
Abstract
Current treatment options against hepatitis B and D virus (HBV/HDV) infections have only limited curative effects. Identification of Na+/taurocholate co-transporting polypeptide (NTCP) as the high-affinity hepatic receptor for both viruses in 2012 enables target-based development of HBV/HDV cell-entry inhibitors. Many studies already identified appropriate NTCP inhibitors. However, most of them interfere with NTCP’s physiological function as a hepatic bile acid transporter. To overcome this drawback, the present study aimed to find compounds that specifically block HBV/HDV binding to NTCP without affecting its transporter function. A novel assay was conceptualized to screen for both in parallel; virus binding to NTCP (measured via binding of a preS1-derived peptide of the large HBV/HDV envelope protein) and bile acid transport via NTCP. Hits were subsequently validated by in vitro HDV infection studies using NTCP-HepG2 cells. Derivatives of the birch-derived pentacyclic lupane-type triterpenoid betulin revealed clear NTCP inhibitory potency and selectivity for the virus receptor function of NTCP. Best performing compounds in both aspects were 2, 6, 19, and 25. In conclusion, betulin derivatives show clear structure–activity relationships for potent and selective inhibition of the HBV/HDV virus receptor function of NTCP without tackling its physiological bile acid transport function and therefore are promising drug candidates.
Similar content being viewed by others

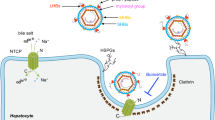
Introduction
Infections with the Hepatitis B (HBV) and D (HDV) viruses are the main cause of hepatocellular carcinoma (HCC) and liver cirrhosis as consequences of chronic hepatitis. Although vaccination is possible, more than 250 million people worldwide suffer from chronic hepatitis based on HBV/HDV infections, and 887,000 deaths can be traced back to this every year1. The genome of the enveloped DNA virus HBV consists of 3.2 kb which encode for three envelope proteins, referred to as small (SHBs), middle (MHBs), and large (LHBs)2. The 2–48 N-terminal amino acids of the myristoylated preS1 domain (myr-preS12-48 lipopeptide) of the LHBs are essential for virus binding to hepatocytes3,4. As HDV is a satellite virus of HBV and bears identical envelope proteins, it also shares this myr-preS12-48 lipopeptide5. About 5% of all chronic HBV carriers are additionally infected with HDV1. This co-infection causes more rapid disease progression, increased mortality rates, and increased incidence of HCC and liver cirrhosis6. To date, there are only few treatment options for HBV and HDV infections available. Chronic hepatitis can usually be controlled by antiviral therapy with nucleoside reverse transcriptase inhibitors and interferon. This is, however, rarely curative. Furthermore, interferon therapy is highly prone to adverse drug reactions and nucleoside reverse transcriptase inhibitors have to be given life-long7,8.
A promising novel drug target to block HBV/HDV virus entry into hepatocytes is represented by the Na+/taurocholate co-transporting polypeptide NTCP (gene symbol SLC10A1), which has been identified as the bona fide hepatic receptor for HBV/HDV9,10. It is generally accepted that HBV/HDV entry into hepatocytes essentially involves attachment of the virus particles to heparan sulfate proteoglycans11, followed by high-affinity binding of the myr-preS1 domain of the large envelope protein to certain domains of NTCP that finally triggers subsequent steps such as endocytosis of the virus-receptor complex12.
It is known for long that an artificial myr-preS12-48 lipopeptide itself can block in vitro HBV/HDV infection in a dose-dependent manner with inhibitory constants (IC50) in the low nanomolar range4. Based on this, a synthetic analogue of the myr-preS12-48 lipopeptide, called bulevirtide (generic name, formerly known as Myrcludex B), was developed as a therapeutic agent against HBV and HDV infections. Several clinical studies have been successfully completed and very recently, bulevirtide was approved as a new drug to treat hepatitis D in Europe and Russia and will be marketed under the brand name Hepcludex13. Furthermore, it was demonstrated that viral DNA (HBV) and RNA (HDV) levels can be reduced significantly in chronically infected patients with a combination of bulevirtide and pegylated interferon alpha (PEG-IFNα)14. These data clearly indicate that NTCP is an appropriate drug target to control hepatic HBV/HDV levels. In addition, an HBV/HDV entry inhibitor addressing NTCP might be beneficial to prevent new infections of hepatocytes during chronic infection with HBV/HDV12.
Physiologically, NTCP is expressed dominantly at the basolateral membrane of hepatocytes and acts as a sodium-dependent uptake carrier of bile salts to enable their enterohepatic recirculation15,16,17. Thus, NTCP has a dual function as bile salt transporter and virus receptor. It is already known that both functions interfere with each other. The myr-preS12-48 lipopeptide can block the bile salt transport in primary human hepatocytes and NTCP-overexpressing HepG2 hepatoma cells in a concentration-dependent manner. Vice versa, bile salts can prevent myr-preS12-48 lipopeptide binding to NTCP as well as in vitro HBV infection10,18. Based on this, it was expected that in clinical trials using bulevirtide as a therapeutic agent, the total plasma bile salt levels would increase significantly. In a current study on healthy volunteers who received 10 mg bulevirtide, which is above the therapeutic dose, total plasma bile salt levels indeed increased 19.2-fold, and single bile salts even higher (e.g. 124-fold in the case of taurocholic acid, further referred to as TC)19. This data can be interpreted as dysregulation of the bile acid homeostasis under bulevirtide treatment.
On the basis of the therapeutically used effects of bulevirtide, a promising second strategy for the development of HBV/HDV entry inhibitors targeting NTCP would be to (I) investigate small molecules with oral bioavailability which (II) block virus binding to NTCP, but (III) without tackling its physiologically important bile salt transporter activity at relevant therapeutic drug concentrations. Currently, several studies identified novel chemical entry inhibitors for HBV and HDV by screening for bile salt transport inhibitors20 or by screening for inhibitors of myr-preS12-48 lipopeptide attachment and/or in vitro HBV/HDV infection21 in appropriate cell culture models overexpressing NTCP. However, none of these studies addressed and fulfilled all of the criteria outlined above. In contrast, the present study aimed to find small molecular HBV/HDV entry inhibitors that specifically block myr-preS12-48 lipopeptide binding to NTCP without affecting its bile salt transport function. We analyzed a set of derivatives of the pentacyclic lupane-type triterpenoid betulin, and indeed found individual compounds that were quite potent and selective for myr-preS12-48 lipopeptide binding inhibition and significantly blocked in vitro HDV infection of NTCP-expressing HepG2 cells. These compounds now can be further optimized for increased potency and then can be forwarded for in vivo infection experiments.
Results
Screening assay for bile acid transport and myr-preS12-48 lipopeptide binding
The major goal of the present study was to analyze in parallel the effect of inhibitors on both, bile acid transport via NTCP and myr-preS12-48 lipopeptide binding to NTCP in a cell culture system. This was achieved by plating NTCP-HEK293 cells onto 96-well plates and by using tritium-labelled analytes, being [3H]taurocholic acid ([3H]TC) and [3H]myr-preS12-48 lipopeptide ([3H]preS1), which were analyzed in a microplate scintillation counter. For validation of this assay, transport of 1 µM [3H]TC first was inhibited by increasing concentrations of TC (concentrations ranging from 0.1 to 1,000 µM) itself and by myr-preS12-48 lipopeptide (concentrations ranging from 0.001 to 10 µM). In addition, binding of 5 nM [3H]preS1 was inhibited by myr-preS12-48 lipopeptide itself (concentrations ranging from 0.001 to 10 µM) and by TC (concentrations ranging from 0.1 to 1,000 µM). As shown in Fig. 1a, TC showed nearly equipotent inhibition of the [3H]TC transport with IC50 of 30 µM and of the [3H]preS1 binding with IC50 of 42 µM. The same was true for myr-preS12-48 lipopeptide as the inhibitor that revealed IC50 of 0.35 µM for [3H]TC transport inhibition and IC50 of 0.55 µM for [3H]preS1 binding inhibition (Fig. 1b). These data clearly confirm the previously shown interference of the transport and receptor function of NTCP10,18 and demonstrate the functionality of this screening assay. Furthermore, these data indicate that the myr-preS12-48 lipopeptide has an about 100-fold higher inhibitory potency (IC50 = 0.35 µM / 0.55 µM) in both assays compared to the physiological transport substrate TC (IC50 = 30 µM / 42 µM). Four reference compounds were analyzed (concentrations ranging from 0.1 to 1,000 µM) which had shown inhibition of the transporter and receptor function of NTCP before, namely ciglitazone22, troglitazone22, cyclosporine A23,24,25, and rapamycin26. Interestingly, all compounds showed comparable inhibition of [3H]TC uptake and [3H]preS1 binding with IC50 values of 1 µM and 1.3 µM for ciglitazone (Fig. 1c), 1.9 µM and 2.5 µM for troglitazone (Fig. 1d), 14 µM and 26 µM for cyclosporine A (Fig. 1e), as well as 34 µM and 21 µM for rapamycin (Fig. 1f), respectively. These compounds therefore can be classified as inhibitors, non-selective for one of the two measured functions of NTCP. Among them, ciglitazone and troglitazone had a tenfold higher inhibitory potency compared to cyclosporine A and rapamycin, demonstrating that diverse compounds differ in their potency as NTCP inhibitors. Cyclosporine A, rapamycin and TC showed comparable inhibitory pattern and potency in both assays, as shown by similar IC50 values.

[3H]TC transport and [3H]preS1 binding screening assay. NTCP-HEK293 cells were seeded onto 96-well plates and were incubated with tetracycline in order to induce expression of the human NTCP protein. Cells without tetracycline treatment were used as 0% controls of [3H]TC uptake and [3H]preS1 binding, respectively. Cells were subjected to bile acid transport experiments with 1 µM [3H]TC and myr-preS12-48 lipopeptide binding experiments with 5 nM [3H]preS1, each over 10 min at 37 °C. Experiments were performed with solvent alone (set to 100%) and with increasing concentrations of the indicated inhibitors. The mean of the 0% control was subtracted to calculate net [3H]TC transport rates (shown in blue) as well as net [3H]preS1 binding rates (shown in red), which are depicted in the diagrams (expressed as % of control at the y-axis). Half maximal inhibitory concentrations (IC50) were calculated by nonlinear regression analysis using the equation log(inhibitor) versus response (GraphPad Prism). In order to validate the assay, (a) TC and (b) myr-preS12-48 lipopeptide were used as inhibitors. Furthermore, the already established NTCP inhibitors (c) ciglitazone, (d) troglitazone, (e) cyclosporine A, and (f) rapamycin were analyzed for [3H]TC transport inhibition and [3H]preS1 binding inhibition. Data represent means ± SD of quadruplicate determinations of representative experiments.
Screening of betulin derivatives for bile acid transport and [3H]preS1 binding inhibition
Next, 30 derivatives of betulin were tested in both assays and IC50 values were determined for [3H]TC transport inhibition and [3H]preS1 binding inhibition. Chemical structures of all compounds are depicted in Table 1. All derive from the betulin core structure by chemical modifications. These include methyl, succinyl, acetyl, bromoacetyl, dimethylglutaryl, caffeoyl, oxime, tetrahydropyranyl, cinnamoyl, nicotinoyl, L-aspartyl- or 1,2,4-triazoline-3,5-dione conjugations mostly at positions 3′ and/or 28′, as well as oxidations of the hydroxyl groups at positions 3′ and/or 28′. Compound 29 represents allobetulin and compound 30 3-oxoallobetulin. All IC50 values for [3H]TC transport and [3H]preS1 binding are listed in Table 2 with their 95% confidence intervals, and selected diagrams are shown in Fig. 2. In the case of [3H]TC uptake inhibition, the IC50 values ranged from 1 µM (4) up to values > 1,000 µM with very low, if any transport inhibition (3, 24, 25, 28 and 30). This clearly indicates structure–activity relationships over three orders of magnitude. Most of the betulin derivatives showed quite potent inhibition of [3H]preS1 binding with IC50 values ranging from 2–3 µM (4, 8, 21) up to 199 µM (24), also demonstrating clear structure–activity relationships over a broad inhibitor concentration range.

Inhibition of [3H]preS1 peptide binding and [3H]TC transport by betulin derivatives. NTCP-HEK293 cells were incubated with tetracycline in order to induce NTCP expression and were used for bile acid transport experiments with 1 µM [3H]TC and for myr-preS12-48 lipopeptide binding experiments with 5 nM [3H]preS1, each over 10 min at 37 °C. Cells without tetracycline treatment were used as 0% controls of [3H]TC uptake and [3H]preS1 binding, respectively. Experiments were performed with solvent alone (set to 100%) and with increasing concentrations of the indicated inhibitor. The mean of the 0% control was subtracted to calculate net [3H]TC transport rates (shown in blue) as well as net [3H]preS1 binding rates (shown in red), both expressed as % of control at the y-axis. Half maximal inhibitory concentrations (IC50) were calculated by nonlinear regression analysis using the equation log(inhibitor) vs. response (GraphPad Prism). The figure shows selected diagrams. Chemical structures of the compounds are depicted in Table 1, the respective IC50 values are listed in Table 2. Data represent means ± SD of quadruplicate determinations (n = 4).
In addition, ratios between the IC50 values for [3H]TC transport inhibition and the IC50 values for [3H]preS1 binding inhibition were calculated and are further referred to as selectivity index. The higher this index, the more selective the respective compound for myr-preS12-48 lipopeptide inhibition. Therefore, the data listed in Table 2 are not only helpful to identify potent myr-preS12-48 lipopeptide binding inhibitors but also to assess their selectivity. These values ranged from an index of 1, including more potent (e.g. 4) and less potent (e.g. 12) non-selective inhibitors, up to an index of > 60, including the highly selective and potent inhibitors 2 and 25. Figure 2 depicts selected diagrams which show the inhibitory curves for the [3H]TC transport inhibition (in blue) and [3H]preS1 binding inhibition (in red). This panel shows compounds with different characteristics. Within the group of potent [3H]preS1 binding inhibitors (IC50 < 10 µM), there were (I) non-selective inhibitors (4, 9, 10, and 20), (II) poorly selective inhibitors (e.g. 21), and highly selective inhibitors, being 2 and 25. Also, from the group of less potent [3H]preS1 binding inhibitors (IC50 = 11–50 µM), some were poorly selective (e.g. 18) and others were highly selective (e.g. 13, 17, 19 and 30). Taken together, these data clearly indicate structure–activity relationships of the betulin derivatives for both, bile acid transport inhibition and myr-preS12-48 lipopeptide binding inhibition.
Most relevant modifications for structure–activity relationships
Closer analysis of the structure–activity relationships of the betulin derivatives revealed several interesting patterns that can be used to further optimize the potency and selectivity index of these compounds in order to end up with an even more suitable HBV/HDV entry inhibitor from the class of betulin derivatives. Of note, betulin (1) itself showed relatively low inhibitory potency for bile acid transport inhibition (IC50 = 748 µM) and [3H]preS1 binding inhibition (IC50 = 109 µM) resulting in a selectivity index of 7. However, relatively moderate modifications at the betulin core structure revealed significantly improved potency and selectivity index of the individual compounds. Some of these modifications are depicted in more detail in Fig. 3a/3b and involve modifications at positions 3′, 28′, 29′, and 30′. While the diacetylated betulin derivative 3,28-di-O-acetylbetulin (24) itself has a relatively low potency for [3H]preS1 binding inhibition (IC50 = 199 µM) and poor selectivity (selectivity index: 5) (Fig. 3, shown in orange), additional C29 hydroxylation (25) improved the potency for [3H]preS1 binding inhibition to IC50 = 8 µM and increased the selectivity index to 125 (Fig. 3, shown in purple). 3-O-caffeoyl (27) conjugation of betulin (1) also reduced the IC50 value for [3H]preS1 binding inhibition to 6 µM and increased the selectivity index to 22 (Fig. 3, shown in light blue). Replacement of the 28-hydroxyl group of betulin by an oxime group (6) increased the inhibitory potency for [3H]preS1 binding by one order of magnitude (109 µM vs. 11 µM) and also improved the selectivity index (7 vs. 48) (Fig. 3, shown in red). The most successful modification, however, was the saturation of the double bond between C20 and C30, which increased the potency for [3H]preS1 binding inhibition to IC50 of 4 µM, while maintaining the low degree of interference with the bile acid transport with IC50 of 260 µM (selectivity index: 65) (Fig. 3, shown in green).

Structure–activity relationships of betulin derivatives for [3H]TC transport inhibition and [3H]preS1 binding inhibition. Chemical modifications at the lupane skeleton of betulin revealed clear structure–activity relationships for the potency (pot., IC50 of [3H]preS1 binding inhibition) and virus selectivity index (sel. ind., ratio IC50 [3H]TC transport inhibition / IC50 [3H]preS1 binding inhibition). (a) 3,28-di-O-acetylation (shown in orange), additional C29-hydroxylation (shown in purple), 3-O-modification with caffeoyl (shown in light-blue), 28-oxime generation (shown in red), and C30 saturation (shown in green) of the betulin core structure improve the potency and/or selectivity index of the respective compounds. The respective inhibition pattern are illustrated by bar graphs for [3H]TC uptake (shown in dark-blue) and [3H]preS1 peptide binding (shown in red), respectively, in the presence or absence of a fix concentration (100 µM) of the respective betulin derivative. Graphs show means ± SD of quadruplicate determinations. *Significantly different with p < 0.01 (two-way ANOVA with Sidak's multiple comparison test); ns, not significant. (b) The diagram shows all IC50 values for [3H]TC uptake inhibition and [3H]preS1 binding inhibition of the respective compounds. Effects (IC50 shifts) of the structural modifications are illustrated by arrows, starting from the core structure betulin (compound 1). Compounds can be divided into three groups regarding their potency to inhibit [3H]preS1 binding: potent inhibitors (IC50 ≤ 10 µM), less potent inhibitors (10 µM < IC50 < 100 µM) and poor inhibitors (IC50 ≥ 100 µM). Compounds with a selectivity index of > 10 are classified as selective inhibitors.
Acidic betulin derivatives are potent, but non-selective inhibitors of NTCP
All potent (IC50 preS1 binding < 10 µM) and non-selective (selectivity index < 10) NTCP inhibitors from the group of betulin derivatives bear at least one acidic group. This applies to compounds 4, 8—12, 20, and 21 (Tab. 1). Based on their pKa values, these compounds are predominantly deprotonated and negatively charged at pH 7.4, similar to bile acids. In order to investigate the role of the pH-dependent negative charge of these compounds, the acidic derivatives 4 (pKa = 4.8) and 8 (pKa = 4.1) were tested for [3H]TC uptake inhibition and [3H]preS1 binding inhibition at pH 7.4 and pH 5.5. Betulin (1) was included as a non-acidic compound for control. In the case of compound 4 a shift of the IC50 value for [3H]TC uptake inhibition from 0.8 to 103 µM was observed (Fig. 4, upper panel). Compound 8 also showed an IC50 shift from 8 to 200 µM by lowering the pH from 7.4 to 5.5. Interestingly, the IC50 values for [3H]preS1 binding inhibition did not change at lower pH (Fig. 4, lower panel). Apart from this effect on the inhibition pattern, acidification of the incubation medium also affected the absolute [3H]TC uptake and [3H]preS1 binding rates (Fig. 4, bar graphs). Both functions of NTCP were significantly lower at pH of 5.5, most likely due to pH sensitive amino acid residues at the sites of substrate/preS1 binding.

Influence of pH on the inhibition pattern of acidic betulin derivatives. NTCP-HEK293 cells were incubated with tetracycline in order to induce NTCP expression and were used for bile acid transport experiments with 1 µM [3H]TC and for myr-preS12-48 lipopeptide binding experiments with 5 nM [3H]preS1, each over 10 min at 37 °C. Cells without tetracycline treatment served as control (set to 0%). Experiments were performed with solvent alone (set to 100%) and with increasing concentrations of the indicated inhibitors in the range of 0.1 to 1,000 µM at pH 7.4 and pH 5.5, respectively. Diagrams show inhibition curves at pH 7.4 (solid lines) and pH 5.5 (dashed lines). The mean of the 0% control was subtracted to calculate net [3H]TC transport rates (shown in blue) as well as net [3H]preS1 binding rates (shown in red), both expressed as % of control at the y-axis. Bar graphs show the relative [3H]TC uptake rates (blue bars) and [3H]preS1 binding rates (red bars) at pH 7.4 (filled bars) and pH 5.5 (striped bars), respectively, expressed as pmol/10 min/well. Half maximal inhibitory concentrations (IC50) were calculated by nonlinear regression analysis using the equation log(inhibitor) vs. response (GraphPad Prism). Data represent means ± SD of quadruplicate determinations (n = 4). *Significantly different with p < 0.01 (unpaired t-test with Welch's correction).
Betulin derivatives inhibit HDV infection
Selected betulin derivatives were investigated for their inhibitory potency of in vitro HDV infection of NTCP-expressing HepG2 hepatoma cells. These experiments were of particular interest as in the [3H]preS1 assays, [3H]preS1 peptide binding is just used as a surrogate parameter for virus binding to NTCP. Of note, these infection experiments were performed in a different cell line as the screening assays and the betulin inhibitors had to be incubated much longer for the in vitro infection experiments (6 h) compared to the [3H]preS1 binding assay (15 min). While NTCP-HepG2 cells without inhibitor were used as control (set to 100% infection rate), co-incubation with 0.5 µM of the myr-preS12-48 lipopeptide completely abolished infection of the cells (set to 0% infection rate). As inhibitors for in vitro HDV infection, betulinic acid (4) was used as a potent but non-selective inhibitor, 20,29-dihydrobetulin (2) as a potent and highly selective inhibitor, as well as lupenone (17) and betulonoyl dimethyl-L-aspartate (19) as less potent but still selective inhibitors. All four compounds significantly blocked in vitro HDV infection of the NTCP-HepG2 cells in a concentration-dependent manner (Fig. 5), clearly indicating that betulin derivatives represent an interesting novel compound class of HBV/HDV entry inhibitors. Of note, none of the compounds induced toxicity in the cells even after long term incubation at high (300 µM/600 µM) inhibitor concentrations (Fig. 5h).

Selected betulin derivatives were used for HDV infection inhibition of NTCP-HepG2 cells. NTCP-HepG2 cells were preincubated for 5 min with the indicated concentrations of (a) betulinic acid (compound 4), (b) lupenone (compound 17), (c) betulonoyl dimethyl-L-aspartate (compound 19), (d) 20,29-dihydrobetulin (compound 2), and (g) the myr-preS12-48 peptide in DMEM at 37 °C. Then, cells were additionally inoculated with 700 genome equivalents/cell of HDV particles at 37 °C. After 6 h, cells were washed and further incubated with inhibitor- and virus-free medium, and medium was changed every 3–4 days. At day 9 post infection, cells were fixed and an immunostaining against the HDAg was performed, as a marker of HDV infection. The number of infected cells per well was determined by fluorescence microscopy. NTCP-HepG2 cells incubated without inhibitor were used as control (set to 100% infection rate). Infection experiments in the presence of 0.5 µM myr-preS12-48 peptide served as control for the 0% infection rate. Scale bars: 100 µm. Data represent means ± SEM of three independent experiments each with triplicate determinations (n = 9). (e, f) Cells were preincubated with 600 µM of compound 4 and compound 17, respectively, for intervals of 15 min and 6 h at 37 °C. Cells treated with solvent (DMEM) alone served as control (set to 100%) and data from cells without induction of NTCP expression were set to 0%. Subsequently, cells were washed three times with tempered PBS and [3H]TC (1 µM) uptake as well as [3H]preS1 (5 nM) binding were analyzed, respectively. Means of the 0% controls were subtracted and net [3H]TC transport rates and net [3H]preS1 binding rates are expressed as % of control at the y-axis. Data were combined from two independent experiments, each with quadruplicate determinations (n = 8). (h) Cell viability was determined with an LDH cytotoxicity assay. HepG2 cells treated with HGM w/o test compound were used as positive control and lysed cells as negative control. Data show means ± SD of three independent experiments each with triplicate determinations (n = 9). *Significantly different from control with p < 0.01 (two-way ANOVA with Dunnett's multiple comparison test).
Inhibition of HDV infection cannot be attributed to drug-induced internalization of NTCP
Compounds 4 and 17 underwent further investigations to clarify, whether their inhibitory effects on in vitro HDV infection may have been attributed to drug-induced internalization of NTCP. Therefore, NTCP-expressing HepG2 hepatoma cells were pretreated with 600 µM of the respective compound for 15 min or 6 h, respectively (Fig. 5e,f). Subsequently, cells were washed three times with tempered PBS, before [3H]TC (1 µM) uptake and [3H]preS1 (5 nM) binding were analyzed for 10 min at 37 °C. Cells pretreated with solvent (DMEM) alone served as 100% controls. Data from HepG2 cells without NTCP expression were set to 0%. Even after three intense washing steps, [3H]TC uptake was significantly reduced after short (15 min) and long (6 h) term preincubation with compound 4. In contrast, [3H]preS1 binding did not change, clearly confirming plasma membrane localization of NTCP even after long term preincubation with high inhibitor concentrations of 600 µM. In the case of compound 17, no differences were observed after short term (15 min) preincubation, while [3H]TC uptake and [3H]preS1 binding slightly changed in opposite directions after long term (6 h) preincubation. These data confirm that also after preincubation with compound 17, NTCP remained at the plasma membrane.
Discussion
The identification of the hepatic bile acid transporter NTCP as the high-affinity receptor for HBV and HDV was the basis for the development of a novel class of anti-HBV/anti-HDV drugs, being cell-entry inhibitors targeting NTCP9. Since then, many studies have identified compounds that are capable to inhibit the virus receptor function of NTCP: Hepcludex13, cyclosporine A and other cyclosporine derivatives23,24,25, ezetimibe10, irbesartan27, ritonavir28, (-)-epigallocatechin-3-gallate29, vanitaracin A30, Ro41-525331, proanthocyanidin32, zafirlukast33, sulfasalazine33, and Chicago Sky Blue 6B (an azo dye)33. Among them, Hepcludex is a peptide-based drug and cannot be administered orally13. Furthermore, it has correspondingly complex manufacturing, storage, and distribution requirements. Therefore, it is still desirable to develop a small molecular drug with HBV/HDV cell-entry inhibitor activity that can be orally applied and would be easier to handle.
Many of the other mentioned compounds, e.g. cyclosporine A, were previously known to inhibit the bile acid transport function of NTCP17. Others were identified as novel HBV/HDV inhibitors and later revealed their interference with the physiological bile acid transport function of NTCP. Overall, this interference indicates that NTCP domains relevant for substrate binding and transport overlap with the domains mediating myr-preS12-48 lipopeptide / virus binding to NTCP. However, there are also some hints that both functions can be separated. In a previous study, we identified amino acid G158 of the human NTCP as absolutely essential for myr-preS12-48 lipopeptide binding and in vitro HBV and HDV infection. This amino acid is located at a domain (amino acids 157–165) that previously was shown to be involved in myr-preS12-48 binding to human NTCP9,18. Accordingly, G158R NTCP mutants were completely insusceptible for in vitro HBV/HDV infection, but still supported transport of bile acids. It was discussed that the larger amino acid side chain of arginine compared to glycine might sterically preclude myr-preS1 peptide binding, while bile acids can still bind to their respective binding pocket34. Furthermore, Shimura et al.25 showed for the cyclosporine derivative SCY995 higher potency against in vitro HBV infection (IC50 < 8 µM) than for bile acid transport inhibition (IC50 > 25 µM).
Based on this, the present study was designed to enable the identification of a selective HBV/HDV cell-entry inhibitor from the group of small molecules. Therefore, all test compounds were a priori tested for both, inhibition of [3H]preS1 binding to NTCP as a surrogate parameter for HBV/HDV infection and inhibition of [3H]TC transport via NTCP. In the present study, a novel chemical class of promising HBV/HDV cell-entry inhibitors was identified, represented by derivatives of the natural pentacyclic lupane-type triterpenoid betulin. Betulin can be isolated from various plant species, belonging to a variety of families such as Betulaceae, Platanaceae, Dilleniaceae, Rhamnaceae, Rosaceae, and Fagaceae35,36. Characteristics of the lupane skeleton of betulin are the five-membered E ring and the isopropylidene group37. Interestingly, nearly all betulin derivatives tested in the present study significantly inhibited [3H]TC transport and [3H]preS1 binding. However, the ratios between the IC50 values for transport inhibition and for [3H]preS1 binding inhibition were quite different and the resulting selectivity indices ranged from 1 up to 65. The latter was the case for compound 2 with an IC50 of 4 µM for [3H]preS1 binding inhibition and an IC50 of 260 µM for bile acid transport inhibition. Some other compounds showed no transport inhibition at all, being 24, 25, 28, and 30, but at least in the case of 25, potent inhibition of [3H]preS1 peptide binding with IC50 of 8 µM. This data clearly indicate structure–activity relationships for both NTCP functions. Of note, only small changes in the molecular structure of the betulin derivatives significantly changed their potency and selectivity index. Moreover, hydroxylation of compound 24 to compound 25, as an example, improved both factors in parallel: potency for [3H]preS1 binding inhibition and the selectivity index. Also other modifications of the betulin molecule (1) such as 3-O-caffeoly conjugation (compound 27) or C20-C30 saturation (compound 2) significantly improved both factors simultaneously.
Inhibition studies at pH 5.5 revealed additional structure–activity relationships regarding the charge of the tested compounds. All potent but non-selective inhibitors (Fig. 3b) bear at least one acidic group at R1 and/or R2 (see Tab. 1) and show pKa values between 4–5. Therefore, compounds from this group are predominantly deprotonated and negatively charged at physiological pH of 7.4, representing the standard condition in the inhibition assays. For comparison, also conjugated bile acids (pKa = 1–4) are predominantly deprotonated and negatively charged at physiological pH38,39,40. Based on this it can be speculated that acidic betulin derivatives may not only be inhibitors but also transporter substrates of NTCP. This would explain, why these compounds (representatively shown for compounds 4 and 8) largely lost their [3H]TC uptake inhibitory potency when the pH was reduced to 5.5 and, thereby reducing the relative charge of the molecules. In contrast, the [3H]preS1 binding inhibitory potency was insensitive to this pH shift and only showed little differences between pH 7.4 and pH 5.5 conditions. This again indicates that the transporter and the receptor function of NTCP can be separated under certain experimental conditions and that blocking of the preS1 peptide binding site of NTCP does not require negative charge of the inhibitor. In contrast to compounds 4 and 8, betulin itself (compound 1) does not bear any acidic group. Consequently, betulin could be less active at the bile acid binding site, what would explain the relatively low potency for TC uptake inhibition (IC50 = 748 µM) at neutral and acidic pH values. However, no uniform legality could be established for the other chemical modifications of betulin, which would precisely predict the potency and selectivity pattern of the respective betulin derivative. Nevertheless, based on the structure–activity relationships described in the present study it is most likely that even more potent betulin derivatives can be identified, while sparing the cross-reactivity on the bile acid transport function of NTCP.
Among the betulin derivatives, betulinic acid (4) particularly underwent intensive toxicological assessment35,41,42,43. In vitro and in vivo model systems consistently categorized betulinic acid (4) as a safe compound with low toxicity. More precisely, no toxic effects were observed in rats that obtained single i.p. application of 400 mg/kg or in mice receiving 100 mg/kg i.p. every 3–4 days for a total of six treatments, or 500 mg/kg i.p. once35,42,43. Unfortunately, betulinic acid (4), although potent in [3H]preS1 binding inhibition (IC50 = 2 µM), is not selective and showed equipotent inhibition of the bile acid transport function of NTCP at least at physiological pH. Therefore, betulinic acid (4) is not regarded as the optimal HBV/HDV entry inhibitor candidate from the betulin class of compounds. Apart from betulinic acid (4), betulin (1) also has a good safety profile. Betulin (1) is approved for medical use in humans and is currently available as a betulin containing gel (Oleogel-S10, Episalvan). It was authorized in 2016 by the European Medicines Agency (EMA) for treatment of partial thickness wounds in adults. This gel contains an extract from birch bark of Betula pendula Roth, Betula pubescens Ehrh., and hybrids of both species, obtained with n-heptane as the extraction solvent. The resulting betulin (1) concentrations are in the range of 72–88 mg betulin (1) per 100 mg extract. The gel itself is composed of 10% birch bark extract in 90% sunflower oil. Moreover, Episalvan is currently undergoing a phase III efficacy and safety study in patients with inherited epidermolysis bullosa (EB) that started in 2017 with an estimated enrollment of 250 participants (NCT03068780). Based on this good safety profile and the established use in human medicine, betulin (1) is an attractive early lead candidate for selective HBV/HDV cell-entry inhibition. However, despite its superior selectivity in comparison to betulinic acid (4), the potency for [3H]preS1 binding inhibition is relatively low (IC50 = 109 µM). Nevertheless, the present study clearly demonstrates that even small modifications of the lupane skeleton of betulin can improve the potency and selectivity of the respective compounds significantly. Therefore, it seems likely that advanced rational drug design can further improve this compound class to end up with a potent and selective HBV/HDV cell-entry inhibitor.
Apart from the above-mentioned clinical application, several previous studies showed that betulin (1) and betulin derivatives have a broad spectrum of pharmacological effects, including anti-cancer, anti-viral, anti-microbial, anti-inflammatory, and anti-fibrotic effects44,45,46,47,48. Interestingly, betulinic acid (4) was also used in HBV infection studies before, which showed inhibition of HBV replication by suppression of manganese superoxide dismutase (SOD2) expression with subsequent mitochondrial reactive oxygen species (ROS) over-generation49. Based on this and the results from the present study, betulinic acid (4) might have additive HBV/HDV antiviral properties by addressing different cellular targets involved in cell-entry (NTCP) and replication (SOD2). Therefore, further studies with betulin derivatives should take selective NTCP inhibition and potential suppression of SOD2 into account, which was beyond the scope of the present study.
Our [3H]preS1 binding assay successfully predicted the inhibitory effect of the betulin derivatives on in vitro HDV infection. A minor but expected limitation is that absolute IC50 values varied between both experimental setups. While the qualitative activity pattern of the betulin derivatives remained the same between the two assays, the absolute IC50 values for in vitro HDV infection inhibition were about one to two orders of magnitude higher than predicted from the [3H]preS1 binding experiments. Most likely, several factors contribute to this effect. One factor could be the different incubation times for [3H]preS1 binding (minutes) and in vitro HDV infection experiments (hours). Another factor might be the use of different cell lines and incubation media. Whereas, the [3H]preS1 binding assays were performed in NTCP-HEK293 cells in pure DMEM, the infection experiments needed to be performed with NTCP-HepG2 cells in HGM containing significant amounts of bovine serum albumin (BSA) as well as polyethylene glycol (PEG). The latter medium conditions potentially alter free drug concentrations due to protein binding. Lowering of the inhibitory potency of betulinic acid (4) due to protein binding has already been identified in a previous study, where this compound was used as inhibitor for the organic anion transporting polypeptide OATP1B350. A further factor could be the different stoichiometry of the single preS1 peptide molecule and the multitude of preS1 domains of the large envelope proteins of HBV/HDV virions. The latter one could need a higher degree of blocked NTCP receptors for successful virus binding and cell-entry inhibition. On this background, it could be beneficial if an NTCP inhibitor would not only be selective, but would also irreversibly block the preS1/virus binding sites at NTCP. However, this was not achieved with the betulin derivatives analyzed in the present study. The data obtained on compounds 4 and 17 more suggest a reversible mode of inhibition for myr-preS12-48 peptide binding to NTCP. Their inhibitory effect on [3H]preS1 binding was completely abrogated by washing the cell surface with inhibitor-free medium (see Fig. 5e,f). But, these observations clearly indicate that the inhibitory effects of compounds 4 and 17 on HDV infection cannot be attributed to drug-induced internalization of NTCP.
Of interest are also the long-term effects of compounds 4 and 17 on the transporter and receptor function of NTCP. In the case of compound 4 preincubation with 600 µM significantly and selectively reduced the transporter function of NTCP, while NTCP still was completely preS1 peptide binding competent. As compound 4 (betulinic acid) belongs to the group of acidic betulin derivatives one can speculate that betulinic acid is transported by NTCP, accumulates inside the cell during preincubation and so by trans-inhibiton blocks substrate binding but not preS1 peptide binding to the outer surface of the NTCP protein. This again would support the idea that bile acid and preS1 peptide binding to NTCP occur at separate, but overlapping domains. In contrast, compound 17 showed a paradoxical effect after long term preincubation and increased the capacity for [3H]preS1 peptide binding to approximately 130%, while the [3H]TC uptake rate was slightly reduced. This could be explained by longer lasting allosteric effects of this compound that differently affects the transport and receptor function of NTCP. The mechanism behind these effects could not be clarified in the present study and needs further investigation, including LC–MS/MS supported direct transport experiments with key betulin derivatives.
In conclusion, betulin derivatives show clear structure–activity relationships for potent and selective inhibition of the HBV/HDV virus receptor function of NTCP without tackling its physiological bile acid transport function. Therefore, betulin derivatives are promising candidates for further development of HBV/HDV cell-entry inhibitors. Further studies with this compound class should additionally focus on the mode of inhibition (reversible vs. irreversible) and should be extended to all relevant HBV genotypes.
Methods
NTCP-expressing cell lines
Human embryonic kidney (HEK293) cells were stably transfected with human NTCP, C-terminally tagged with the FLAG epitope (further referred to as NTCP-HEK293 cells) as reported before51. Cells were maintained at 37 °C, 5% CO2 and 95% humidity in DMEM/F-12 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal calf serum (Sigma-Aldrich, St. Louis, MO, USA), 4 mM L-glutamine (PAA, Cölbe, Germany) and penicillin/streptomycin (PAA). HepG2 cells stably transfected with NTCP-FLAG (further referred to as NTCP-HepG210) were cultured under the same conditions in DMEM with all supplements listed above, except for L-glutamine. For induction of the transgene, the medium was supplemented with 1 µg/ml tetracycline (Roth, Karlsruhe, Germany) in the case of the NTCP-HEK293 cells or with 2 µg/ml doxycycline (Sigma-Aldrich) in the case of the NTCP-HepG2 cells.
Inhibitory concentrations (IC50) for [3H]preS1 binding and [3H]TC transport
Bile acid transport measurements were performed in the NTCP-HEK293 cells with [3H]TC (20 Ci/mmol, 0.09 mCi/ml, Perkin Elmer, Waltham, USA) as reported before51. In parallel, [3H]preS1 peptide binding experiments were performed with a tritium-labelled myr-preS12-48 lipopeptide -HBV subgenotype D3- that was purchased from Pharmaron (120 Ci/mmol, 1 mCi/ml, Cardiff, UK). Briefly, cells were seeded onto polylysine-coated 96-well plates, induced with 1 µg tetracycline per ml, and grown to confluence over 72 h at 37 °C. Then, cells were washed once with tempered phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 7.3 mM Na2HPO4, pH 7.4) at 37 °C and preincubated with 80 µl DMEM for 5 min at 37 °C. The medium was replaced by 80 µl DMEM containing the respective betulin derivative (concentrations ranging from 0.1 to 1,000 µM) or solvent alone (100% uptake/binding control), and cells were further incubated for 5 min at 37 °C. After this preincubation, bile acid transport experiments were started by adding 20 µl DMEM containing 5 µM [3H]TC (final concentration: 1 µM). Binding of [3H]preS1 was initiated by adding 20 µl DMEM containing 25 nM [3H]preS1 (final concentration: 5 nM). Experiments were stopped after 10 min by two-times washing with ice-cold PBS. For 0% uptake/binding control, the NTCP-HEK293 cells were not induced with tetracycline (-tet). Cell-associated radioactivity of [3H]TC or [3H]preS1 was quantified by liquid scintillation counting in a Packard Microplate Scintillation Counter TopCount NXT (Packard Instrument Company, Meriden, USA). Transport rates and [3H]preS1 binding were determined in counts per minute (cpm). The mean of the 0% control was subtracted and the net [3H]TC transport rates and net [3H]preS1 binding rates, respectively, were expressed as % of control. Experiments at pH 5.5 were performed the same way, using DMEM acidified with hydrochloric acid (32%, Roth). A set of betulin derivatives was purchased from Adipogen AG (Liestal, Switzerland), including the compounds betulin (1), betulinic acid (4), lupeol (3), betulonaldehyde (18), lupenone (17), 3-O-caffeoylbetulin (27), 3,28-di-O-acetylbetulin (24), 28-O-(3,3-dimethylglutaroyl)betulin (10), 3,28-di-O-(3,3-dimethylglutaroyl)betulin (12), 28-O-succinoylbetulin (8), 3,28-di-O-succinoylbetulin (9), 3-O-(3,3-dimethylglutaroyl)betulinic acid (11), and 3-O-acetylbetulinic acid (21) (TargoSet). All other betulin derivatives were synthesized as outlined elsewhere52,53,54. IC50 values were calculated from quadruplicate determinations by GraphPad Prism 6 (GraphPad, San Diego, CA, USA).
HDV infection experiments
For infection experiments NTCP-HepG2 cells were cultured in 96-well plates in Hepatocyte Growth Medium (HGM) consisting of William’s E Medium (Thermo Fisher Scientific) containing 2% bovine serum albumin (BSA, Roth), 2 mM L-Glutamin (Thermo Fisher Scientific), 100 µg/ml gentamicin (Thermo Fisher Scientific), 10 nM dexamethasone (Sigma-Aldrich), 1 mM sodium pyruvate (Thermo Fisher Scientific), 1 × Insulin-Transferrin-Selen (Thermo Fisher Scientific), 2% DMSO (Merck, Darmstadt, Germany), 4% polyethylene glycol (Sigma-Aldrich) and 2 µg/ml doxycycline (Sigma-Aldrich) as reported10. Subsequently, cells were washed with DMEM and cultured in HGM supplemented with 2% DMSO, 2% BSA and 2 µg/ml doxycycline. NTCP-HepG2 cells were preincubated for 5 min with inhibitors solved in 80 µl HGM per well in concentrations ranging from 5 µM to 300 µM (compounds 2 and 19) or from 5 µM to 600 µM (compounds 4 and 17). HDV stock solved in 20 µl HGM per well was added for infection and cells were incubated for 6 h with final concentration of 700 genome equivalents/cell of HDV particles. HDV production was done in vitro as described before55,56. RT-qPCR was performed to determine genome equivalents. Every three days medium was changed until cells were fixed at 9 days post infection with 3% paraformaldehyde (Sigma-Aldrich) in PBS, for 30 min at room temperature (RT). Cells were permeabilized with 0.2% Triton X 100 (Roth) in PBS for 30 min at RT, and blocked by incubation with 5% bovine serum albumin (Roth) in PBS, for 30 min at RT. Then, cells were immunostained with purified human anti-HDV-positive serum at 37 °C for 1 h (1:400 dilution). Goat anti-human IgG secondary antibody coupled to Alexa Fluor fluorophore (1:400 dilution, Thermo Fisher Scientific) was added for 1 h at 37 °C for detection of Hepatitis Delta antigen (HDAg) as described before34. Nuclei were stained with Hoechst 33342 (1 µg/ml, Thermo Fisher Scientific).
Preincubation studies in NTCP-HepG2 cells
NTCP-expressing HepG2 hepatoma cells were seeded onto rat tail collagen coated 24-well plates, induced with 2 µg doxycycline per ml, and grown to confluence over 72 h at 37 °C. For experiments, cells were preincubated with a fix concentration (600 µM) of compound 4 and compound 17, respectively, solved in 300 µl DMEM for intervals of 15 min or 6 h at 37 °C. Cells treated with solvent alone (DMEM) were used as 100% controls. Cells without induction of NTCP expression by doxycycline were used as 0% controls. Subsequently, cells were washed three times with tempered PBS. Then, [3H]TC uptake and [3H]preS1 binding experiments were started by adding 300 µl of 1 µM [3H]TC or 5 nM [3H]preS1, respectively. After 10 min, experiments were stopped by washing the cells 5 times with ice-cold PBS. Cell-associated radioactivity was quantified by liquid scintillation counting as described above. [3H]TC uptake and [3H]preS1 binding rates were determined in pmol/mg protein/10 min. Means of the 0% controls were subtracted and the net [3H]TC uptake and net [3H]preS1 binding rates, respectively, were expressed as % of control. Data were calculated from two independent experiments, each with quadruplicate determinations by GraphPad Prism 6 (GraphPad, San Diego, CA, USA).
Cytotoxicity assay
Pierce lactate dehydrogenase (LDH) cytotoxicity assay (Thermo Fisher Scientific) was performed according to the manufacturer’s protocol. Briefly, NTCP-HepG2 cells were incubated with 300 µM or 600 µM of the respective betulin derivatives over 6 h at 37 °C. 45 min before the end of incubation, lysis buffer was added to the maximal LDH control. After 6 h, 50 µl of each sample and control medium were transferred to a new well plate, 50 µl of the reaction mixture were added and incubation was performed over 30 min at room temperature. Finally, the reaction was stopped by adding 50 µl of stop solution and samples were measured by ELISA reader (GloMax-Multi Detection System, Promega, Madison, WI, USA).
Structure modeling and pKa determination
2D compound structures were generated using the MAESTRO 12.2 Molecular Modeling Interface of SCHRÖDINGER, Inc. (New York City, NY, USA). The Jaguar pKa prediction module of the SCHRÖDINGER software was used to calculate pKa values of compounds 4 and 8.
Statistics
Determination of IC50 values was done by nonlinear regression analysis using the equation log(inhibitor) vs. response settings of the GraphPad Prism 6.0 software (GraphPad). Data of bile acid transport and [3H]preS1 binding are expressed as means ± SD from quadruplicate determinations. IC50 values are listed with their 95% confidence intervals in Table 2. In addition, mean IC50 values were calculated for each experiment. Infection studies show data from three independent experiments, each with triplicate determinations represented as means ± SEM. Statistical analyses of the HDV infection experiments and NTCP-HepG2 preincubation studies were performed by two-way ANOVA, followed by Dunnett's multiple comparison test by GraphPad Prism 6.0, considering p < 0.01 as statistically significant.
Data availability
Obtained and analyzed data of this study are available from the corresponding author on request.
References
World Health Organization. Global hepatitis report 2017. Geneva 2017. Licence:CC BY-NC-SA 3.0 IGO. (2017).
Glebe, D. & Bremer, C. M. The molecular virology of hepatitis B virus. Semin. Liver. Dis. 33, 103–112. https://doi.org/10.1055/s-0033-1345717 (2013).
Glebe, D. et al. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 129, 234–245. https://doi.org/10.1053/j.gastro.2005.03.090 (2005).
Gripon, P., Cannie, I. & Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 79, 1613–1622. https://doi.org/10.1128/JVI.79.3.1613-1622.2005 (2005).
Hughes, S. A., Wedemeyer, H. & Harrison, P. M. Hepatitis delta virus. Lancet 378, 73–85. https://doi.org/10.1016/S0140-6736(10)61931-9 (2011).
Sureau, C. & Negro, F. The hepatitis delta virus: Replication and pathogenesis. J. Hepatol. 64, S102–S116. https://doi.org/10.1016/j.jhep.2016.02.013 (2016).
Martinez, M. G., Villeret, F., Testoni, B. & Zoulim, F. Can we cure hepatitis B virus with novel direct-acting antivirals?. Liver Int. 40(Suppl 1), 27–34. https://doi.org/10.1111/liv.14364 (2020).
World Health Organization. Guidelines for the prevention, care and treatment of persons with chronic hepatitis b infection. (2015).
Yan, H. et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife https://doi.org/10.7554/eLife.00049.
König, A. et al. Kinetics of the bile acid transporter and hepatitis B virus receptor Na+/taurocholate cotransporting polypeptide (NTCP) in hepatocytes. J. Hepatol. 61, 867–875. https://doi.org/10.1016/j.jhep.2014.05.018 (2014).
Lamas Longarela, O. et al. Proteoglycans act as cellular hepatitis delta virus attachment receptors. PLoS ONE 8, e58340. https://doi.org/10.1371/journal.pone.0058340 (2013).
Fukano, K., Tsukuda, S., Watashi, K. & Wakita, T. Concept of Viral Inhibitors via NTCP. Semin. Liver Dis. 39, 78–85. https://doi.org/10.1055/s-0038-1676804 (2019).
MYR Pharmaceuticals. http://myr-pharma.com/. (2020).
Bogomolov, P. et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J. Hepatol. 65, 490–498. https://doi.org/10.1016/j.jhep.2016.04.016 (2016).
Geyer, J., Wilke, T. & Petzinger, E. The solute carrier family SLC10: more than a family of bile acid transporters regarding function and phylogenetic relationships. Arch. Pharmacol. 372, 413–431. https://doi.org/10.1007/s00210-006-0043-8 (2006).
Stieger, B. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb. Exp. Pharmacol. https://doi.org/10.1007/978-3-642-14541-4_5 (2011).
Döring, B., Lütteke, T., Geyer, J. & Petzinger, E. The SLC10 carrier family: transport functions and molecular structure. Curr. Top. Membr. 70, 105–168. https://doi.org/10.1016/B978-0-12-394316-3.00004-1 (2012).
Ni, Y. et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146, 1070–1083. https://doi.org/10.1053/j.gastro.2013.12.024 (2014).
Blank, A. et al. The NTCP-inhibitor Myrcludex B: effects on Bile Acid Disposition and Tenofovir Pharmacokinetics. Clin. Pharmacol. Ther. 103, 341–348. https://doi.org/10.1002/cpt.744 (2018).
Goh, B. et al. Development of a mass spectrometric screening assay for hepatitis B virus entry inhibitors. J. Pharm. Biomed. Anal. 178, 112959. https://doi.org/10.1016/j.jpba.2019.112959 (2020).
Iwamoto, M. et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun. 443, 808–813. https://doi.org/10.1016/j.bbrc.2013.12.052 (2014).
Fukano, K. et al. Troglitazone impedes the oligomerization of sodium taurocholate cotransporting polypeptide and entry of hepatitis B virus into hepatocytes. Front. Microbiol. 9, 3257. https://doi.org/10.3389/fmicb.2018.03257 (2018).
Watashi, K. et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 59, 1726–1737. https://doi.org/10.1002/hep.26982 (2014).
Nkongolo, S. et al. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J. Hepatol. 60, 723–731. https://doi.org/10.1016/j.jhep.2013.11.022 (2014).
Shimura, S. et al. Cyclosporin derivatives inhibit hepatitis B virus entry without interfering with NTCP transporter activity. J. Hepatol. 66, 685–692. https://doi.org/10.1016/j.jhep.2016.11.009 (2017).
Saso, W. et al. A new strategy to identify hepatitis B virus entry inhibitors by AlphaScreen technology targeting the envelope-receptor interaction. Biochem. Biophys. Res. Commun. https://doi.org/10.1016/j.bbrc.2018.04.187 (2018).
Wang, X. J. et al. Irbesartan, an FDA approved drug for hypertension and diabetic nephropathy, is a potent inhibitor for hepatitis B virus entry by disturbing Na(+)-dependent taurocholate cotransporting polypeptide activity. Antiviral Res. 120, 140–146. https://doi.org/10.1016/j.antiviral.2015.06.007 (2015).
Blanchet, M., Sureau, C. & Labonte, P. Use of FDA approved therapeutics with hNTCP metabolic inhibitory properties to impair the HDV lifecycle. Antiviral Res. 106, 111–115. https://doi.org/10.1016/j.antiviral.2014.03.017 (2014).
Huang, H. C. et al. (-)-Epigallocatechin-3-gallate inhibits entry of hepatitis B virus into hepatocytes. Antiviral Res. 111, 100–111. https://doi.org/10.1016/j.antiviral.2014.09.009 (2014).
Kaneko, M. et al. A novel tricyclic polyketide, vanitaracin A, specifically inhibits the entry of hepatitis B and D viruses by targeting sodium taurocholate cotransporting polypeptide. J. Virol. 89, 11945–11953. https://doi.org/10.1128/JVI.01855-15 (2015).
Tsukuda, S. et al. Dysregulation of retinoic acid receptor diminishes hepatocyte permissiveness to hepatitis B virus infection through modulation of sodium taurocholate cotransporting polypeptide (NTCP) expression. J. Biol. Chem. 290, 5673–5684. https://doi.org/10.1074/jbc.M114.602540 (2015).
Tsukuda, S. et al. A new class of hepatitis B and D virus entry inhibitors, proanthocyanidin and its analogs, that directly act on the viral large surface proteins. Hepatology 65, 1104–1116. https://doi.org/10.1002/hep.28952 (2017).
Donkers, J. M. et al. Reduced hepatitis B and D viral entry using clinically applied drugs as novel inhibitors of the bile acid transporter NTCP. Sci. Rep. 7, 15307. https://doi.org/10.1038/s41598-017-15338-0 (2017).
Müller, S. F., König, A., Döring, B., Glebe, D. & Geyer, J. Characterisation of the hepatitis B virus cross-species transmission pattern via Na+/taurocholate co-transporting polypeptides from 11 New World and Old World primate species. PLoS ONE 13, e0199200. https://doi.org/10.1371/journal.pone.0199200 (2018).
Cichewicz, R. H. & Kouzi, S. A. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med. Res. Rev. 24, 90–114. https://doi.org/10.1002/med.10053 (2004).
Moghaddam, M.G., Ahmad, F.B., Samzadeh-Kermani, A. Biological Activity of Betulinic Acid: A Review. Pharmacol. Pharm. 03, 119–123, https://doi.org/10.4236/pp.2012.32018 (2012)
Zhang, D. M. et al. Betulinic acid and its derivatives as potential antitumor agents. Med. Res. Rev. 35, 1127–1155. https://doi.org/10.1002/med.21353 (2015).
Greupink, R. et al. In silico identification of potential cholestasis-inducing agents via modeling of Na(+)-dependent taurocholate cotransporting polypeptide substrate specificity. Toxicol. Sci. 129, 35–48. https://doi.org/10.1093/toxsci/kfs188 (2012).
Kim, R. B. et al. Modulation by drugs of human hepatic sodium-dependent bile acid transporter (sodium taurocholate cotransporting polypeptide) activity. J. Pharmacol. Exp. Ther. 291, 1204–1209 (1999).
Kouzuki, H., Suzuki, H., Stieger, B., Meier, P. J. & Sugiyama, Y. Characterization of the transport properties of organic anion transporting polypeptide 1 (oatp1) and Na(+)/taurocholate cotransporting polypeptide (Ntcp): comparative studies on the inhibitory effect of their possible substrates in hepatocytes and cDNA-transfected COS-7 cells. J. Pharmacol. Exp. Ther. 292, 505–511 (2000).
Kessler, J. H., Mullauer, F. B., de Roo, G. M. & Medema, J. P. Broad in vitro efficacy of plant-derived betulinic acid against cell lines derived from the most prevalent human cancer types. Cancer Lett. 251, 132–145. https://doi.org/10.1016/j.canlet.2006.11.003 (2007).
Zuco, V. et al. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 175, 17–25. https://doi.org/10.1016/s0304-3835(01)00718-2 (2002).
Pisha, E. et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat. Med. 1, 1046–1051. https://doi.org/10.1038/nm1095-1046 (1995).
Zhan, X. K., Li, J. L., Zhang, S., Xing, P. Y. & Xia, M. F. Betulinic acid exerts potent antitumor effects on paclitaxel-resistant human lung carcinoma cells (H460) via G2/M phase cell cycle arrest and induction of mitochondrial apoptosis. Oncol. Lett. 16, 3628–3634. https://doi.org/10.3892/ol.2018.9097 (2018).
Chen, Y. et al. The design, synthesis and structure-activity relationships associated with C28 amine-based betulinic acid derivatives as inhibitors of HIV-1 maturation. Bioorg. Med. Chem. Lett. 28, 1550–1557. https://doi.org/10.1016/j.bmcl.2018.03.067 (2018).
Adeleke, G. E. & Adaramoye, O. A. Betulinic acid protects against N-nitrosodimethylamine-induced redox imbalance in testes of rats. Redox Rep. 22, 556–562. https://doi.org/10.1080/13510002.2017.1322750 (2017).
Laavola, M. et al. Betulin derivatives effectively suppress inflammation in vitro and in vivo. J. Nat. Prod. 79, 274–280. https://doi.org/10.1021/acs.jnatprod.5b00709 (2016).
Wan, Y. et al. The anti-fibrotic effect of betulinic acid is mediated through the inhibition of NF-kappaB nuclear protein translocation. Chem. Biol. Interact. 195, 215–223. https://doi.org/10.1016/j.cbi.2012.01.002 (2012).
Yao, D. et al. Betulinic acid-mediated inhibitory effect on hepatitis B virus by suppression of manganese superoxide dismutase expression. FEBS J. 276, 2599–2614. https://doi.org/10.1111/j.1742-4658.2009.06988.x (2009).
Oh, Y. et al. Inhibition of organic anion transporting polypeptide 1B1 and 1B3 by betulinic acid: effects of preincubation and albumin in the media. J. Pharm. Sci. 107, 1713–1723. https://doi.org/10.1016/j.xphs.2018.02.010 (2018).
Geyer, J. et al. Cloning and functional characterization of human sodium-dependent organic anion transporter (SLC10A6). J. Biol. Chem. 282, 19728–19741. https://doi.org/10.1074/jbc.M702663200 (2007).
Alakurtti, S. et al. Synthesis and anti-leishmanial activity of heterocyclic betulin derivatives. Bioorg. Med. Chem. 18, 1573–1582. https://doi.org/10.1016/j.bmc.2010.01.003 (2010).
Pohjala, L., Alakurtti, S., Ahola, T., Yli-Kauhaluoma, J. & Tammela, P. Betulin-derived compounds as inhibitors of alphavirus replication. J. Nat. Prod. 72, 1917–1926. https://doi.org/10.1021/np9003245 (2009).
Salin, O. et al. Inhibitory effect of the natural product betulin and its derivatives against the intracellular bacterium Chlamydia pneumoniae. Biochem. Pharmacol. 80, 1141–1151. https://doi.org/10.1016/j.bcp.2010.06.051 (2010).
Rasche, A. et al. Highly diversified shrew hepatitis B viruses corroborate ancient origins and divergent infection patterns of mammalian hepadnaviruses. Proc. Natl. Acad. Sci. U. S. A. 116, 17007–17012. https://doi.org/10.1073/pnas.1908072116 (2019).
de Carvalho Dominguez Souza, B. F. et al. A novel hepatitis B virus species discovered in capuchin monkeys sheds new light on the evolution of primate hepadnaviruses. J. Hepatol. 68, 1114–1122, https://doi.org/10.1016/j.jhep.2018.01.029 (2018).
Acknowledgements
The authors want to acknowledge Edda Wacker for critical reading of the manuscript and Anita Neubauer for her competent support in carrying out the experiments. We also thank Dr. Gary Grosser for his excellent scientific support regarding the setup of inhibition studies. This study was supported by Flex Funds from the LOEWE-Center DRUID (Novel Drug Targets against Poverty-related and Neglected Tropical Infectious Diseases) and in part by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 197785619 – SFB 1021 and the Foundation for Research of Natural Resources in Finland, Marjatta ja Eino Kollin Säätiö, and the COST Action CM-0801 (New drugs for neglected diseases).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
M.K., S.F.M., K.A.A.T.L. and J.G. conceived the experiments, M.K., S.F.M. and K.A.A.T.L. performed the experiments, M.K., S.F.M., K.A.A.T.L. and J.G. analyzed and interpreted the results, N.G., F.L. and D.G. provided materials and laboratories for infection studies, S.A. and J.Y.K. provided derivatives of betulin, M.K., S.F.M., K.A.A.T.L. and J.G. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kirstgen, M., Lowjaga, K.A.A.T., Müller, S.F. et al. Selective hepatitis B and D virus entry inhibitors from the group of pentacyclic lupane-type betulin-derived triterpenoids. Sci Rep 10, 21772 (2020). https://doi.org/10.1038/s41598-020-78618-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78618-2
This article is cited by
-
Structure of antiviral drug bulevirtide bound to hepatitis B and D virus receptor protein NTCP
Nature Communications (2024)
-
Structural basis of hepatitis B virus receptor binding
Nature Structural & Molecular Biology (2024)
-
Synthesis of New Amidoethanesulfonamides of Betulonic Acid
Chemistry of Natural Compounds (2023)
-
Structural insights into the HBV receptor and bile acid transporter NTCP
Nature (2022)
-
Structure of the bile acid transporter and HBV receptor NTCP
Nature (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
