are potentially used for medicinal purposes10,11. N. cadamba is exploited for antimicrobial, wound healing and antioxidant activities12,13,14 and for traditionally curing a number of diseases, such as diabetes, anaemia and infectious diseases6. The species as a medicinal plant is appreciated in South Asia15,16 and shows enormous medical implications.
Although N. cadamba is a miraculous tree, the absence of reference genome limits the molecular and evolutionary studies of this species. Current genetic studies of this species cover broad areas, including provenance trials17,18, propagation through tissue culture19,20, transcriptome analysis of gene expressions21,22, single nucleotide polymorphisms (SNPs) and SNP-trait association23,24, expressed sequence tags (ESTs) of xylem tissues25 and gene discovery in the developing xylem tissue26. Nevertheless, few studies with molecular markers have been reported on population genetic structure, phylogeography and molecular systematics27. This necessitates determination of the genomic sequences to understand the genetic basis of these characters (rapid growth, quality timber, secondary metabolites, etc.), to develop appropriate molecular markers for breeding program, and to gain insights into the evolutionary history of this species.
To develop markers for population genetics and phylogenetic analysis, we here sequenced and reported the mitochondrial genome of this species. The well-known features of mitochondrial DNA (mtDNA) in plants include (i) maternal inheritance in angiosperms, (ii) haplotype per cell, (iii) intra-molecular recombination between repeats28, and (iv) the number of females as its population size (Nf). These features differ from those of nuclear genomes, which correspondingly exhibits (i) biparental inheritance, (ii) diploid per cell, (iii) inter-chromosome recombination and relatively high mutation rates, and (iv) large effective population sizes (2Ne) of nuclear genes (\(2N_{e} = 4N_{f}\) under 1:1 sexual ratio)29. Compared with chloroplast and nuclear DNAs, mtDNA has generally a lower mutation rate in plants30. Thus, mtDNA sequences are useful for studying the long-term phylogenetic relationships at the level of species or higher order, and also for studying other perspectives of evolutionary relationships, such as lineage sorting, hybridization and cytonuclear interactions31.
Although three major subfamilies in Rubiaceae are delineated, including Rubioideae32, Ixoroideae33 and Cinchonoideae34, systematic position of N. cadamba remains to be evident. From the morphological characters, N. cadamba is classified into subfamily Cinchonoideae, tribe Naucleeae1. Based on the cytogenetic study35,36, N. cadamba has 44 chromosomes (2n) and belongs to subfamily Cinchonoideae, tribe Naucleeae and Subtribes Neolamarckinae37. In this study, we determined the complete mtDNA sequence of N. cadamba and detailed its characteristics. Based on the mtDNA sequence, we then evaluated the phylogenetic relationships among families and genera of Rubiaceae to gain insights into the taxonomic position of N. cadamba.
Results and discussion
Assembly of mitochondrial genome
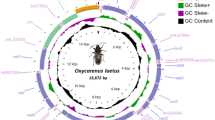
MtDNA sequence of N. cadamba was determined using PacBio sequencing technique and was successfully assembled in two genome circles. This probably reflects the feature of rapid evolution of structure of plant mitochondrial genomes38,39,40. Figure 1 shows two parts of circular structure of the mitochondrial genome, designated as genomes 1 and 2. The genome 1 has 109,836 bp (GenBank Access No. MT320890). It contains 14 genes (Table 1), including 7 protein-coding genes (PCGs), 5 transfer RNA genes, and 2 other genes (ccmFc, ccmFn). The PCGs are 1 NADH dehydrogenase genes (nad7), 2 ATP synthase genes (atp6, atp9), 2 ribosomal proteins genes (rpl18, rps3), 1 maturases gene, and 1 ORF. Four PCGs (nad7, rpl16, rps3, atp9), 4 tRNA genes, and 1 other gene (ccmFc) are on the N-strand, and genes of 1 PCG atp6, 1 tRNA gene and 1 other gene (ccmFn) are on the J-strand. There is only one overlapping region (110 bp in length) between rpl16 and rps3 in genome 1.

Two circular maps of the mitochondrial genome of Neolamarckia cadamba.
The mitochondrial genome 2 is 305,144 bp in length (GenBank Access No. MT364442). The genome 2 contains 69 genes (Table 2), including 33 PCGs, 26 transfer RNA genes, 6 ribosomal RNA genes, and 4 other genes (ccmC, mttB, ccmB, ccmC). The PCGs are 8 NADH dehydrogenase genes (nad4L, nad2, nad3, nad5, nad6, nad3, nad9, nad4), 1 succinate dehydrogenase genes (sdh4), 2 ubichinol cytochrome reductase genes, 4 cytochrome c oxidase genes, 5 ATP synthase genes (atp4, atp1, atp9, atp1, atp8), 10 ribosomal proteins genes (6 rps, 4 rpl), and 3 ORFs. The 15 PCGs (nad4L, nad2, nad3, nad9, atp4, atp1, cob, cox2, rpl10, rpl5, rps13, rps12, rps4, orf954, orf108), 10 tRNA genes, and 3 rRNA genes and 1 other gene (ccmC) are located on the N-strand, and the remaining 18 PCG genes, 16 tRNA genes, 3 rRNA genes and 3 other genes (mttB, ccmB, ccmC) are on the J-strand. There are two overlapping regions, with 73 bp overlapping between cox3 and sdh4 and 817 bp overlapping between rps4 and tRNA-Leu. There are certain intergenic sequences among adjacent genes in the remaining genes, indicating relatively low density of gene distribution along the genome. This is consistent with the patterns of other plants where non-coding regions are the important parts in consisting of mitochondrial genome40,41,42.
Characteristics of nucleotide composition
The two genome circles slightly differ in nucleotide composition (SI Table 1). Genome 1 has a high content of the T base but a low content of the G base. The AT content is 54.45% and the four types of bases are 29,521 bp of A (26.88%), 30,287 bp of T (27.57%), 25,616 bp (23.32%), and 24,412 bp of G (22.23%). Genome 2 has a high content of the T base but a low content of the C base. The AT content is 54.94%, and the four bases are 83,584 bp of A (27.39%), 84,075 bp of T (27.55%), 68,286 bp of C (22.38%), and 69,089 bp of G (22.64%). The AT content is slightly higher than the GC content. The relatively high AT content was also reported in other plant species43 or animal species44.
Besides the AT or GC content, the AT-and GC-skews are often used to assess the nucleotide-compositional differences in mitochondrial genomes45. From SI Table 1, both AT- and GC-skews in genome 1 are negative (AT-skew = −0.0128 and GC-skew = −0.0241), indicating that genome 1 has a higher percentage of T and C than A and G, respectively. Both AT- and GC-skews are negative in PCG sequences (AT-skew = −0.0408 and GC-skew = −0.0501). However, the AT-skew in tRNAs is positive (0.0430), indicating that these genes have a higher percentage of A than T. The GC-skew in tRNAs is negative (−0.1135), indicating that these genes have a higher percentage of G than C.
In genome 2, the AT-skew (−0.0029) is negative but the GC-skew (0.0058) is positive (SI Table 1), indicating that genome 2 has a higher percentage of T and G than A and C, respectively. The GC-skews in both PCGs (−0.0115) and rRNAs (−0.1242) are negative, but positive in tRNAs (0.0449). The AT-skews are negative in PCGs (−0.0569), tRNA (−0.0289) and rRNAs (−0.0864). The extents of both AT- and GC-skews are greater in rRNAs than in PCGs and tRNAs. Generally, the extents of AT-and GC-skews in both genomes 1 and 2 are small, comparable to the pattern in mitochondrial genomes of Pyrus pyrifolia (AT-skew = 0.004, GC-skew = 0)46 but different from that of animal species Ledra auditura 44 (AT-skew = 0.22 and GC-skew = 0.12).
Protein-coding genes and codon usage
Codon usage bias is an important character of a genome since it is associated with gene expression47,48, the base composition of genes49, amino acid composition50, GC content51, the length of a gene52 and tRNA richness53,54. Large differences in the codon usage of genes often occur among different species and organisms52.
The mitochondrial genome of N. cadamba harbors a total of 83 coding genes and 45,639 bp in length, accounting for about 11% of the entire mitochondrial genome. This density is greater than those of watermelon (Citrullus lanatus; 10.3% of 379,236 bp), zucchini (Cucurbita pepo; 3.9% of 982,833 bp)55 and neem (Azadirachta indica A. Juss; 7.7% of 266,430 bp)56 mitochondrial genomes. The base composition of the whole mtDNA of N. cadamba is 27.26% for A, 22.63% for C, 22.53% for G and 27.56% for T, exhibiting a AT-biased pattern, with the A + T content of 54.82%. The AT-biased pattern is frequently observed in both plant and animal mitochondrial genomes57.
The mitochondrial genomic protein-coding genes of N. cadamba are 37,521 bp in length, accounting for 83.03% of all coding genes. The 40 protein-coding genes encode a total of 12,507 codons. Figure 2 shows the frequencies of different amino acids in the protein-coding genes where the amino acid Leu is most frequently used, followed by Ser, Ile and Gly. From the values of relative synonymous codon usage (RSCU), there are 32 optimal codons (RSCU > 1): TAA, GCT, TAT, CAA, CAT, GGA, TTA, TCT, CCT, AGA, CGA, GAA, GAT, ACT, AAT, ATT, GGT, TGT, GTT, CTT, GTA, CGT, TTG, TCA, AAA, TTT, CCA, AGT, ACC, GCA, ATG, and TGG. The remaining 32 codons are non-optimal (RSCU < 1). The most frequently used codons are TTT (Phe), ATT (lle), GAA (Glu) and GCT (Ala). Reasons for the bias synonymous codon usage probably arise from different processes (e.g., distinct levels of gene expression, the base composition of genes, gene length and tRNA richness).

Amino acid frequency and RSCU value of protein-coding genes in mitochondrial genome of Neolamarckia cadamba.
According to the RSCU values, codons are classified into optimal codons (RSCU > 1) and non-optimal codon (RSCU < 1). From Fig. 2 and SI Table 3, each amino acid has its preferred codon, with exception of amino acids Met (ATG) and Trp (TGG) that have only one codon and no preference.
A universal genetic code is used for all mitochondrial genes in angiosperms, and the third codon tends to be A or T58. A typical translation initiation codon is ATG, but alternative initiation codons occur in translation of rpl1659, mttB52, and matB genes. The initiation codon of the protein-coding genes in the mitochondrial genome of N. cadamba is ATG, except for rps10 and rpl16 where ACG is the initiation codon.
Transfer RNA and ribosomal RNA genes
There are 5 tRNA genes in genome 1, with a total length of 371 bp (Table 1). The five tRNA genes range from 71 (tRNA-Arg) to 84 bp (tRNA-Tyr) in length, of which four genes are on the N-strand and one gene is on the J-strand. There are 26 tRNA genes in genome 2, with a total length of 1,909 bp (Table 1). These genes range from 60 bp (tRNA-Val) to 88 bp (tRNA-Ser) in length, of which ten genes are on the N-strand and sixteen genes are on the J-strand.
The secondary structure map of tRNA was predicted and generated using tRNAscan-SE 2.0 (http://lowelab.ucsc.edu/tRNAscan-SE/) 60 and ARWEN (Version1.2, http://mbio-serv2.mbioekol.lu.se/ARWEN/) 61. Structurally, tRNA-Ser (GCT), tRNA-Ser (TGA) and tRNA-Tyr (GTA) have a group of stem-loop structure on the extra loop between the TψC loop and the anti-codon stem, but the remaining tRNA genes are the typical clover-type secondary structure (SI Fig. 1).
In the secondary structure of tRNA, besides three classic base matches (A-T, G-C and G-T), there are also mismatches, such as G-A, A-C, T-T, T-C and A-A. The T-C and A-A mismatch pairs are only in the anti-codon stems. Three G-A pairs are in the amino acid acceptor stems, and the other three G-A mismatch pairs are in the TψC stems. Two A-C pairs are in the TψC stems, and the other five A-C pairs are in the amino acid acceptor stems. One T-T pair is in the amino acid acceptor stems, and the other four T-T pairs are in the anti-codon stems.
The mitochondrial genome of N. cadamba has 3 rRNA genes in total (rrn18, rrn5, and rrn26), ranging from 116 bp to 3,429 bp in length, and all rRNA genes are on the N-strand.
Repetitive sequences
SI Table 2 indicates that both genome 1 (~ 0.08%) and genome 2(~ 0.16%) have small proportions of repetitive sequences, with the repeat length of 579 bp in total. Most repetitive sequences (microsatellites) consist of single- and di-nucleotide repeats, with more numbers of (A)n and (T)n (14) than (G)n and (C)n (2), and more numbers of (AT)n and (TA)n (8) than others (2 (GA)n). Three minisatellites are present in genome 2. All these repeats are not located in protein-coding regions except (T)10 in orf309 of genome 1 and (A)10 in Atp1 of genome 2. The small proportion of repeats implies that repetitive sequences do not play an important role in contributing to mitochondrial genome size of N. cadamba, different from the patterns of Nymphaea colorata 42 and other plants 40. However, these repetitive sequences could be used to develop molecular markers for population genetic structure analysis in the future.
Phylogenetic analyses
To assess the taxonomic position of N. cadamba, we analyzed the phylogenetic trees species based on complete mitochondrial genomes. Twenty-three species of the asterids-lamiids classification with complete mitochondrial genomes were selected, and Helianthus annuus of the non-lamiids classification of the asterids was selected as outgroup. This selection of 22 species of Astragalus was based on tandem sequences of 24 protein-coding genes. The 24 protein-coding genes were 3 adenosine triphosphate synthase genes (atp1, atp6, atp9), 3 cytochrome c oxidase genes (cox1, cox2, cox3), and 1 cytochrome b protein gene (cytB), 9 nicotinamide adenine dinucleotide (NADH) dehydrogenase protein genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9), 4 ribosomal proteins genes (rps12, rps13, rps3, rps4) and 4 other genes (ccmb, ccmc, ccmfc, ccmfn).
JModelTest2.1.7 was used to test the nucleic acid model of the selected sequence DNA62, and the best model was GTR + I + G. Maximum likelihood phylogenetic tree was constructed with RAxML8.1.5 software63. The clade with N. cadamba in Gentianales has two families (Fig. 3): Rubiaceae and Apocynacceae. Rhazya stricta, Asclepias syriaca, Cynanchum auriculatum and C. wilfordii in the neighbor branches belong to Apocynacceae, and have closer genetic relationships. N. cadamba as the species in family Rubiaceae was earlier differentiated from Apocynacceae. This phylogenetic relationship among the 22 species is consistent with taxonomic groups based on morphological studies.

Maximum likelihood tree based on the sequences of 24 PCGs from the mitochondrial genomes of 23 species. The values on branch nodes represent the supporting rates (percentages) derived from 1000 bootstrapping analyses.
The rps3 gene sequence of 60 species of Rubiaceae was available from NCBI GenBank. Phylogenetic genetic relationships based on this single gene was constructed using the maximum likelihood method. SI Fig. 2 shows that N. cadamba is genetically close to Cephalanthus occidentalis and Hymenodictyon parvifolium. These three species together with Cubanola domingensis, Hillia triflora and Rondeletia odorata provide evidence that they belong to the Cinchonoideae subfamily although Deppea grandiflora (Ixoroideae subfamily) and Guettarda scabra (Rubioideae subfamily) were incompletely sorted in this clade. Using cpDNA segments (rbcL, rsp16 intron, nadhF, atpB–rbcL spacer) and nuclear ribosomal ITS, Rydin et al.64 showed that five species (C. occidentalis, H. parvifolium, C. domingensis, H. triflora and R. odorata) belong to Cinchonoideae subfamily. The whole phylogenetic relationships indicate that large genetic divergence and incomplete linage sorting occurred among the three subfamilies of Rubiaceae in terms of the rps3 gene sequence.
Conclusions
In this study, we sequenced the mitochondrial genome of N. cadamba and successfully assembled the genome in two maps of circular molecule structure. Genome 1 has 109,836 bp and contains 14 genes. Genome 2 has 305,144 bp and contains 69 genes. The whole genome has slightly high AT content (54.82%). Genome 1 shows negative AT- and GC-skews, while genome 2 shows a negative AT-skew but a positive GC-skew. All protein-coding genes are initiated by the start codon ATG, except for a few genes initiated by alternative codons. The termination codes are TAA for most genes but TGA or TAG for a few genes. Each amino acid has its preferred codon except amino acids Met (ATG) and Trp (TGG) that have only one codon and no preference. The tRNA genes exhibit a typical clover-type secondary structure except tRNA-Ser (GCT), tRNA-Ser (TGA) and tRNA-Tyr (GTA) that have an extra loop between the TψC loop and the anti-codon stem. Tandem repeat sequences are minor, accounting for ~ 0.14% of the whole genome. Phylogenetic analysis with the DNA sequences of 24 PCGs confirms that N. cadamba belongs to order Gentianales. Analysis with a single gene rps3 of 60 species shows that N. cadamba is genetically closer to Cephalanthus accidentalis and Hymenodictyon parvifolium and belongs to the Cinchonoideae subfamily.
Methods
Sample collection and DNA extraction
The leaf sample used in this study was collected from a wild tree (Specimen ID: SCAUNC20190110) on January 10th, 2019. This tree grows on University Campus (23°16′N 113°35′E), South China Agricultural University (SCAU), Guangzhou, Guangdong Province, China. Figure 4 shows the sample tree growing in the fertile and humid soil. XW and XSH identified the voucher specimen and collected leaf samples. The specimen was stored for records in Guangdong Key Laboratory for Innovative Development and Utilization of Forest Plant Germplasm, SCAU, Guangdong Province, China. The use of plant leaves in this study complies with institutional guidelines. Collection of the plant specimen was permitted by the University. Total genomic DNA was extracted from fresh leaves using CTAB method65. Then the quality of the extracted DNA samples was tested using (1) 0.8% agarose electrophoresis to detect DNA samples for degradation and impurities, and to estimate the DNA concentration; (2) Nanodrop spectrophotometer to detect the concentration and purity of samples; and (3) Qubit 2.0 Flurometer (Life Technologies, USA) to detect the concentration of samples.

The tree of Neolamarckia cadamba from which young leaves were sampled for mtDNA sequencing. The tree grows on campus of South China Agricultural University (23°16′N 113°35′E), Guangzhou, China. It is about 14.5 m in height and 49.04 cm in diameter at the breast height in eleven years.
Library construction and high-throughput sequencing
High-quality genomic DNA of 50 μg was used to generate a 40-kb SMRTbell library, with the size selection on the BluePippin (Sage Science, USA). The genomic DNA library was sequenced on the PacBio sequel platform (Pacific Biosciences, USA). SMRTbell DNA library preparation and sequencing were performed in accordance with the manufacturer’s protocols (Pacific Biosciences, USA), and totally 2 Gb subreads were generated. In order to check the correction of PacBio assembly, an insert size of 500 bp pair-end genomic DNA library for Illumina Hiseq 4000 (Illumina, USA), was constructed by Science Corporation of Gene according to the standard protocol of Illumina. DNA library was constructed after quality control with Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Four gigabytes DNA data were sequenced by Illumina Hiseq 4000 (Illumina, USA).
Different sequencing methods were used in this study because lengths of PacBio sequencing reads were up to 40 kb, which was more suitable for complex genome assembly. However, the PacBio long reads potentially had much more sequencing errors, and the Illumina short reads were then used to fix the errors.
Genome assembly and annotations
The mitochondrial genome sequence was assembled using Canu (version 2.1, https://github.com/marbl/canu) 66 with default parameters on PacBio CLR subreads, and mitochondrial genome sequences were identified with blastn (version 2.10.1 + , https://blast.ncbi.nlm.nih.gov/Blast.cgi) and NCBI nucleotide sequence database. To make improvements of assembly genome with Pilon (version 1.24, https://github.com/broadinstitute/pilon) 67, the final PacBio CLR subreads and Illumina clean reads were remapped to mitochondrial genome with bwa (version 0.7.17-r1188, http://bio-bwa.sourceforge.net/) 68 and IGV (version 2.9.4, https://igv.org/) 69 to confirm. Genome was annotated using DOGMA (http://dogma.ccbb.utexas.edu/) 70 and ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/). For the preliminary results of the annotations, the methods of Blastn and Blastp were used to compare the encoded proteins and rRNA of the reported mitochondrial genome of related species, verify the accuracy of the results and modify them. TRNA was annotated by tRNAscan-SE 2.0 (http://lowelab.ucsc.edu/tRNAscan-SE/) 71 and ARWEN (Version1.2, http://mbio-serv2.mbioekol.lu.se/ARWEN/) 61, leaving out the tRNA with unreasonable length and incomplete structure, and generating the tRNA secondary structure diagram. Microsatellite identification tool (MISA v2.1) 72 and tandem repeat finder (TRF) 73 were used to search for repetitive sequences.
Comparative analysis of mitochondrial genomes
The use of mitochondrial codons had a preference, which would affect the expression of genes and reflect the evolutionary relationship of species to a certain extent. The calculation of relative synonymous codon usage was analyzed with a reference to the formula mentioned in Sharp and Li74. The relative synonymous codon usage (RSCU) was calculated as the ratio of the frequency of a focal codon to the mean frequency of all synonymous codons in a given protein-coding sequence. The usage bias of one synonymous codon is indicated when RSCU is not equal to 1; no usage bias is present when RSCU is equal to 1.
In most bacterial genomes, mitochondrial and plastid genomes, there are significant differences in base composition between heavy and light chains, which are called AT-skew and GC-skew. Calculations of the AT- and GC-skews are as follows75:
where A%, T%, G% and C% represent the percentages of A, T, G and C in a given sequence, respectively.
Phylogenetic analyses
MUSCLE v.3.8.31 (http://www.drive5.com/muscle/) software76 was used to compare individual genes among multiple species, and then the genes of each species were aligned in a certain order. The protein-encoding gene sequence set of each species was generated by catenating 24 PCG sequences in the same gene order for further analysis. jModelTest2.1.7 (https://code.google.com/p/jmodeltest2/) was used to test the nucleic acid model of the selected sequence DNA62, and the best model has the minimum AIC (Akaike Information Criterion) value. Phylogeny tree was constructed with RAxML8.1.5 software (https://sco.h-its.org/exelixis/web/software/raxml/index.html)63 using the maximum likelihood (ML) method for both the catenated sequences of 23 species and the rps3 gene sequences of 60 species. The bootstrap value was set to be 1000 for each phylogenetic tree analysis.
Data availability
mtDNA sequences of Neolamarckia cadamba in NCBI GenBank: Genome 1:https://www.ncbi.nlm.nih.gov/nuccore/MT320890.1. Genome 2: https://www.ncbi.nlm.nih.gov/nuccore/MT364442.1.
References
Lo, H.S., Ko, W.C., Chen, W.C., Hsue, H.H. & Wu, H. Flora Reipublicae Popularis Sinicae: Tomus 71(1): Angiospermae Dictotyledoneae, Rubiaceae (1), 260–261. (Science Press, Beijing, 1999) (in Chinese).
Mojiol, AR, Lintangah, W, Maid, M. & Julius, K, Neolamarckia cadamba (Roxb.) Bosser, 1984, 1–12 (2014).
Ho, W.S., et al. Applications of genomics to plantation forestry with kelampayan in Sarawak in Sustaining Tropical Natural Resources Through Innovations, Technologies and Practices (eds. Wasli, M.E., et al.). 4th Regional Conference on Natural Resources in the Tropics, 103–111. (Universiti Malaysia Sarawak, 2012).
Pandey, A., Chauhan, A. S., Haware, D. J. & Negi, P. S. Proximate and mineral composition of Kadamba (Neolamarckia cadamba) fruit and its use in the development of nutraceutical enriched beverage. J. Food Sci. Technol. 55(10), 4330–4336. https://doi.org/10.1007/s13197-018-3382-9 (2018).
He, L. et al. Effect of applying lactic acid bacteria and cellulase on the fermentation quality, nutritive value, tannins profile and in vitro digestibility of Neolamarckia cadamba leaves silage. J. Anim. Physiol. Anim. Nutr. 102, 1429–1436. https://doi.org/10.1111/jpn.12965 (2018).
Pandey, A. & Negi, P. S. Traditional uses, phytochemistry and pharmacological properties of Neolamarckia cadamba: A review. J. Ethnopharmacol. 181, 118–135. https://doi.org/10.1016/j.jep.2016.01.036 (2016).
Santiarworn, D., Liawruangrath, S., Baramee, A., Takayama, H. & Liawruangrath, B. Bioactivity screening of crude alkaloidal extracts from some Rubiaceae. Chiang Mai Univ. J. 4, 59–64 (2005).
Dwevedi, A., Sharma, K. & Sharma, Y. K. Cadamba: A miraculous tree having enormous pharmacological implications. Pharmacogn. Rev. 9(18), 107. https://doi.org/10.4103/0973-7847.162110 (2014).
Chandel, M. et al. Isolation and characterization of flavanols from Anthocephalus cadamba and evaluation of their antioxidant, antigenotoxic, cytotoxic and COX-2 inhibitory activities. Rev. Bras. Farmacogn. 26, 474–483. https://doi.org/10.1016/j.bjp.2016.02.007 (2016).
Dubey, A., Nayak, S. & Goupale, D. A review on phytochemical, pharmacological and toxicological studies on Neolamarckia cadamba. Pharm. Lett. 3, 45–54 (2011).
Mishra, D. P. et al. Monoterpene indole alkaloids from Anthocephalus cadamba fruits exhibiting anticancer activity in human lung cancer cell line H1299. Chem. Sel. 3, 8468–8472. https://doi.org/10.1002/slct.201801475 (2018).
Umachigi, S. P. et al. Antimicrobial, wound healing and antioxidant activities of Anthocephalus cadamba. Afr. J. Tradit. Complement. Alternat. Med. 4(4), 481–487. https://doi.org/10.4314/ajtcam.v4i4.31241 (2007).
Patel Divyakant, A., Dirji, V., Bariya, A., Patel, K. & Sonpal, R. Evaluation of antifungal activity of Neolamarckia cadamba (roxb.) bosser leaf and bark extract. Int. Res. J. Pharm. 2, 192–193 (2011).
Acharyya, S., Rathore, D., Kumar, H. & Panda, N. Screening of Anthocephalus cadamba (Roxb.) Miq. root for antimicrobial and anthelmintic activities. Int. J. Res. Pharm. Biomed. Sci. 2(1), 297–300 (2011).
Dogra, S. C. Antimicrobial agents used in ancient India. Indian J. Hist. Sci. 22(2), 164–169 (1987).
Khare, C. P. & Khare, C. P. Indian Herbal Remedies: Rational Western Therapy, Ayurvedic and Other Traditional Usage, Botany (Springer, 2004).
Que, Q. M. et al. Genetic variation of young forest growth traits of Neolamarckia cadamba. Subtrop. Plant Sci. 46, 248–253 (2017) ((in Chinese)).
Parthiban, K. T., Thirunirai-Selvan, R., Palanikumaran, B. & Krishnakumar, N. Variability and genetic diversity studies on Neolamarckia cadamba genetic resources. J. Trop. Res. Sci. 31, 90–98. https://doi.org/10.26525/jtfs2019.31.1.090098 (2019).
Li, J. J., Zhang, D., Ouyang, K. X. & Chen, X. Y. High frequency plant regeneration from leaf culture of Neolamarckia cadamba. Plant Biotechnol. 36, 13–19. https://doi.org/10.5511/plantbiotechnology.18.1119a (2019).
Mok, P. K. & Ho, W. S. Rapid in vitro propagation and efficient acclimatisation protocols of Neolamarckia cadamba. Asian J. Plant Sci. 18, 153–163. https://doi.org/10.3923/ajps.2019.153.163 (2019).
Ouyang, K. et al. Transcriptomic analysis of multipurpose timber yielding tree Neolamarckia cadamba during xylogenesis using RNA-seq. PLoS ONE 11, e159407. https://doi.org/10.1371/journal.pone.0159407 (2016).
Huang, T. et al. Selection and validation of reference genes for mRNA expression by quantitative real-time PCR analysis in Neolamarckia cadamba. Sci. Rep. 8, 9311. https://doi.org/10.1038/s41598-018-27633-5 (2018).
Tchin, B. L., Ho, W. S., Pang, S. L. & Ismail, J. Association genetics of the cinnamyl alcohol dehydrogenase (CAD) and cinnamate 4-hydroxylase (C4H) genes with basic wood density in Neolamarckia cadamba. Biotechnology 11, 307–317. https://doi.org/10.3923/biotech.2012.307.317 (2012).
Tiong, S. Y., Ho, W. S., Pang, S. L. & Ismail, J. Nucleotide diversity and association genetics of xyloglucan endotransglycosylase/hydrolase (XTH) and cellulose synthase (CesA) genes in Neolamarckia cadamba. J. Biol. Sci. 14, 267–275. https://doi.org/10.3923/jbs.2014.267.275 (2014).
Ho, W.-S., Pang, S.-L. & Abdullah, J. Identification and analysis of expressed sequence tags present in xylem tissues of kelampayan (Neolamarckia cadamba (Roxb.) Bosser). Physiol. Mol. Biol. Plants 20, 393–397. https://doi.org/10.1007/s12298-014-0230-x (2014).
Pang, S. L., Ho, W. S., Mat-Isa, M. N. & Abdullah, J. Gene discovery in the developing xylem tissue of a tropical timber tree species: Neolamarckia cadamba (Roxb.) Bosser (kelampayan). Tree Genet Genomes 11, 47. https://doi.org/10.1007/s11295-015-0873-y (2015).
Ying, T. S., Fu, C. S., Seng, H. W. & Ling, P. S. Genetic diversity of Neolamarckia cadamba using dominant DNA markers based on inter-simple sequence repeats (ISSRs) in Sarawak. Adv. Appl. Sci. Res. 5, 458–463 (2014).
Morley, S. A. & Nielsen, B. L. Plant mitochondrial DNA. Front. Biosci. 22, 1023–1032. https://doi.org/10.2741/4531 (2017).
Wright, S. Evolution and the Genetics of Populations. The Theory of Gene Frequencies Vol. 2 (University of Chicago Press, Chicago, 1969).
Wolfe, K. H., Li, W. H. & Sharp, P. M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 84, 9054–9058. https://doi.org/10.2307/30764 (1987).
Wang, X. et al. Assessing the ecological and evolutionary processes underlying cytonuclear interactions. Sci. Sin. Vitae 49, 951–964. https://doi.org/10.1360/SSV-2019-0049 (2019).
Bremer, B. & Manen, J. F. Phylogeny and classification of the subfamily Rubioideae (Rubiaceae). Plant Syst. Evol. 225, 43–72. https://doi.org/10.1007/BF00985458 (2000).
Andreasen, K. & Bremer, B. Combined phylogenetic analysis in the Rubiaceae–Ixoroideae: morphology, nuclear and chloroplast DNA data. Am. J. Bot. 87, 1731–1748. https://doi.org/10.2307/2656750 (2000).
Andersson, L. & Antonelli, A. Phylogeny of the tribe Cinchoneae (Rubiaceae), its position in Cinchonoideae, and description of a new genus, Ciliosemina. Taxon 54, 17–28. https://doi.org/10.2307/25065299 (2005).
Mathew, P. M. & Philip, O. The distribution and systematic significance of pollen nuclear number in the Rubiaceae. Cytologia 51, 117–124 (1986).
Lee, Y. S. Remarks on chromosome number in Rubiaceae. Korean J. Plant Taxon 9(1), 57–66. https://doi.org/10.11110/kjpt.1979.9.1.057 (1979).
Eng, W. H., Ho, W. S. & Ling, K. H. Cytogenetic, chromosome count optimization and automation of Neolamarckia cadamba (Rubiaceae) root tips derived from in vitro mutagenesis. Notulae Sci. Biol. 13(3), 10995. https://doi.org/10.15835/nsb13310995 (2021).
Palmer, J. D. & Herbon, L. A. Plant mitochondrial-DNA evolves rapidly in structure, but slowly in sequence. J. Mol. Evol. 28, 87–97. https://doi.org/10.1007/BF02143500 (1988).
Morley, S. A., Ahmad, N. & Nielsen, B. L. Plant organelle genome replication. Plants 8, 358. https://doi.org/10.3390/plants8100358 (2019).
Mower, J. P., Sloan, D. B. & Alverson, A. J. Plant mitochondrial genome diversity: the genomics revolution. In Plant Genome Diversity Vol. 1 (eds Wendel, J. F. et al.) 123–144 (Springer, Wien, 2012). https://doi.org/10.1007/978-3-7091-7_9.
Guo, W., Zhu, A., Fan, W. & Mower, J. P. Complete mitochondrial genomes from the ferns Ophioglossum californicum and Psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 213(1), 391–403. https://doi.org/10.1111/nph.14135 (2017).
Dong, S. et al. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genomics 19, 614. https://doi.org/10.1186/s12864-018-4991-4 (2018).
Shi, Y. et al. Assembly and comparative analysis of the complete mitochondrial genome sequence of Sophora japonica ‘JinhuaiJ2’. PLoS ONE 13(8), 0202. https://doi.org/10.1371/journal.pone.0202485 (2018).
Wang, J. J., Li, D. F., Li, H., Yang, M. F. & Dai, R. H. Structural and phylogenetic implications of the complete mitochondrial genome of Ledra auditura. Sci. Rep. 9, 15746. https://doi.org/10.1038/s41598-019-52337-9 (2019).
Alexandre, H., Nelly, L. & Jean, D. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst. Biol. 54, 277–298. https://doi.org/10.1080/10635150590947843 (2005).
Chung, H., Won, S., Kang, S., Sohn, S. & Kim, J. S. The complete mitochondrial genome of Wonwhang (Pyrus pyrifolia). Mitochondrial DNA Part B Resources 2, 902–903. https://doi.org/10.1080/23802359.2017.1413300 (2017).
Gupta, S. K. & Ghosh, T. C. Gene expressivity is the main factor indicating the codon usage variation among the genes in Pseudomonas aeruginosa. Gene 273, 63–70. https://doi.org/10.1016/S0378-1119(01)00576-5 (2001).
Carlini, D. B., Ying, C. & Stephan, W. The relationship between third-codon position nucleotide content, codon bias, mRNA secondary structure and gene expression in the drosophilid alcohol dehydrogenase genes Adh and Adhr. Genetics 159, 623–633. https://doi.org/10.1103/PhysRevA.86.013626 (2001).
Alexei, F., Serge, S. & Walter, G. Regularities of context-dependent codon bias in eukaryotic genes. Nucleic Acids Res. 30, 1192–1197. https://doi.org/10.1093/nar/30.5.1192 (2002).
Onofrio, G. D., Mouchiroud, D., Aïssani, B., Gautier, C. & Bernardi, G. Correlations between the compositional properties of human genes, codon usage, and amino acid composition of proteins. J. Mol. Evol. 32, 504–510. https://doi.org/10.1007/BF02102652 (1991).
Knight, R. D., Freeland, S. J. & Landweber, L. F. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2, 2001. https://doi.org/10.1186/gb-2001-2-4-research0010 (2001).
Marias, G. & Duret, L. Synonymous codon usage, accuracy of translation and gene length in Caenorhabditis elegans. J. Mol. Evol. 52, 275–280. https://doi.org/10.1007/s002390010155 (2001).
Moriyama, E. N. & Powell, J. R. Gene length and codon usage bias in Drosophila melanogaster, Saccharomyces cerevisiae and Escherichia coli. Nucleic Acids Res. 26, 3188–3193. https://doi.org/10.1093/nar/26.13.3188 (1998).
Moriyama, E. N. & Powell, J. R. Codon usage bias and tRNA abundance in Drosophila. J. Mol. Evol. 45, 514–523. https://doi.org/10.1007/PL00006256 (1997).
Alverson, J. A. et al. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27, 1436–1448. https://doi.org/10.1093/molbev/msq029 (2010).
Kuravadi, N. A. et al. Comprehensive analyses of genomes, transcriptomes and metabolites of neem tree. Peer J 3, 341–345. https://doi.org/10.7717/peerj.1066 (2015).
Bi, Q. X. et al. Complete mitochondrial genome of Quercus variabilis (Fagales, Fagaceae). Mitochond. DNA Part B 4, 3927–3928. https://doi.org/10.1080/23802359.2019.1687027 (2019).
Sugiyama, Y. et al. The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: comparative analysis of mitochondrial genomes in higher plants. Mol Genet Genomics 272, 603–615. https://doi.org/10.1007/s00438-004-1075-8 (2005).
Rowen, L., Mahairas, G. & Hood, L. Sequencing the human genomes. Science 278, 605–607. https://doi.org/10.2165/00128413-199208300-00010 (1997).
Lowe, T. M. & Chan, P. P. tRNAscan-SE on-line: search and contextual analysis of transfer RNA genes. Nucleic Acids Res. 44, 54–57. https://doi.org/10.1093/nar/gkw413 (2016).
Laslett, D. & Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175. https://doi.org/10.1093/bioinformatics/btm573 (2008).
Posada, D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. https://doi.org/10.1093/molbev/msn083 (2008).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. https://doi.org/10.1093/bioinformatics/btl446 (2006).
Rydin, C., Kainulainen, K., Razafimandimbison, S. G., Smedmark, J. E. E. & Bremer, B. Deep divergences in the coffee family and the systematic position of Acranthera. Plant Syst. Evol. 278, 101–123. https://doi.org/10.1007/s00606-008-0138-4 (2009).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15. http://irc.igd.cornell.edu/Protocols/DoyleProtocol.pdf (1987).
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R. & Phillippy, A. M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. https://doi.org/10.1101/gr.215087.116 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963. https://doi.org/10.1371/journal.pone.0112963 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. https://doi.org/10.1038/nbt.1754 (2011).
Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. https://doi.org/10.1093/bioinformatics/bth352 (2004).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. https://doi.org/10.1093/nar/25.5.955 (1997).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. https://doi.org/10.1093/bioinformatics/btx198 (2017).
Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Sharp, P. M. & Li, W. H. The codon Adaptation Index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. https://doi.org/10.1093/nar/15.3.1281 (1987).
Hassanin, A., Léger, N. & Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 54, 277–298. https://doi.org/10.1080/10635150590947843 (2005).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. https://doi.org/10.1093/nar/gkh340 (2004).
Acknowledgements
We appreciate associate editor and two anonymous reviewers for helpful comments that substantially improved this article. This work is financially supported by the Central Finance Forestry Reform and Development Fund (2018-GDTK-08) and the funding from South China Agricultural University (4400-K16013).
Author information
Authors and Affiliations
Contributions
X.S.H. and X.W. conceived and designed the study; X.W. conducted the experiment and drafted the manuscript; L.L.L. and Y.X. participated in experiment, X.Y.C. provided experimental supports, J.H.C. participated in DNA sequencing; X.S.H. revised and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Li, LL., Xiao, Y. et al. A complete sequence of mitochondrial genome of Neolamarckia cadamba and its use for systematic analysis. Sci Rep 11, 21452 (2021). https://doi.org/10.1038/s41598-021-01040-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-01040-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
