
Abstract
Although genome-wide association studies have identified single nucleotide polymorphisms (SNPs) associated with the susceptibility to Mycobacterium avium subsp. paratuberculosis (MAP) infection, only a few functional mutations for bovine paratuberculosis (PTB) have been characterized. Expression quantitative trait loci (eQTLs) are genetic variants typically located in gene regulatory regions that alter gene expression in an allele-specific manner. eQTLs can be considered as functional links between genomic variants, gene expression, and ultimately phenotype. In the current study, peripheral blood (PB) and ileocecal valve (ICV) gene expression was quantified by RNA-Seq from fourteen Holstein cattle with no lesions and with PTB-associated histopathological lesions in gut tissues. Genotypes were generated from the Illumina LD EuroG10K BeadChip. The associations between gene expression levels (normalized read counts) and genetic variants were analyzed by a linear regression analysis using R Matrix eQTL 2.2. This approach allowed the identification of 192 and 48 cis-eQTLs associated with the expression of 145 and 43 genes in the PB and ICV samples, respectively. To investigate potential relationships between these cis-eQTLs and MAP infection, a case–control study was performed using the genotypes for all the identified cis-eQTLs and phenotypical data (histopathology, ELISA for MAP-antibodies detection, tissue PCR, and bacteriological culture) of 986 culled cows. Our results suggested that the heterozygous genotype in the cis-eQTL-rs43744169 (T/C) was associated with the up-regulation of the MDS1 and EVI1 complex (MECOM) expression, with positive ELISA, PCR, and bacteriological culture results, and with increased risk of progression to clinical PTB. As supporting evidence, the presence of the minor allele was associated with higher MECOM levels in plasma samples from infected cows and with increased MAP survival in an ex-vivo macrophage killing assay. Moreover, the presence of the two minor alleles in the cis-eQTL-rs110345285 (C/C) was associated with the dysregulation of the eukaryotic elongation factor 1-α2 (eEF1A2) expression and with increased ELISA (OD) values. Finally, the presence of the minor allele in the cis-eQTL rs109859270 (C/T) was associated with the up-regulation of the U1 spliceosomal RNA expression and with an increased risk of progression to clinical PTB. The introduction of these novel functional variants into marker-assisted breeding programs is expected to have a relevant effect on PTB control.
Similar content being viewed by others

Introduction
Paratuberculosis (PTB) is a gastrointestinal disease of domestic and wild ruminants caused by Mycobacterium avium susbp. paratuberculosis (MAP). PTB causes direct economic losses to the dairy farms due to decreased milk production, replacement cost, and premature culling or death from the clinical disease1,2. The most common clinical signs are progressive weight loss, diarrhea, and decreased milk yield3. PTB is a major problem for animal health and must be notified to the World Organization for Animal Health. In addition, MAP is a suspected cause of Crohn’s disease (CD) in humans4. Colorectal cancer is a complication of the two forms of idiopathic inflammatory bowel disease (IBD); colonic CD and ulcerative colitis. Interestingly, MAP bacilli have been detected in the intestines of patients with CD, ulcerative colitis, and IBD-associated colorectal cancer5,6.
Improving host genetics through selective breeding could enhance natural resistance to MAP infection and complement existing control strategies7,8,9. Over the last decade, different strategies including case–control studies in candidate genes and genome-wide association studies (GWAS) identified single nucleotide polymorphisms (SNPs) significantly associated with susceptibility to bovine PTB10,11,12,13,14,15,16. However, few SNPs were identified with consistent association across studies and the molecular mechanisms by which they contribute to disease remain largely unknown. Recent studies using RNA next-generation sequence technology (RNA-Seq) have been conducted to gain a better understanding of the molecular mechanisms involved in the circulating and local response against a natural MAP infection17,18,19. While transcriptomic studies provide insight into the genes and mechanisms involved in the response to MAP infection, they do not reveal whether there is variation in the transcriptional response among individuals and how this might influence PTB outcome.
Jansen and Nap20 introduced the concept of “genetical genomics” where genomic variants were associated with transcript abundance which acts as a heritable endophenotype. This concept supported the idea that integrating genomic variants and gene expression data could contribute to a better understanding of the genetic architecture underlying disease outcome (an endpoint phenotype)21,22. Polymorphisms associated with gene expression are known as expression quantitative trait loci (eQTLs). In a cis position (within 1 Mb upstream of the transcription start site of a gene locus), cis-eQTLs might alter transcription regulation, mRNA stability, or mRNA splicing23,24. This leads to changes in the level, timing, and/or localization of gene expression which can significantly influence variations in individual phenotype. In the current study, we hypothesized that individual genetic variation in the transcriptional response to PTB infection might have implications in disease outcome. The main objective was to identify cis-eQTLs significantly associated with changes in gene expression levels in peripheral blood (PB) and ileocecal valve (ICV) samples collected from Holstein cattle with no lesions and with PTB-associated lesions in gut tissues. Next, we assessed whether the genes with cis-eQTLs were DE in PTB-infected cows versus control cows. Finally, to test whether the identified cis-eQTLs affected not just gene expression but also the PTB risk, a case–control study using the genotypes of all the identified cis-eQTLs and the phenotypical data of a cohort of 986 culled cattle was performed. The workflow of the study is presented in Fig. 1.

Study design. The approach starts from the individual expression levels, then identifies genetic variants controlling differences in gene expression, and finally checks that the identified cis-eQTLs are indeed associated with disease outcome.
Results
RNA-Seq data and genotypes
Samples from the animals included in this study were tested by ELISA for the detection of MAP-specific antibodies, qPCR for MAP DNA detection, bacteriological culture, and histopathological analysis, as previously described (Table 1)18. All control animals (N = 4) were negative for all the diagnostic tests. In contrast, the infected cows had detectable lesions in gut tissues with distinct severity; focal/multifocal (N = 6) or diffuse (N = 4). RNA-extraction from PB and ICV samples of the fourteen cows, RNA-Seq libraries preparation, and sequencing using an Illumina NextSeq500 sequencer were performed as previously described18. The raw reads were filtered by their length (minimum size 75 bp long) and percentage of ambiguous base N less than 10% using Prinseq-lite 0.20.425. Then, the trimmed sequences were mapped to the Bos Taurus reference genome (Bos_taurus.UMD3.1. version 87) with TopHat 2.1.1.26. In the current study, the mapped reads were quantified with RSubread to calculate gene expression levels (read counts)27. In addition, genomic DNA was extracted from PB samples of the fourteen animals and subsequently genotyped using the Illumina MD EuroG10K BeadChip (54,609 SNPs) as previously described28. Altogether the study is comprised of 54,609 genetic variants and expression values of 24,616 genes in 28 samples.
cis-eQTL analysis
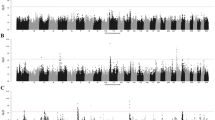
Gene expression data (read counts) were normalized with the mean-of-ratios method included in the DESeq 2 package29. Our integrated genomic-transcriptomic dataset was analyzed for associations between SNPs-normalized read counts using R Matrix eQTL 2.230 including the age of slaughter as a covariate. Matrix eQTL tests for associations between SNPs-normalized read counts by modeling the effect of genotype as additive linear (least square model). cis-eQTLs were identified by including all the variants on the same chromosome that are located 1 Mb upstream of the transcription start site (TSS) of a gene locus. A genetic variant was regarded as a significant cis-eQTL for a given gene if PFDR ≤ 0.05. For the PB samples, two analyses were performed including all the tested genetic variants located within 0.5 and 1 Mb upstream of the TSS of a gene locus; Fig. 2a,b respectively. Using the 24,616 mapped genes, we identified 96 and 192 cis-eQTLs which were associated with 76 and 145 genes, respectively. As expected, all the cis-eQTLs identified in the first analysis were validated when the analysis was extended 1 Mb upstream of the TSS of a gene locus. Using the ICV datasets, the SNPs-reads association analysis allowed the identification of 48 cis-eQTLs located within 0.5 Mb upstream of a TSS and associated with the expression of 43 genes (Fig. 2c). As expected, all the identified cis-eQTLs were located on the same chromosome as the associated gene. Most of the identified cis-eQTLs were located in intronic or intergenic regions (Fig. 2d–f). The complete cis-eQTL discovery in the PB and ICV samples is presented in Tables S1–S3.

cis-eQTL analysis. (a,b) PB cis-eQTLs identified within 0.5 (a) and 1 Mb (b) upstream of a TSS. (c) ICV cis-eQTLs identified within 0.5 Mb upstream of a TSS. The plots show the − log10 (P-values) of the most significant variants for each of the 24,616 analyzed genes. The red line represents the threshold of P = 2.03E−05 (a), P = 2.14E-05 (b) and P = 1.02E−05 (c). (d,e) The chart depicts the genomic distribution of the PB-cis-eQTLs identified within 0.5 (d) and 1 Mb (e) upstream a TSS according to the Ensembl Variant Effect Predictor (VEP). (f) Genomic distribution of the ICV cis-eQTLs according to the VEP.
The cis-eQTL (rs109475758) with the lowest PFDR (PFDR = 4593E−14) in the PB dataset was associated with the obscurin (OBSC) gene (Table S1), a highly mutated gene in different types of cancer. The rs109475758 heterozygous genotype (T/G) resulted in increased counts of the OBSC gene while the most frequent homozygous genotype (T/T) correlated with low levels of OBSC gene expression (Fig. 3a). In the ICV samples, the gene with the strongest cis-eQTL (rs41753850) association was the apolipoprotein A4 gene (APOA4 ; PFDR = 8413E−11) (Table S3). APOA4 is associated with circulating high-density lipoproteins (HDL) and plays an important role in cholesterol transport and lipid metabolism. Interestingly, the rs41753850 was also associated with the apolipoprotein C3 (APOC3) gene expression (PFDR = 0.0031). APOC3 plays an important role in regulating the metabolism of triglyceride-rich lipoproteins and has been regarded as a main component of chylomicrons and very-low-density lipoproteins (VLDL). The heterozygous genotype in the rs41753850 (C/A) resulted in increased levels of APOA4 and APOC3 expression, while the most frequent homozygous (C/C) correlated with lower levels of APOA4 and APOC3 gene expression (Fig. 3b,c).

Boxplots for nine significant cis-eQTLs associations with gene expression. The rs109475758 (a), rs41753850 (b,c), rs41591969 (d), rs108986746 (e), rs110435196 (f), rs43744169 (g), rs110345285 (h) and rs109859270 (i) were associated with the OBSC, APOA4, APOC3, C3, S100B, SELENOS, MECOM, eEF1A2 and U1 spliceosomal RNA expression, respectively. Gene variants are presented on the horizontal axis and gene expression levels, normalized gene counts, on the vertical axes. Samples were grouped according to the absence of lesions (negative) or the presence of focal/multifocal and diffuse lesions in gut tissues. The median and the upper and lower quartiles of each group are represented by horizontal and vertical lines, respectively. The lowest point is the minimum of the data set and the highest point is the maximum of the data set.
Association between the genes with cis-eQTLs and MAP infection
We assessed whether the genes with cis- eQTLs were DE in PTB-infected cows versus controls (Table 2) using the differential expression information generated with the Cufflinks package 2.2.1. in our previous study18 and with DESeq2 in the current study. Despite the small number of control cows, we found that 24 of the genes with cis-eQTLs were DE in the PB samples from MAP-infected cows with focal/multifocal (N = 6) or diffuse lesions (N = 4) versus control cows (N = 4) (Table 2). Some of these cis-eQTLs-associated genes play key roles in tumorigenicity (OBSC, GSTO1), the apoptotic process (CD5L, BNIP3), transcription regulation (PERM1), cell adhesion (NRCAM), platelet aggregation (GP1Bβ, MRVI1), G-protein coupled receptor signaling pathway (ADCY8, ACKR1), lymphocyte activation (HS1BP3), adhesion of blood neutrophils in cytokine-activated endothelium (Selectin E) and in the regulation of hypoxia-induced autophagy (ANKRD37) in MAP-infected cattle. In addition, we found that fourteen genes with cis-eQTLs were DE in the ICV samples from MAP-infected cows with focal/multifocal or diffuse lesions versus control cows (Table 2). Some of these cis-eQTLs-associated genes play key roles in the extracellular matrix structure (ACAN), negative regulation of NF-kβ (TRIM40), innate immune response (NEURL3, ARSB, Duosenase-1 like), tumorigenicity (PROM2), G-protein coupled receptor signaling pathway (ACKR1), and oxidative stress (PRDX1) in MAP-infected cattle.
To identify common biological functions, enrichment analysis of biological processes was performed with the cluster Profiler Bioconductor package 3.10.1.31 using all genes with cis-eQTLs. The rationale is that any change to a single gene involved in a process could alter many genes in a biological process and thus the phenotype of the animal. Gene ontology enrichment analysis indicated that three genes (C3, S100B, SELENOS) with cis- eQTLs were associated with the regulation of acute inflammatory response (GO:0002673) which was significantly enriched (PFDR ≤ 0.05). The heterozygous genotype in the cis-eQTLs of the C3 (Fig. 3d), S100B (Fig. 3e), and SELENOS (Fig. 3f) genes resulted in higher gene expression levels; PFDR = 0.0254, PFDR = 0.0315 and PFDR = 0.0320, respectively. In addition, the functional analysis revealed that the cholesterol metabolic route (bta04979) was significantly enriched, with the APOA4 and APOC3 genes involved in this route (PFDR = 0.009, Gene ratio: 2/46). This finding reinforced the idea that the APOA4-APOC3 gene cluster may be associated with the PTB risk.
Case–control association study
If the expression levels of a cis-eQTL have any effect on the PTB risk, we should observe differences in disease phenotypes among the different genotypes. To address this hypothesis, a case–control association study using the genotypes for the 235 identified cis-eQTLs and phenotypical data of 986 cull Holstein Friesian cows was performed. The infection status of these cows was previously determined by histopathological analysis, ELISA, PCR, and bacteriological culture32. Cattle were classified as cases if they were positive for ELISA, PCR culture, and/or if they had PTB-associated lesions in gut tissues. The 986 cows were genotyped for the identified cis-eQTLs and cows with a genotyping rate < 80% were excluded. SNPs deviated significantly from the Hardy–Weinberg equilibrium were removed33. Case–control associations were analyzed with the WGassociation function of SNPassoc 1.9.2 under five different genetic models (co-dominant, dominant, recessive, over-dominant, and log-additive)34. P-values were adjusted for multiple comparisons using the Bonferroni correction. As seen in Table 3, the rs43744169, rs110345285, and rs109859270 showed significant associations with PTB phenotypes. The association analysis using the cis-eQTLs identified with the PB dataset and a minor allele frequency (MAF) > 20% revealed significant associations between the cis-eQTL-rs43744169 (T/C) located on chromosome 1 and several disease phenotypes including histopathology, ELISA (OD), and ELISA, PCR, and bacteriological culture results. The heterozygous genotype (T/C) in the cis-eQTL-rs43744169 was associated with the up-regulation of the MDS1 and EVII complex (MECOM) expression (Fig. 3g; PFDR = 0.00003).
Using a MAF > 10%, an association between the cis-eQTL-rs110345285 (T/C) and mean ELISA (OD) values was observed under the codominant and recessive genetic models. The presence of the two minor alleles in the cis-eQTL-rs110345285 (C/C) regulating the eukaryotic elongation factor 1-α2 (eEF1A2) expression was associated with a significant increase in mean OD values (OD = 0.80) when compared with cows with the most frequent homozygous genotype (T/T) (OD = 0.29) or with the heterozygous genotype (T/C) (OD = 0.26). The heterozygous genotype in the cis-eQTL-rs110345285 (T/C) was associated with the up-regulation of the eEF1A2 expression when compared with cows with the most frequent homozygous genotype (T/T) (Fig. 3h; PFDR = 1.23E−10). Using the ICV-identified cis-eQTLs and a MAF > 10%, a significant association between the cis-eQTL-rs109859270 (C/T) and the absence or presence of PTB-associated lesions was observed. The heterozygous genotype in the cis-eQTL-rs109859270 (C/T) located in the bovine chromosome 8 was associated with higher expression levels of the U1 spliceosomal RNA (Fig. 3i; PFDR = 0.0068).
The distribution of the rs43744169 and rs109859270 genotypes and odds ratio (OR) for each statistically significant genetic model and binary phenotype are presented in Table 4. For the rs43744169, the OR with 95% confidence interval (CI) under the codominant genetic model for the T/C and C/C genotypes versus the T/T genotype were 2.71 (95% CI 1.50–4.88) and 5.36 (95% CI 1.99–14.41) respectively. These results indicated that the proportion of T/C heterozygotes and C/C homozygotes with a positive ELISA, culture, and PCR result were significantly higher in the cases as compared to control cows. Under the dominant model, the genotypes T/C and C/C increased the risk of PTB compared to the genotype T/T; OR = 2.98 (95% CI 1.70–5.24). For the rs109859270 locus, the OR for the C/T and T/T genotypes versus the C/C genotype was 1.64 (95% CI 1.24–2.16), which indicated that the proportion of animals with one or two copies of the minor allele was significantly higher in animals with PTB-associated lesions as compared to control animals without lesions. Overall, the presence of the minor alleles in the rs43744169 and rs109859270 increased the PTB risk. Consequently, selecting against the rs43744169 and rs109859270 minor alleles may reduce the risk of PTB in cattle.
The presence of the minor allele in the cis-eQTL-rs43744169 (T/C) resulted in increased MECOM protein levels
The heterozygous genotype (T/C) in the cis-eQTL-rs43744169 was associated with higher MECOM gene expression levels (Fig. 3g, PFDR = 0.00003). Using a bovine MECOM quantitative sandwich ELISA, we tested the MECOM protein levels in plasma samples from cows with focal lesions in gut tissues and with the three different rs43744169 genotypes (T/T, T/C, C/C). As seen in Fig. 4, the heterozygous (T/C) genotype and the minor allele homozygous (C/C) genotype were associated with significantly higher MECOM protein expression (19.60 and 8.62 ng/ml, respectively) compared with the concentration of MECOM in plasmas from cows with the major allele homozygous genotype (T/T, 3.90 ng/ml). This finding confirmed that increases in MECOM gene counts did correlate with higher levels of protein expression.

The MECOM expression is associated with the rs43744169. MECOM protein levels in plasmas from homozygous and heterozygous cows in the rs43744169 were measured with a bovine ELISA. Samples were selected from the case–control population and grouped according to the rs43744169 genotypes. Cows with the alleles T/C (N = 13) and C/C (N = 12) were associated with elevated MECOM protein levels when compared with the MECOM levels in the plasmas from cows with the two major alleles T/T (N = 11); P = 0.05 and P = 0.03, respectively. The median and the upper and lower quartiles of each group are represented by horizontal and vertical lines, respectively. The lowest point is the minimum of the data set and the highest point is the maximum of the data set. P-values were calculated using an unpaired t-test. *P-value ≤ 0.05. Each sample is represented by a circle on the dot plot.
Association between MAP survival and the cis-eQTL-rs43744169 genotypes using a macrophage killing assay
The association between susceptibility to MAP and the cis-eQTL-rs43744169 genotypes (T/T and T/C) was tested using an ex vivo monocyte-derived macrophages (MDM) killing assay. MDM were purified from peripheral blood of uninfected Holstein cattle with the cis-eQTL-rs43744169 genotypes T/T (N = 8) and T/C (N = 8) and infected in vitro with a virulent strain of MAP (K10 isolate). Bacterial load (log colony-forming units, log CFU) at 2 h and 7 days post-infection (p. i.) was estimated as previously described35. The genotype T/T showed higher MAP uptake at 2 h p.i. (P = 0.0031) and more successful MAP killing at 7 days p.i. when compared with the T/C genotype (P = 0.020) (Table 5). These results suggested a significant effect of the cis-eQTL-rs43744169 heterozygous genotype on bacterial survival within macrophages and confirmed that cis-regulatory variation modulates susceptibility to MAP infection.
Discussion
The association between expression and genotype can be tested using linear regression and ANOVA models, as well as non-linear techniques including generalized linear and mixed models, Bayesian regression, and models accounting for pedigree30. In the current study, we integrated gene expression data quantified by RNA-Seq and SNP data for a cohort of cows naturally exposed to MAP infection and maintained in the same environment. Using linear regression analysis, we identified 192 and 48 cis-eQTLs associated with the expression of 145 and 43 genes in PB and ICV, respectively. Only one of the identified cis-eQTLs, the rs109604269 (PFDR = 0.011), was previously found to be associated with susceptibility to MAP infection36. Although the number of individual samples in this first part of the study was limited (N = 28), three of the identified cis- eQTLs were associated with PTB susceptibility in a larger cattle population (N = 986). Our study provides first insights into the role of cis-eQTLs in gene transcription regulation and PTB susceptibility. Since RNA-Seq technology is becoming increasingly affordable and provides direct information on the presence of genetic variations at the sequence level, further studies should be directed to call SNPs from RNA-Seq data. This approach would provide a secondary confirmation of the cis-eQTLs identified in the current study.
Functional analysis of the 235 genes under cis-regulation revealed that there was a significant enrichment of the regulation of the acute inflammatory response (GO:0002673) with three genes (C3, S100B, SELENOS) involved in this biological process. Heterozygosis in the cis-eQTLs regulating these three genes resulted in increased C3, S100B, and SELENOS expression. Higher C3 levels may allow increased opsonophagocytosis and effective bacterial clearance of several microorganisms including mycobacteria37. SELENOS is involved in the degradation process of misfolded proteins in the endoplasmic reticulum, and may also have a role in inflammation control38. Since inflammation control depends largely on the recruitment of granulocytes (i.e., neutrophils, eosinophils, and basophils) to the inflammatory site, chemokine signaling is involved in this process. Neutrophils then secrete antibacterial proteins such as the S100 family to destroy microbes. It is well recognized that chronic inflammatory diseases develop only when prompt and active innate immunity mechanisms, like granulocytes recruitment to the sites of infection, are ineffective or defective39. In fact, our recent RNA-Seq analysis from PB samples of Holstein cattle infected with MAP revealed down-regulation of the C3, S100B, SELENOS genes, and of the CXCL8/IL8 signaling pathway, a pathway involved in neutrophils recruitment and resolution of inflammation18. Similarly, patients with CD have decreased secretion of CXCL8/IL8 and reduced neutrophils migration to the sites of infection compared with healthy individuals40. The results presented in the current study provide evidence that genetic variants in regulatory regions (cis-eQTLs) can up-regulate the C3, S100B, and SELENOS gene expression which might activate a prompt resolution of the infection. However, we did not find any association between the cis-eQTLs regulating the C3, S100B, and SELENOS gene expression and PTB resistance in our case–control study. Further studies are needed to make sure that the regulation of the C3, S100B, and SELENOS gene expression has not a functional role in PTB resistance or tolerance.
When a functional analysis with the identified cis-regulated genes was performed, the cholesterol metabolic route (bta04979) was significantly enriched with the apolipoproteins APOA4 y APOC3 involved in this route (PFDR = 0.009, Gene ratio: 2/46). The genes coding for the APOA1 and APOC3 are closely linked on the bovine chromosome 15 and have synergistic functions. We found that the heterozygous genotype in the rs41753850 (C/A) resulted in higher levels of APOA4 and APOC3 expression, while its absence correlated with lower levels of gene expression. Recent reports have demonstrated that MAP can manipulate the host lipid metabolism and accumulate cholesterol within macrophages which might favor MAP persistence41. Our results reinforce the notion that the lipoprotein metabolism plays an important role in PTB pathogenesis and that PTB-susceptible cows might carry alleles that increase lipoprotein production and transport. Consequently, cis-eQTLs associated with the APOA4 and APOC3 gene expression may be associated with the PTB risk. In humans, genetic variants in the APOA1 –APOC3–APOA4 cluster has been associated with the risk of Alzheimer disease, polycythemia induced gastric injury, and metabolic syndrome42,43,44. However, we did not find any association between the cis-eQTLs regulating APOA4 and APOC3 gene expression and PTB risk in our case–control study.
Although the number of normalized reads was low, Matrix eQTL found a significant association between the rs43744169 (T/C) and the MECOM gene expression (PFDR = 0.00003). Next, we found that the presence of the minor allele in the rs43744169 (T/C) was associated with positive ELISA, PCR, and bacteriological culture results, and with increased risk of clinical PTB. Since our findings provided compelling evidence for the critical role of the MECOM in the progression to clinical PTB, it was prioritized in functional studies. Using a MECOM quantitative ELISA, we observed that the presence of the minor allele in the rs43744169 was associated with higher levels of the MECOM protein in plasma samples from infected cows and with increased MAP survival in an ex vivo macrophage killing assay. These findings should be confirmed in a larger population study and, additionally, by performing in vivo challenges. Allelic variants affecting the human MECOM gene have been associated with human inflammatory bowel disease45. Mutations resulting in aberrant expression of the MECOM have also been found in many types of solid cancers, including colorectal cancer, as well as in acute myeloid leukemia46,47. Previous scientific evidence showed that the MECOM acts as an oncogene regulating signaling pathways that lead to increased tumor proliferation48. More specifically, cell line assays have demonstrated a role for the MECOM in the transcriptional control of c-fos, transforming growth factor β (TGF-β) and the activator protein 1 (AP1) proliferative pathway49. The MECOM also binds to the promoter regions of the majority of genes involved in the JAK-STAT signaling pathway50. Excessive JAK-STAT signaling activation results in numerous inflammatory and hematopoietic disorders. Recent findings indicate that the MECOM is up-regulated by inflammatory stimuli, including bacteria, and that mutations in the MECOM made mice more susceptible to bacterial infections51. The MECOM has also a recognized function as a crucial regulator of the nuclear factor-kappa β (NF-κβ)-mediated inflammatory response52. NF-κβ is a critical factor in the gut immune responses to pathogens and in promoting inflammation-associated carcinoma in the gastrointestinal tract53. Our findings suggest that genetic variants in a cis-eQTL affecting the expression of the MECOM, a transcriptional regulator of the NF-κβ inflammatory response, may cause an uncontrolled and aberrant inflammatory response which might exacerbate tissue injury in PTB-infected cattle. This is in agreement with Kiser et al., who found an association between seven genes linked to the NF-κβ pathway (DUSP10, IKBKB, NR4A1, PRKCA, SLC2A5, TGFB2, and PIK3R1) and MAP tissue infection in two Holstein populations using gene set enrichment analysis-SNP (GSEA-SNP)16.
Our case–control association study revealed a second cis-eQTL, the rs110345285 (T/C), associated with the up-regulation of the eEF1A2 gene expression and with high levels of MAP antibodies. More specifically, increased ELISA (OD) values were associated with the less frequent homozygous genotype (C/C) in the cis-eQTL-rs110345285. The eEF1A2 is a protein translation factor involved in protein synthesis and with important anti-apoptotic, migration and pro-metastasis functions on cancer development54,55. Mechanistic studies revealed that the PI3K/Akt /NF-κβ and JAK/STAT signaling pathways play an important role in mediating the effects of the eEF1A256. eEF1A2 expression increases the formation of filopodia structures that have an important role in driving cell migration and invasion57. High eEF1A2 expression is associated with severe tumor grades and metastasis in several cancer cell lines and with some cases of multiple myeloma, a plasma cell neoplasm of humans58. Furthermore, the eEF1A2 blocks apoptosis and favors viral replication59. In agreement with this, our results suggested that PTB-infected cows carrying the risk allele in the rs110345285 (C) and exhibiting high levels of eEF1A2 might have a reduced probability of survival compared to their non-eEF1A2-expressing counterparts.
Our case–control association analysis revealed a third significant association between the cis-eQTL-rs109859270 (C/T) and the absence or presence of PTB-histopathological lesions. For the rs109859270 locus, the OR for the C/T and T/T genotypes versus the C/C genotype was 1.64 (95% CI 1.24–2.16), which indicated that the proportion of animals with one or two copies of the minor allele were significantly higher in animals with PTB-associated lesions as compared to control animals without lesions. In addition, the heterozygous genotype in the cis-eQTL-rs109859270 was associated with the up-regulation of the U1 spliceosomal RNA expression. Splicing is performed by a spliceosome that assembles on each intron and predominantly comprises U1, U2, U4, and U5, and U6 uridyl-rich small nuclear RNPs (snRNPs) in equal stoichiometry60,61. U1 snRNPs plays an essential role in defining the 5′ end splice site by RNA-RNA base pairing. Besides its splicing role, U1 snRNPs protects pre-mRNAs from drastic premature termination by premature cleavage and polyadenylation (PCPA) at more proximal alternative polyadenylation sites, which has already been proved in activated immune, neuronal, and cancer cells62. U1 snRNA abnormalities can cause defects in pre-mRNA splicing, which are considered as a primary cause of several diseases63. In other words, mammalian cells require U1 snRNA with expression levels in a restricted range to prevent drastic premature termination of the nascent pol II transcripts by PCPA64. In human macrophages, U1 snRNA over-expression has been associated with impairment of autophagosome-lysosome fusion65. Recent studies reported that the infection of human macrophages with Mycobacterium tuberculosis (MTb) results in massive alterations in the pattern of RNA splicing in the host66,67. Furthermore, two genes related to macrophage function, the monocyte to macrophage differentiation gene (MDM) and adenosine deaminase (ADA) genes, were differentially spliced in MAP-infected ileum suggesting a possible mechanism by which MAP escapes the host innate immune response68. Our findings suggested that the heterozygous genotype in the cis-eQTL-rs109859270 (C/T) resulted in increased U1 spliceosomal RNA expression levels. In contrast, the most frequent homozygous genotype (C/C) maintained the U1 snRNA expression levels in a restricted range and was associated with a more controlled risk of infection. Further studies are required to understand the mechanisms of splicing and alternative splicing regulation upon MAP infection and how they can impact immune responses in cattle.
Conclusions
The identification of functional variants (cis-eQTLs) followed by a case–control association analysis allowed the identification of three cis-eQTLs regulating the expression of the MECOM, eEF1A2, and U1 spliceosomal RNA expression and their validation as significantly associated with PTB susceptibility in a larger cattle population. It should be emphasized that MECOM, eEF1A2, and U1 risk alleles have shown detrimental effects in other inflammatory diseases including human IBD and colorectal cancer. Furthermore, the introduction of the cis-eQTLs described in the current study into marker-assisted breeding programs might reduce the disease through selection, reduce economic losses, and improve animal health.
Materials and methods
Ethics statement
Experimental procedures were approved by the Animal Ethics Committee of the Servicio Regional de Investigation y Desarrollo Agroalimentario (SERIDA) and authorized by the Regional Consejeria de Agroganadería y Recursos Autoctonos of the Principality of Asturias (authorization code PROAE 29/2015). All the procedures were conducted following 2012/63/EU of the European Parliament. Blood, fecal, and tissue samples were collected by trained personnel and following good veterinary practice. Animals used in the case–control study were sampled under pertinent legislation for safeguarding animal welfare (Basque Government Decree 454/1994, Spanish Government Law 32/2007, Royal decree 731/2007, and European council regulation number 1099/2009).
Animals and PTB diagnosis
PB and ICV samples were collected from fourteen Holstein Friesian cows from a single commercial dairy farm in Asturias (Spain) at the time of slaughter. This farm was selected for sample collection based on PTB clinical cases, with some cows confirmed by necropsy examination. The mean prevalence of the disease in the farm (period 2016 to 2019) was estimated at 6.29% by ELISA for the detection of MAP antibodies. Infection status was determined by histopathological analysis of gut tissues, ELISA, bacteriological culture, and PCR assays as previously described28. For the case–control study, the infection status of 986 Holstein Friesian cattle from eight Northeast Spain regions was determined by histopathological analysis of gut tissues, ELISA for MAP antibodies detection, and tissue culture and PCR for MAP detection32. Animals were 5.5 years old on average. Except for six animals that were aged between 18 and 23.2 months, the majority of the animals were adults (2 years or older).
Genotyping
Blood samples were collected from the coccygeal (tail) vein into EDTA Vacutainer tubes (BD Vacutainer System, Becton, Dickinson, and Company, Sparks, MD, USA). Blood samples were centrifuged at 2500×g for 20 min at 4 °C and buffy layers containing white blood cells were collected. Genomic DNA was extracted from the blood buffy coat using the QIAmp DNA Blood Mini Kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Purified genomic DNA was quantified spectrophotometrically and subsequently genotyped on the EuroG10K BeadChip at the molecular genetic laboratory service of the Spanish Federation of Holstein Cattle (CONAFE) using the Infinium iScan software for allele assignation (Illumina, San Diego, CA) as previously described28. The EuroG10K BeadChip is a development by EuroGenomics and its collaborators.
Gene expression data
In the current study, the alignment files from our previous RNA-Seq study were used18. Briefly, PB samples were collected by venipuncture of the coccygeal vein using PAXgene Blood RNA tubes (2.5 ml) (Qiagen, Hilden, Germany). Total RNA was purified from the PB samples using the PAXgene blood RNA kit according to the manufacturer´s instructions (Qiagen, Hilden, Germany). For RNA isolation, 150–200 mg of ICV were harvested and immediately submerged in 2 ml of RNA later (Sigma, St. Louis, MO). Purification of RNA was performed using the RNeasy Mini Kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Residual genomic DNA was removed using DNase digestion with RNase-free DNase I amplification grade following the recommended protocol (Invitrogen, Spain). The concentration and quality of the total RNAs were measured using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, US). All samples had an RNA integrity value of 7 or greater. Approximately 250 ng of RNA were used for RNA-Seq library preparation using the Illumina NEBNext Ultra Directional RNA Library preparation kit following the manufacturer´s instructions (Illumina, San Diego, CA, US). Libraries quality was assessed using an Agilent Bioanalyzer and Agilent high sensitivity DNA chip to confirm that the insert sizes were 188–274 bp for all the individual libraries. RNA-Seq libraries were single-end sequenced in a 1 × 75 bp format using an Illumina NextSeq 500 sequencer at the Genomic Unit of the Scientific Park of Madrid, Spain. All the libraries were sequenced generating an average of 22.31 million raw reads per library with a Phred quality score > 30. The raw reads were filtered by their length (minimum size 75 bp long) and percentage of ambiguous base N less than 10% using Prinseq-lite 0.20.425. After quality control, reads were subsequently mapped to the Bos Taurus reference genome (Bos_taurus.UMD3.1. version 87) with TopHat 2.1.126. The resulting alignment files (.bam) were provided to the Cufflinks package 2.2.169 to calculate gene expression levels and genes differentially expressed in the infected animals with focal/multifocal or diffuse lesions versus the control group.
In the current study, the alignment files (.bam) from our previous RNA-Seq study18 were used to generate a table of reads for each sample using Rsubread27. The number of reads mapped to 24,616 genes was counted using the function feature counts in the Rsubread package. Next, the analysis of differential expression between the cows with focal/multifocal or diffuse lesions versus control cows was conducted using DESeq229. The differentially expressed genes were selected as those with an adjusted P- value (PFDR) ≤ 0.05.
cis-eQTL analysis
Gene expression data (read counts) were normalized with the mean-of-ratios method included in the DESeq2 package29. Then, R Matrix eQTL 2.2 was used to test for associations between each SNP-normalized counts by modeling the effect of genotype as additive linear (least square model), where the null hypothesis is no association between genotype and gene expression. This tool implements matrix covariance calculation and efficiently runs linear regression analysis for cis-eQTL discovery30. cis-eQTLs were calculated by selecting gene variants on the same chromosome that are located within 1 Mb upstream of the TSS of a gene locus. Specific details regarding the matrix structure in Matrix eQTL have been previously described70. Briefly, S and G denote the genotype matrix and the gene expression matrix, respectively. Each row of these matrices holds different measurements for a single SNP among samples and a single gene across samples, respectively. Data matrices are sliced into blocks of up 10,000 variables. Then, gene expression and genotype matrices are standardized. For each pair of blocks, the correlation matrix for a relevant block is calculated and Matrix eQTL checks if the absolute value of any correlation exceeds a predefined threshold. Then, Matrix eQTL calculates the P-values only for those SNP-normalized counts associations selected based on their correlations being larger than the threshold. Matrix eQTL uses the false discovery rate (FDR)71 to adjust for multiple hypotheses testing and to identify significant cis-eQTLs (PFDR ≤ 0.05). FDR is the expected proportion of false positives among all the significant tests.
Functional enrichment analysis
Genes with significant cis-eQTLs were investigated for enrichment of biological processes using gene ontology categories72 within the cluster Profiler Bioconductor package 3.10.131 and Panther database73. A threshold for significant enrichment was set at PFDR ≤ 0.05 after adjustment with the Benjamini–Hochberg procedure71.
Case–control study
Associations between genotypes-phenotypes were assessed by logistic regression analysis using the WGassociation function of SNPassoc 1.9.2 under five different genetic models (co-dominant, dominant, recessive, over-dominant, and log-additive)34. The response variable can be binary (positive or negative diagnostic result) or continuous (ELISA OD values and PTB-associated lesions classified according to their severity in five grades). The genotypes of the cis- eQTLs were included as possible explanatory variables. The WGassociation function fits individual logistic regression models to each of the class variables (genotypes) using age as a covariate. P-values were adjusted using the Bonferroni correction (based on the total number of markers tested) for multiple comparisons. For each cis-eQTL and genetic model, the function WGstats of SNPassoc 1.9.2. provided genotype frequencies, OR (or mean differences for quantitative traits), and 95% CI with the major homozygous genotype deemed as the baseline.
Bovine MECOM ELISA kit
The levels of MECOM protein in plasma samples (50 µl) were assessed by using a quantitative sandwich ELISA according to the manufacturer’s instructions (MyBioSource, San Diego, US). The sensitivity of the kit is 1.0 ng/ml and the detection range is 3.12–100 ng/ml. Briefly, standards and samples (50 µl) were added in duplicate into an appropriate ELISA plate. One hundred microliters of the horseradish peroxidase-conjugated antibody was added to each well. After incubation for 60 min at 37 °C, the plate was washed four times with 350 µl of wash solution and incubated with 50 µl of 3, 3′, 5, 5′-Tetramethylbenzidine for 15 min at 37 °C in the dark. After adding 50 µl of stop solution into each well, the OD values were measured in an ELISA reader at 450 nm (Thermo Scientific Multiskan, US). We average the duplicate readings for each standard and sample to subtract the average OD of the blank. A standard curve was generated by plotting the mean OD values of each standard on the vertical axis and the corresponding concentration on the horizontal axis. The concentration level of the MECOM in each sample was interpolated from the standard curve. Statistical analysis was performed using an unpaired t-test for comparison between two groups (GraphPad Prism 8, San Diego, California, US). Differences were considered significant when P ≤ 0.05.
Macrophage killing assay
Peripheral blood was drawn from healthy Holstein cows into heparinized Vacutainer tubes (Becton, Dickinson and Company, Sparks, MD), diluted 1:2 in Hanks balanced salt solution (HBSS), layered over 10 ml of Ficoll-Paque gradient (1.084 g/cm3) (GE HelthCare, Uppsala, Sweden) and centrifuged at 900×g for 30 min. The cell interphase was collected and centrifuged at 400×g for 10 min to remove platelets from peripheral blood mononuclear cells (PBMC). PBMC were resuspended in RPMI-1640 supplemented with 20 mM l-glutamine, 10% heat-inactivated bovine serum, 100 U/ml penicillin G, and 100 mg/ml streptomycin sulfate (Lonza, Spain) and seeded at a density of 1 × 106 PBMC/ml into 24-well tissue culture plates. After 2 h at 37 °C in a humidified 5% CO2 incubator, non-adherent cells were removed. Adherent cells were incubated for 7 days at 37 °C to allow differentiation to MDM before infection. Differentiated MDM were inoculated in triplicate with a single-cell suspension of MAP reference strain K10 at a multiplicity of infection (bacteria:cells) of 10:1. After 2 h, the supernatant was removed, and the cells were washed twice with HBSS to remove extracellular bacteria. Infected MDM were lysed at this time (2 h p.i.) or cultured at 37 °C for 7 days in fresh medium. At each time point, the supernatant was aspirated and infected MDM were lysed by vigorous pipetting with 0.5 ml of 0.1% Triton X-100 (Sigma-Aldrich) in sterile water for 10 min. Supplemented Mycobacteria Growth indicator tubes (MGIT) (Becton, Dickinson and Company, Sparks, MD, US) were inoculated with 0.1 ml of each initial bacterial suspension and with the 2 h and 7 d p. i. cell lysates. The tubes were incubated at 37 ± 2 °C for up to 41 days in a Bactec MGIT 960 instrument (Becton, Dickinson and Company). The earliest instrumental indication of positivity (i.e., time to detection [TTD]) for each tube was recorded. The predicted number of bacteria in each positive tube was calculated using previously generated mathematical equations that relate TTD (in days) to the estimated log CFUs35. The CFU ratios between 2 h and 7 days p.i. were calculated by dividing the estimated log CFUs at day 7 by that at 2 h p.i. MAP survival index was estimated as the square root CFU ratio × 100. Higher values of the MAP survival index reflect higher susceptibility to infection. A student’s t-test was performed for comparison of the survival indexes between two groups (GraphPad Prism 8, San Diego, CA, US). Differences were considered significant when P-values were ≤ 0.05.
Data availability
RNA-Seq raw data have been deposited in the NCBI Gene Expression Omnibus (GEO) database under the accession number GSE137395. SNPs datasets will not be made publically available as the data were generated from private-owned cattle, with a legal commitment to confidentiality. Researchers may request access to the data and consideration will be given to individuals following the conclusion of a confidentiality agreement.
Change history
25 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-00840-3
References
Ott, S. L., Wells, S. J. & Wagner, B. A. Herd-level economic losses associated with Johne’s disease on US dairy operations. Prev. Vet. Med. 40, 179–192. https://doi.org/10.1016/S0167-5877(99)00037-9 (1999).
Hasonova, L. & Pavlik, I. Economic impact of paratuberculosis in dairy cattle herds: A review. Vet. Med. 51, 193–211. https://doi.org/10.17221/5539-VETMED (2006).
Sweeney, R. W. Pathogenesis of paratuberculosis. Vet. Clin. N. Am. Food Anim. Pract. 27, 537–546. https://doi.org/10.1016/j.cvfa.2011.07.001 (2011).
Juste, R. A. et al. Association between Mycobacterium avium subsp. paratuberculosis DNA in blood and cellular and humoral immune response in inflammatory bowel disease patients and controls. Int. J. Infect. Dis. 13, 247–254. https://doi.org/10.1016/j.ijid.2008.06.034 (2009).
Jeyanathan, M. et al. Visualization of Mycobacterium avium in Crohn’s tissue by oil-immersion microscopy. Microbes Infect. 9, 1567–1573. https://doi.org/10.1016/j.micinf.2007.09.001 (2007).
Pierce, E. S. Could Mycobacterium avium subspecies paratuberculosis cause Crohn’s disease, ulcerative colitis and colorectal cancer?. Infect. Agent Cancer 13, 1–6. https://doi.org/10.1186/s13027-017-0172-3 (2018).
Groenendaal, H. et al. A simulation of Johne’s disease control. Prev. Vet. Med. 54, 225–245. https://doi.org/10.1016/S0167-5877(02)00027-2 (2002).
Garry, F. Control of paratuberculosis in dairy herds. Vet. Clin. N. Am. Food Anim. Pract. 27, 599–607. https://doi.org/10.1016/j.cvfa.2011.07.006 (2011).
Alonso-Hearn, M. et al. Immunization of adult dairy cattle with a new heat-killed vaccine is associated with longer productive life prior to cows being sent to slaughter with suspected paratuberculosis. J. Dairy Sci. 95, 618–629. https://doi.org/10.3168/jds.2009-2860 (2012).
Purdie, A. C., Plain, K. M., Begg, D. J., de Silva, K. & Whittington, R. J. Candidate gene and genome-wide association studies of Mycobacterium avium subsp paratuberculosis infection in cattle and sheep: A review. Comp. Immunol. Microbiol. Infect. Dis. 34, 197–208. https://doi.org/10.1016/j.cimid.2010.12.003 (2011).
Ruiz-Larrañaga, O. et al. Genetic association between bovine NOD2 polymorphisms and infection by Mycobacterium avium subsp. paratuberculosis in Holstein-Friesian cattle. Anim. Genet. 41, 652–655. https://doi.org/10.1111/j.1365-2052.2010.02055.x (2010).
Ruiz-Larrañaga, O. et al. SP110 as a novel susceptibility gene for Mycobacterium avium subspecies paratuberculosis infection in cattle. J. Dairy Sci. 93, 5950–5958. https://doi.org/10.3168/jds.2010-3340 (2010).
Ruiz-Larrañaga, O. et al. Identification of single nucleotide polymorphisms in the bovine solute carrier family 11 member 1 (SLC11A1) gene and their association with infection by Mycobacterium avium subspecies paratuberculosis. J. Dairy Sci. 93, 1713–1721. https://doi.org/10.3168/jds.2009-2438 (2010).
Ruiz-Larrañaga, O. et al. Genetic variation of toll-like receptor genes and infection by Mycobacterium avium ssp. paratuberculosis in Holstein-Friesian cattle. J. Dairy Sci. 94, 3635–3641. https://doi.org/10.3168/jds.2010-3788 (2011).
Ruiz-Larrañaga, O. et al. Single-nucleotide polymorphisms in the bovine CD209 candidate gene for susceptibility to infection by Mycobacterium avium subsp. paratuberculosis. Anim. Genet. 43, 646–647. https://doi.org/10.1111/j.1365-2052.2011.02316.x (2012).
Kiser, J. N., Neupane, M., White, S. N. & Neibergs, H. L. Identification of genes associated with susceptibility to Mycobacterium avium ssp. paratuberculosis (MAP) tissue infection in Holstein cattle using gene set enrichment analysis–SNP. Mamm. Genome 29, 539–549. https://doi.org/10.1007/s00335-017-9725-4 (2018).
Hempel, R. J., Bannantine, J. P. & Stabel, J. R. Transcriptional profiling of ileocecal valve of Holstein dairy cows infected with Mycobacterium avium subsp. paratuberculosis. PLoS ONE 11, 1–19. https://doi.org/10.1371/journal.pone.0153932 (2016).
Alonso-Hearn, M. et al. RNA-Seq analysis of ileocecal valve and peripheral blood from Holstein cattle infected with Mycobacterium avium subsp. paratuberculosis revealed dysregulation of the CXCL8/IL8 signaling pathway. Sci. Rep. 9, 1–17. https://doi.org/10.1038/s41598-019-51328-0 (2019).
Ariel, O. et al. Transcriptome Profiling of bovine macrophages infected by Mycobacterium avium spp. paratuberculosis depicts foam cell and innate immune tolerance phenotypes. Front. Immunol. 10, 1–27. https://doi.org/10.3389/fimmu.2019.02874 (2020).
Jansen, R. C. & Nap, J. P. Genetical genomics: The added value from segregation. Trends Genet. 17, 388–391. https://doi.org/10.1016/S0168-9525(01)02310-1 (2001).
Fairfax, B. P. & Knight, J. C. Genetics of gene expression in immunity to infection. Curr. Opin. Immunol. 30, 63–71. https://doi.org/10.1016/j.coi.2014.07.001 (2014).
Cloney, R. Complex traits: Integrating gene variation and expression to understand complex traits. Nat. Rev. Genet. 17, 194. https://doi.org/10.1038/nrg.2016.18 (2016).
Wittkopp, P. J. Genomic sources of regulatory variation in cis and in trans. Cell. Mol. Life Sci. 62, 1779–1783. https://doi.org/10.1007/s00018-005-5064-9 (2005).
Westra, H. J. & Franke, L. From genome to function by studying eQTLs. Biochim. Biophys. Acta Mol. Basis Dis. 1896–1902, 2014. https://doi.org/10.1016/j.bbadis.2014.04.024 (1842).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. https://doi.org/10.1093/bioinformatics/btr026 (2011).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. https://doi.org/10.1093/bioinformatics/btp120 (2009).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt214 (2013).
Canive, M. et al. Correlations between single nucleotide polymorphisms in bovine CD209, SLC11A1, SP110 and TLR2 genes and estimated breeding values for several traits in Spanish Holstein cattle. Heliyon 6, e04254. https://doi.org/10.1016/j.heliyon.2020.e04254 (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Shabalin, A. A. Matrix eQTL: Ultra-fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358. https://doi.org/10.1093/bioinformatics/bts163 (2012).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Vázquez, P. et al. Genetic association analysis of paratuberculosis forms in Holstein-Friesian cattle. Vet. Med. Int. https://doi.org/10.1155/2014/321327 (2014).
Wigginton, J. E., Cutler, D. J. & Abecasis, G. R. A note on exact tests of Hardy-Weinberg equilibrium. Am. J. Hum. Genet. 76, 887–893. https://doi.org/10.1086/429864 (2005).
González, J. R. et al. SNPassoc: An R package to perform whole genome association studies. Bioinformatics 23, 644–645. https://doi.org/10.1093/bioinformatics/btm02 (2007).
Abendaño, N. et al. Quantification of Mycobacterium avium subsp. paratuberculosis strains representing distinct genotypes and isolated from domestic and wildlife animal species by use of an automatic liquid culture system. J. Clin. Microbiol. 50, 2609–2617. https://doi.org/10.1128/JCM.00441-12 (2012).
Gao, Y. et al. Genome-wide association study of Mycobacterium avium subspecies paratuberculosis infection in Chinese Holstein. BMC Genomics 19, 1–10. https://doi.org/10.1186/s12864-018-5385-3 (2018).
De La Fuente, J., Gortázar, C. & Juste, R. Complement component 3: A new paradigm in tuberculosis vaccine. Expert Rev. Vaccines 15, 275–277. https://doi.org/10.1586/14760584.2016.1125294 (2016).
Curran, J. E. et al. Genetic variation in selenoprotein S influences inflammatory response. Nat. Genet. 37, 1234–1241. https://doi.org/10.1038/ng1655 (2005).
Rogler, G. Resolution of inflammation in inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 2, 521–530. https://doi.org/10.1016/S2468-1253(17)30031-6 (2017).
Marks, D. J. B. et al. Defective acute inflammation in Crohn’s disease: A clinical investigation. Lancet 367, 668–678. https://doi.org/10.1016/S0140-6736(06)68265-2 (2006).
Johansen, M. D., de Silva, K., Plain, K. M., Whittington, R. J. & Purdie, A. C. Mycobacterium avium subspecies paratuberculosis is able to manipulate host lipid metabolism and accumulate cholesterol within macrophages. Microb. Pathog. 130, 44–53. https://doi.org/10.1016/j.micpath.2019.02.031 (2019).
Li, K. et al. Transcriptome reveals 1400-fold upregulation of APOA4-APOC3 and 1100-Fold downregulation of GIF in the patients with polycythemia-induced gastric injury. PLoS ONE https://doi.org/10.1371/journal.pone.0140534 (2005).
Lin, Q. Decreased expression of the APOA1–APOC3–APOA4 gene cluster is associated with risk of Alzheimer’s. Drug Des. Dev. Therapy 9, 5421–5431. https://doi.org/10.2147/DDDT.S89279 (2015).
Wu, Y. et al. Interactions of environmental factors and APOA1-APOC3-APOA4-APOA5 gene cluster gene polymorphisms with metabolic syndrome. PLoS ONE 11, 1–17. https://doi.org/10.1371/journal.pone.0147946 (2016).
Naito, T. et al. Clinical and genetic risk factors for decreased bone mineral density in Japanese patients with inflammatory bowel disease. J. Gastroenterol. Hepatol. 33, 1873–1881. https://doi.org/10.1111/jgh.14149 (2018).
Fehringer, G. et al. Cross-cancer genome-wide analysis of lung, ovary, breast, prostate, and colorectal cancer reveals novel pleiotropic associations. Cancer Res. 76, 5103–5114. https://doi.org/10.1158/0008-5472.CAN-15-2980 (2016).
Glass, C. et al. Global identification of EVI1 target genes in acute myeloid leukemia. PLoS ONE https://doi.org/10.1371/journal.pone.0067134483 (2013).
Yoshimi, A. & Kurokawa, M. Evi1 forms a bridge between the epigenetic machinery and signaling pathways. Oncotarget 2 (7), 575–586. https://doi.org/10.18632/oncotarget.304 (2011).
Tanaka, T. et al. Evi-1 raises AP-1 activity and stimulates c-fos promoter transactivation with dependence on the second zinc finger domain. J. Biol. Chem. 269, 24020–24026. https://doi.org/10.1038/382816a0 (1994).
Kiu, H. & Nicholson, S. E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 30, 88–106. https://doi.org/10.3109/08977194.2012.660936 (2012).
Hood, D. et al. A new model for non-typeable Haemophilus influenzae middle ear infection in the Junbo mutant mouse. DMM Dis. Model. Mech. 9, 69–79. https://doi.org/10.1242/dmm.021659 (2016).
Xu, X. et al. EVI1 acts as an inducible negative-feedback regulator of NF-κB by inhibiting p65 acetylation. J. Immunol. 188, 6371–6380. https://doi.org/10.4049/jimmunol.1103527 (2012).
Liu, T., Zhang, L., Joo, D. & Sun, S. C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. https://doi.org/10.1038/sigtrans.2017.23 (2017).
Xu, C., Hu, D. M. & Zhu, Q. EEF1A2 promotes cell migration, invasion and metastasis in pancreatic cancer by upregulating MMP-9 expression through Akt activation. Clin. Exp. Metast. 30, 933–944. https://doi.org/10.1007/s10585-013-9593-6 (2013).
Sun, Y. et al. Up-regulation of eEF1A2 promotes proliferation and inhibits apoptosis in prostate cancer. Biochem. Biophys. Res. Commun. 450, 1–6. https://doi.org/10.1016/j.bbrc.2014.05.045 (2014).
Li, Z. et al. Eef1a2 promotes cell growth, inhibits apoptosis and activates JAK/STAT and AKT signaling in mouse plasmacytomas. PLoS ONE. https://doi.org/10.1371/journal.pone.0010755 (2010).
Amiri, A. et al. eEF1A2 activates Akt and stimulates Akt-dependent actin remodeling, invasion and migration. Oncogene 26, 3027–3040. https://doi.org/10.1038/sj.onc.1210101 (2007).
Lee, M. H. & Surh, Y. J. eEF1A2 as a putative oncogene. Ann. N. Y. Acad. Sci. 1171, 87–93. https://doi.org/10.1111/j.1749-6632.2009.04909.x (2009).
Abbas, K. A. & Herbein, G. The eEF1A proteins: At the crossroads of oncogenesis, apoptosis, and viral infections. Front. Oncol. 5, 1–10. https://doi.org/10.3389/fonc.2015.00075 (2015).
Nilsen, T. W. The spliceosome: The most complex macromolecular machine in the cell? BioEssays 25, 1147–1149. https://doi.org/10.1002/bies.10394 (2003).
Wahl, M. C., Will, C. L. & Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 136, 701–718. https://doi.org/10.1016/j.cell.2009.02.009 (2009).
Kaida, D. et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468, 664–668. https://doi.org/10.1038/nature09479 (2010).
Faustino, N. A. & Cooper, T. A. Pre-mRNA splicing and human disease. Genes Dev. 17, 419–437. https://doi.org/10.1101/gad.1048803 (2003).
Cheng, Z., Shang, Y., Gao, S. & Zhang, T. Overexpression of U1 snRNA induces decrease of U1 spliceosome function associated with Alzheimer’s disease. J. Neurogenet. 31, 337–343. https://doi.org/10.1080/01677063.2017.1395425 (2017).
Cheng, Z., Du, Z., Zhai, B., Yang, Z. & Zhang, T. U1 small nuclear RNA overexpression implicates autophagic-lysosomal system associated with AD. Neurosci. Res. 136, 48–55. https://doi.org/10.1016/j.neures.2018.01.006 (2018).
Kalam, H., Fontana, M. F. & Kumar, D. Alternate splicing of transcripts shape macrophage response to Mycobacterium tuberculosis infection. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1006236 (2017).
Kalam, H., Singh, K., Chauhan, K., Fontana, M. F. & Kumar, D. Alternate splicing of transcripts upon Mycobacterium tuberculosis infection impacts the expression of functional protein domains. IUBMB Life 70, 845–854. https://doi.org/10.1002/iub.1887 (2018).
Liang, G. et al. Altered microRNA expression and pre-mRNA splicing events reveal new mechanisms associated with early stage Mycobacterium avium subspecies paratuberculosis infection. Sci. Rep. 6, 1–12. https://doi.org/10.1038/srep24964 (2016).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. https://doi.org/10.1038/nprot.2012.016 (2012).
Moradi, F., Hajihosseini, M., Khodayari-Moez, E. & Dinu, I. An application of matrix eQTL to billions hypothesis testing to identify expression quantitative trait loci in genome wide association studies of inflammatory bowel disease. J. Cancer Prev. Curr. Res. 11, 41–46. https://doi.org/10.15406/jcpcr.2020.11.00424 (2020).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x (1995).
Ashburner, M. et al. The gene ontology consortium. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556.Gene (2000).
Thomas, P. D. et al. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141. https://doi.org/10.1101/gr.772403 (2003).
Acknowledgements
Financial support for this study was provided by a grant from the Spanish Ministry of Science, Innovation, and Universities (MICINN, https://sede.micinn.gob.es/, project code: RTI2018-094192-R-C21) and by European Regional Development Funds (FEDER) to MAH. This study was co-funded by a grant from the Plan of Science, Technology, and Innovation of the Principality of Asturias, Regional funds PCTI 2018–2020 (www.ficyt.es/pcti/), project code: IDI2018-000237. MC and CBV have been awarded fellowships from the National Institute for Agricultural Research (INIA) program. Special thanks are due to the dairy producers and veterinarians for their collaboration. We are also grateful to Medelin Ocejo from NEIKER for assistance with data analysis. We are grateful to Kyle Hearn for the careful editing of the manuscript.
Author information
Authors and Affiliations
Contributions
M.C. generated and analyzed the datasets and wrote the manuscript. P.V., R.C., C.B.V., J.G., R.A.J. and M.A.H. participated in sample collection and phenotypic characterization of the animals. N.F. and J.M.B. supervised the analysis and contributed to the interpretation of the results. J.L.L. performed differential expression and functional analysis. R.A.J. helped with the statistical analysis of the data. M.A.H. conceived and coordinated the project, supervised the data analysis, and wrote the manuscript. All authors revised and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article omitted an affiliation for Maria Canive. The correct affiliations are ‘Department of Animal Health, NEIKER- Basque Research and Technology Alliance (BRTA), Derio, Bizkaia, Spain’ and ‘Doctoral Program in Immunology, Microbiology and Parasitology, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), Leioa, Bizkaia, Spain.’
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Canive, M., Fernandez-Jimenez, N., Casais, R. et al. Identification of loci associated with susceptibility to bovine paratuberculosis and with the dysregulation of the MECOM, eEF1A2, and U1 spliceosomal RNA expression. Sci Rep 11, 313 (2021). https://doi.org/10.1038/s41598-020-79619-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-79619-x
This article is cited by
-
Robust identification of regulatory variants (eQTLs) using a differential expression framework developed for RNA-sequencing
Journal of Animal Science and Biotechnology (2023)
-
Summary-data based Mendelian randomization identifies gene expression regulatory polymorphisms associated with bovine paratuberculosis by modulation of the nuclear factor Kappa β (NF-κß)-mediated inflammatory response
BMC Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
