
Abstract
Malaria continues to be an important health problem in Honduras despite major progress achieved reducing its incidence in the last two decades. In a context of case reduction, continuing surveillance of parasite diversity and drug resistance is an important component to assist effective malaria control strategies and support risk assessments. In this study, we employed next generation sequencing on collected Plasmodium vivax and P. falciparum samples from the Hospital Escuela (University Hospital) in Honduras between 2005 and 2017. Hospital Escuela is the main public health hospital in Honduras and receives suspected malaria cases from endemic regions within the country. The resulting sequencing data was used to assess complexity of infections, parasite population structure, parasite diversity and drug resistance profiling. All P. vivax samples and all autochtonous P. falciparum samples were monoclonal and presented a low intra population diversity (π = 0.25 and 0.07, respectively). Genotyping of drug resistance markers showed that three P. falciparum samples presented the chloroquine resistant haplotype SVMNT on pfcrtr (positions 72–76). Epidemiological data suggested that two of these samples were imported cases from Africa whereas the third one was a local case. Three suspected imported cases (two of which were also pfcrt mutants) presented the pfmdr1 86Y mutation that further enhances the CQ resistant genotype. No evidence was found for kelch13 artemisinin resistance associated mutations nor parasite genetic background mutations. Discriminant analysis of principal components and phylogenetic analysis showed two P. vivax and two P. falciparum parasite sub-populations with limited recombination between them. It also confirmed the closer relationship of the three imported cases with African strains. Our findings showed that local Honduras P. falciparum strains do not hold CQ resistance polymorphisms which aligns with clinical data reported by the country and supports the continuity of CQ based treatment in Honduras. In addition, our findings highlight the need of using genomic approaches to provide key information about parasite biology including drug resistance, population structure and HRP2/HRP3 deletions which are becoming relevant as the country move towards elimination.
Similar content being viewed by others

Introduction
Malaria remains as a major public health threat throughout tropical endemic regions causing more than 219 million cases in 20171. Although malaria incidence has recently increased in South America, most countries in the Central America region have experienced a decrease of more than 20% since 20161. This progress has fostered efforts towards malaria elimination in Central America and the island of Hispaniola by 20302. In Honduras, malaria prevalence has consistently dropped in more than 86% since 2010 resulting in less than 2000 cases reported in 20171. Out of those, Plasmodium vivax and P. falciparum are responsible of 90% and 10% of all malaria cases reported, respectively1. Treatment for uncomplicated infections still relies on the use of a combined therapy of chloroquine in addition to primaquine that is used for P. falciparum gametocytes and P. vivax hypnozoites. These drugs are still active despite more than 60 years of use and the widespread prevalence of chloroquine (CQ) resistance reported in most endemic regions of South America3.
It is known that genomic plasticity and diversity of Plasmodium constitute a barrier for malaria elimination programs4 since they can result in the rapid development of resistance against antimalarial drugs4. Furthermore, human migration and commercial activities can provide means for the introduction of drug resistance or virulent genotypes into areas with no indigenous resistant parasites5,6,7.
Therefore, monitoring changes in parasite genotypes can help identify early the emergence of resistant parasites and thereby informing the local health authorities to implement appropriate responses to prevent spread of resistant strains. This is especially important in the context of Honduras given previous reports of CQ resistance in neighboring countries such as Nicaragua8 and the lack of recent studies on genetic diversity and drug resistance9.
Furthermore, genomic studies can provide useful information for malaria elimination which is especially important for Honduras such as assess changes in circulating parasite populations and transmission dynamics, explore the prevalence of HRP2/HRP3 deletions and study molecular markers that can be used to determine the introduction or re-introduction of malaria parasites as the country move towards elimination10.
In this study, we performed a comparative analysis of 16 P. falciparum and 23 P. vivax samples collected in the University Hospital (Hospital La Escuela, HE) in Tegucigalpa Honduras between 2005 and 2017. Genomic data was used to explore the population structure of P. falciparum and P. vivax and identification of drug resistance genotypes.
Materials and methods
Sample collection
The study used de-identified specimens and data collected at the University Hospital in Tegucigalpa, Honduras between 2005 and 2017. The HE is the main public health hospital in Honduras and received suspected malaria cases from multiple regions within the country. As part of regular laboratory activities, blood spots were collected on a 3 mm Whatmann filter paper from each malaria microscopy confirmed patient at the time of diagnosis along with clinical and basic epidemiological data.
The protocol for this study (NAMRU6.2018.0002) was reviewed and approved by the Research Administration Program of the Naval Medical Research Unit-6 (NAMRU-6). Informed consent was not required for this secondary research since it does not meet the definition of research involving human subjects per US Code of Federal Regulations, 32 CFR Part 219-PROTECTION OF HUMAN SUBJECTS, section 219.104 Exempt research. Additionally, all methods were carried out in accordance with relevant guidelines, regulations and good laboratory practices.
DNA extraction and LAMP PCR
DNA from each filter paper was extracted using the DNeasy Blood & Tissue kit (Qiagen) according to the manufacturer’s protocol. The resulting parasite DNA was screened for malaria by a Malachite green LAMP PCR (MG-LAMP) using a previously described method with slight modifications11,12. Briefly, MG-LAMP was performed in a 20 µL reaction volume that contained 5 µL of template DNA in 2× in-house reaction buffer (40 mM Tris–HCl pH8.8, 20 mM KCl, 16 mM MgSO4 , 20 mM (NH4)2SO4 , 0.2% Tween-20, 1.6 M Betaine, 2 mM of dNTP’s each), 0.25 µL of 1:400 SYTO 9 dye, 8 units of Bst Polymerase (New England Biolabs, Ipswich, MA) and 0.004% Malachite Green dye (MG). The genus specific LAMP assay targeted the Plasmodium mitochondria, the P. falciparum specific LAMP assay targeted the Plasmodium 18S subunit ribosomal RNA (ssrRNA) gene whereas the P. vivax specific assay targeted the Pvr64 gene (Table 1).
The amplification reaction was performed at 63 °C for 60 min using a mini heat block (Gene Mate, Bio Express, Utah, US) and results were visually inspected by two independent readers after 15 min post amplification. All samples were processed first by the genus-screening LAMP assay and then by the species-specific assays for P. falciparum and P. vivax.
Sequencing and SNP genotyping
Genotypes for drug resistance loci, genetic barcodes, and parasite speciation were derived from multiplexed amplicon sequencing performed at the Wellcome Sanger Institute. In brief, extracted DNA was selectively whole genome amplified (sWGA)16,17 and multiplex PCRs were performed on the amplified DNA. For. P. falciparum, two multiplexes of 69 and 68 amplicons targeted drug resistance loci and genetic barcodes while a third multiplex contained two amplicons used for parasites speciation (https://www.malariagen.net/resource/29). For P. vivax, a multiplex PCR with 114 targets was performed on the amplified DNA. A second round of PCR was done on both P. falciparum and P. vivax multiplexes which incorporated Illumina flow-cell adapters and sample indexing barcodes. Products were size selected by SPRI-beads and then multiplexes were pooled before sequencing on an Illumina MiSeq. Reads were binned by sample index and genotypes were called per sample by aligning to a P. falciparum 3d7 or P. vivax Sal-1 amplicon reference. For P. falciparum, barcodes were created by concatenated genotypes at 101 SNPs spread across the genome. For P. vivax, the resulting reads were used to create sample barcodes derived from a previously published list of loci18. SNPs within the barcodes were represented by their respective nucleotides (A,T,C and G), as missing (X) or as heterozygous (N).
Data analysis and complexity of infections
Barcoding data was cleaned using the “poppr” R package prior to genetic analysis in order to secure that only high quality data remains19. The parameters used comprised excluding samples with greater than 20% missing SNPs and excluding positions with greater than 20% missing calls.
Complexity of infections were assessed using the COIL and Real McCOIL tools in each sample20,21. These methods use the proportion of heterozygous calls in order to estimate COI under a Markov Chain Monte Carlo (MCMC) framework. COI is represented as the estimated number of individual parasites within a single infection and serves to classify them as monogenomic or polygenomic.
Population diversity
The population barcode diversity (π) was calculated as previously described18. Briefly, barcode diversity was calculated as the mean of differences between each pair of samples across the population divided by the total number of assayed SNPs. The variability of barcode π values was assessed with 10,000 iterations of nonparametric bootstrapping. This method allows to estimate population diversity values ranging from low to high differentiation (0 to 1).
Discriminant analysis of principal components and phylogenetic analysis
In order to provide an additional estimation of the parasite subpopulation, the resulting barcode was used for discriminant analysis of principal components using the R adegenet package22. Based on data from the cases that suggested that some of these samples were imported from Africa, we conducted a phylogenetic analysis. For this purpose, we employed jModelTest 2.1.523 to carry out statistical selection of the best-fit models according to the Bayesian information criterion. Phylogenetic reconstruction was carried out using a maximum likelihood approach implemented in PhyML v3.024 with 1000 bootstrap under the TPM2 substitution model selected by jModelTest 2.1.523. Two African P. falciparum strains were used to root the tree in order to explore the relatedness of the “imported” and local cases. The phylogenetic tree was visualized in Figtree (http://tree.bio.ed.ac.uk/software/figtree/).
Additionally, a phylogenetic network was constructed including 25 sequences from Colombia25, 20 from one of our study sites in Peru and 20 from Sudan26 in order to assess the relationship among haplotypes of the Honduras samples. These analyses were carried out in PopART27 using the median-joining algorithm28.
Disclaimer
The views expressed in this article are those of the author and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the U.S. Government. Some authors of this manuscript are military service members and employees of the U.S. Government. This work was prepared as part of their official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this Title is not available for any work of the United States Government”. Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties.
Results
Sample collection and data cleaning
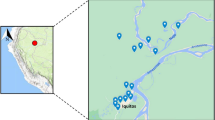
Blood spot samples were collected from 28 patients with P. vivax and 20 patients with P. falciparum monoinfection. From the 2005–2017 study period, nearly 50% of all P. vivax samples were collected between 2013 and 2014 whereas 50% of P. falciparum were collected between 2008 and 2010. Epidemiological data indicate that 21 P. vivax cases came from the regions of El Paraiso and Francisco Morazán whereas in P. falciparum 10 cases came from the departments of Olancho and Gracias a Dios (Fig. 1). In addition, epidemiological data from 3 P. falciparum cases suggests that they have been imported from Africa (Ghana, Congo and Kenya).

Distribution map of study cases. The colors on the map correspond to the Honduras States were patients came from and the numbers to the cases from each of the states. Most samples came from the department of Francisco Morazan where University Hospital is located. The map was created using open data obtained from GADM database of Global Administrative Areas, version 3.6. URL: http://www.gadm.org.
In P. vivax, barcode coverage for 23 out of the 28 samples were observed and data cleaning resulted in 7 out of 38 loci been removed due to missing values higher than 20%. In P. falciparum, barcode coverage for 16 out of the 20 samples were observed. However, 9 samples were removed for population structure and genetic analysis due to higher than 20% missing calls and also because three of them were suspected imported cases from Africa. Further data cleaning resulted in 16 out of 101 P. falciparum loci been removed due to missing values higher than 20%.
Population diversity and complexity of infection
All P. vivax samples and all autochtonous P. falciparum samples were found to be monoclonal by both COIL and Real McCOIL methods. The median bootstrapping values of barcode intra population diversity (π) for P. vivax and P. falciparum were 0.25 and 0.07, respectively, which is indicative of a low parasite diversity.
Drug resistance polymorphisms
In P. vivax, SNP genotyping showed that three samples isolated during different years presented the 976F mutation on pvmdr1 that has been associated with CQ resistance29 whereas the remaining 20 were wildtype (976Y). Two of these subjects were recurrent cases previously diagnosed in the hospital. In addition, all samples presented the wild type haplotype 57F + 61S + 117T for pvdhfr and the wild type haplotype 383A + 553A for pvdhps. Mutations on these genes have been associated with pyrimethamine and sulfadoxine resistance, respectively30,31.
In P. falciparum, SNP genotyping showed that three samples (two imported and one local case) presented the CQ resistant haplotype SVMNT on pfcrtr (positions 72–76) (Table 2). The three samples have heterozygous SNPs on position 72 (C/S) and two of them were heterozygous at positions 74 and 75 (M/I and N/E) (Table 2). The first imported case was from a sample from 2008 from a Japanese subject who likely got the infection in Ghana (Africa) and cleared parasites at day 7 after mefloquine plus primaquine treatment.
The second imported case was from a sample collected in 2012 from a Honduran who likely got the infection in the Republic of Congo or Democratic Republic of Congo. This imported case also presented the pfmdr1 86Y mutation that further enhances the CQ resistant genotype32,33,34 (Table 2). This patient cleared parasites at day 13th post treatment with COARTEM. The local case with a CQ pfcrt resistant haplotype corresponded to a sample collected in 2013 from a subject from the region of Choluteca in Honduras whose therapeutic status was not assessed at that time.
Two samples were quintuple mutants carrying pfdhfr mutations at positions 51, 59 and 108 (IRN haplotype) and at positions 437 and 540 in pfdhps (GE haplotype) which have been strongly associated with sulfadoxine treatment failure35 (Table 2). One of these samples was from the Congo patient which held the pfcrt and pfmdr1 resistant mutations as described. The other sample was from a child who was previously in Kenya and who cleared parasites on the fifth day after standard treatment.
All samples were wildtype for EXO which is associated with piperaquine resistance36 as well as for kelch13 artemisinin resistance associated mutations37. In addition, no evidence was found for parasite genetic background mutations (arps10, ferredoxin, pfmdr2 and pfcrt: 326, 356) that could allow the emergence of k13 mutations38 (Table 2).
Population structure
Discriminant analysis of principal components did not found evidence of distribution according to geographical location nor time of collection for P. vivax (Supplementary figure 1). In the case of P. falciparum, the sample size was limited to see any patterns in the distribution. K-means clustering revealed the presence of two P. vivax and two P. falciparum parasite sub-populations with limited recombination between them (Fig. 2). In P. falciparum, only seven autochthonous samples were analyzed by this method due to the high rate of missing genotypes.

Clustering results of P. vivax and P. falciparum. The figure shows the population structure of 23 P. vivax samples (inset A) and 7 P. falciparum samples (inset B). The y-axis denotes the membership probability of each sample to a cluster whereas the color shows the two clusters that were identified for P. vivax and P. falciparum. The color scheme corresponds to the two clusters that k-means clustering assigned for each of the species.
Phylogenetic analyses showed that the three imported cases (CDR10124, CDR10209 and CDR10057) were genetically closer to African strains rather than to autochthonous cases which clustered in one clade (Fig. 3). The median-joining network showed that samples from Honduras, Colombia, Peru and Africa clustered according to their geographical location (Supplementary figure 2) as indicated by the single mutational-step separation. The three suspected imported samples from Honduras (CDR10124, CDR10209 and CDR10057) clustered separately from the rest of the Honduras samples and were closer to the African strains (Supplementary figure 2).

Maximum likelihood phylogenetic analysis. The figure shows that the three imported cases (CDR10124, CDR10209 and CDR10057) are closely related to the African and 3D7 strains rather than to Honduras autochthonous cases. Node numbers indicate bootstrap support.
Discussion
In malaria, polyclonal infections may arise from multiple infections from different mosquito bites with genetically unrelated parasites or infections with related parasites originated from a single meiosis event39. The genetic diversity that can result from these polyclonal infections could constitute a barrier for malaria elimination39. This is particularly important in a low transmission region like Honduras where these infections can lead to novel parasite variants that could potentially express more virulent phenotypes or contribute to the emergence and spread of drug resistance40.
In this regard, our results points towards a low parasite diversity with near 100% prevalence of monogenomic infections and a low barcode π (π = 0.25 and 0.07) for P. vivax and P. falciparum, respectively. This low level of genetic diversity might have been caused by a population bottleneck as a result of the highly effective intervention campaigns conducted in Honduras during the last two decades in the context of malaria elimination or by natural events such as hurricanes like Mitch in 1998 which swept most of Honduras. However, our sample sizes are too small to ensure an accurate estimate of genetic diversity.
In this regard, a previous study analyzed three markers for P. vivax (pvama, pvcsp and pvmsp1) and two markers for P. falciparum (pfmsp1 and pfsmp2) on samples collected between 2010 and 2011 and showed a high genetic diversity for both species41. In P. vivax, that study reported 41, 23 and 23 genotypes for pvama, pvcsp and pvmsp1, respectively, whereas P. falciparum samples presented all three allelic families for pfmsp1 (K1, MAD20, and RO33) and two allelic families for pfmsp2 (3D7 and FC27). However, their sample size was limited to accurately reflect the overall population diversity, especially for P. falciparum.
In contrast, another study based on neutral microsatellite markers in 110 P. falciparum isolates collected between 2009 and 2012 in Honduras and Nicaragua showed a low genetic diversity in both countries (0.35 and 0.38, respectively)5. Furthermore that study showed no evidence of P. falciparum population structure in Honduras with only one parasite cluster.
This apparent contradictory results might be explained by the different methods used by both studies and the distinct geographical regions that they covered. Nevertheless, the information provided by our and the previous studies highlights the need for more robust assessments including a larger sample size and the analysis of recent specimens given the reduction of malaria cases in Honduras during the last decade.
Our population structure analysis defined two putative clusters for P. vivax and P. falciparum which were not related to temporal nor spatial distribution of the isolates. In the case of the imported P. falciparum cases, our results showed that these strains were closely related with African circulating strains. This result match with the data collected from these cases which showed recent travel history to Africa and the presence of pfdhfr and pfdhps quintuple mutant genotypes associated with SP resistance.
Furthermore, our analyses of drug resistant genotypes showed that four P. falciparum samples presented either the CQ resistant haplotype SVMNT on pfcrtr or the pfmdr1 86Y mutation. Three of these samples were imported cases, one from an Honduran who worked in Africa, the other from a school-aged child42 and the third from a Japanese migrant with travel history to Africa. Due to their particular characteristics, these imported cases were treated with alternative drugs to chloroquine. The forth sample was a local case that harbored the SVMNT pfcrt CQ resistant haplotype whose therapeutic outcome was not assessed.
Conversely, 92% (12 out of 13) autochthonous P. falciparum cases were classified as wild type for all the tested drug resistance loci (Table 2). The evidence of wildtype genotypes in CQ drug resistant loci in our samples supports previous studies that showed that P. falciparum CQ resistance has not emerged nor been introduced in the country5,9,43. Furthermore, our findings are in alignment with clinical data from the country and indicate that CQ is still efficacious for the treatment of local P. falciparum cases in Honduras. However, the finding of a local case and three imported cases with drug resistant genotypes shows a potential risk for introduction of resistant strains and underscores the need for continuing surveillance.
It is important to mention that our study presented some weaknesses that need to be stated. The study sample size was rather small, focused on a specific site and limited time frame. Therefore, our results do not necessarily represent the pattern that could be seen in the country nor on the regions where the participants got infected.
Therefore, active and continuous monitoring of circulating drug resistance genotypes and phenotypes is critical to safeguard the progress that Honduras has achieved towards malaria elimination through early detection and monitor the risk of introduction of resistant strains in the country as well as for the Central America sub-region due to the wide interchange among the countries.
Data availability
The dataset generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. Raw sequence data has been deposited at the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home) under ENA accession numbers ERS3475952 to ERS3475971 for P. falciparum and ERS3516634 to ERS3516661 for P. vivax.
References
Organization, W. H. (2018).
Organization, W. H. Global Technical Strategy for Malaria 2016–2030. (World Health Organization, Geneva, 2015).
Mejiatorres, R. E. et al. Efficacy of chloroquine for the treatment of uncomplicated Plasmodium falciparum malaria in Honduras. Am. J. Trop. Med. Hygiene 88, 850–854. https://doi.org/10.4269/ajtmh.12-0671 (2013).
Rathod, P. K., McErlean, T. & Lee, P. C. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 94, 9389–9393 (1997).
Larranaga, N. et al. Genetic structure of Plasmodium falciparum populations across the Honduras-Nicaragua border. Malaria J. 12, 354. https://doi.org/10.1186/1475-2875-12-354 (2013).
Baldeviano, G. C. et al. Molecular epidemiology of Plasmodium falciparum malaria outbreak, Tumbes, Peru, 2010–2012. Emerg. Infect. Dis. 21, 797–803. https://doi.org/10.3201/eid2105.141427 (2015).
Okoth, S. A. et al. Molecular investigation into a malaria outbreak in Cusco, Peru: Plasmodium falciparum BV1 lineage is linked to a second outbreak in recent times. Am. J. Trop. Med. Hyg. 94, 128–131. https://doi.org/10.4269/ajtmh.15-0442 (2016).
Sridaran, S., Rodriguez, B., Soto, A. M., Macedo De Oliveira, A. & Udhayakumar, V. Molecular analysis of chloroquine and sulfadoxine-pyrimethamine resistance-associated alleles in Plasmodium falciparum isolates from Nicaragua. Am. J. Trop. Med. Hyg. 90, 840–845. https://doi.org/10.4269/ajtmh.13-0214 (2014).
Fontecha, G. A., Sanchez, A. L., Mendoza, M., Banegas, E. & Mejia-Torres, R. E. A four-year surveillance program for detection of Plasmodium falciparum chloroquine resistance in Honduras. Mem. Inst. Oswaldo Cruz 109, 492–493. https://doi.org/10.1590/0074-0276140067 (2014).
Neafsey, D. E. & Volkman, S. K. Malaria genomics in the era of eradication. Cold Spring Harb. Perspect. Med. 7, a025544. https://doi.org/10.1101/cshperspect.a025544 (2017).
Lucchi, N. W., Ljolje, D., Silva-Flannery, L. & Udhayakumar, V. Use of malachite green-loop mediated isothermal amplification for detection of Plasmodium spp. parasites. PLoS ONE 11, e0151437. https://doi.org/10.1371/journal.pone.0151437 (2016).
Barazorda, K. A., Salas, C. J., Bishop, D. K., Lucchi, N. & Valdivia, H. O. Comparison of real time and malachite-green based loop-mediated isothermal amplification assays for the detection of Plasmodium vivax and P. falciparum. PLoS ONE 15, e0234263. https://doi.org/10.1371/journal.pone.0234263 (2020).
Polley, S. D. et al. Mitochondrial DNA targets increase sensitivity of malaria detection using loop-mediated isothermal amplification. J. Clin. Microbiol. 48, 2866–2871. https://doi.org/10.1128/JCM.00355-10 (2010).
Yamamura, M., Makimura, K. & Ota, Y. Evaluation of a new rapid molecular diagnostic system for Plasmodium falciparum combined with DNA filter paper, loop-mediated isothermal amplification, and melting curve analysis. Jpn. J. Infect. Dis. 62, 20–25 (2009).
Patel, J. C. et al. Real-time loop-mediated isothermal amplification (RealAmp) for the species-specific identification of Plasmodium vivax. PLoS ONE 8, e54986. https://doi.org/10.1371/journal.pone.0054986 (2013).
Oyola, S. O. et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malaria J. 15, 597. https://doi.org/10.1186/s12936-016-1641-7 (2016).
Zhang, L. et al. Whole genome amplification from a single cell: Implications for genetic analysis. Proc Natl Acad Sci U S A 89, 5847–5851. https://doi.org/10.1073/pnas.89.13.5847 (1992).
Baniecki, M. L. et al. Development of a single nucleotide polymorphism barcode to genotype Plasmodium vivax infections. PLoS Negl. Trop. Dis. 9, e0003539. https://doi.org/10.1371/journal.pntd.0003539 (2015).
Kamvar, Z. N., Tabima, J. F. & Grunwald, N. J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281. https://doi.org/10.7717/peerj.281 (2014).
Chang, H. H. et al. THE REAL McCOIL: A method for the concurrent estimation of the complexity of infection and SNP allele frequency for malaria parasites. PLoS Comput. Biol. 13, e1005348. https://doi.org/10.1371/journal.pcbi.1005348 (2017).
Galinsky, K. et al. COIL: A methodology for evaluating malarial complexity of infection using likelihood from single nucleotide polymorphism data. Malaria J. 14, 4. https://doi.org/10.1186/1475-2875-14-4 (2015).
Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. https://doi.org/10.1093/bioinformatics/btn129 (2008).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 9, 772. https://doi.org/10.1038/nmeth.2109 (2012).
Criscuolo, A. morePhyML: Improving the phylogenetic tree space exploration with PhyML 3. Mol. Phylogenet. Evol. 61, 944–948. https://doi.org/10.1016/j.ympev.2011.08.029 (2011).
Knudson, A. et al. Spatio-temporal dynamics of Plasmodium falciparum transmission within a spatial unit on the Colombian Pacific Coast. Sci. Rep. 10, 3756. https://doi.org/10.1038/s41598-020-60676-1 (2020).
Hussien, M. et al. Antimalarial drug resistance molecular makers of Plasmodium falciparum isolates from Sudan during 2015–2017. PLoS ONE 15, e0235401. https://doi.org/10.1371/journal.pone.0235401 (2020).
Leigh, J. W. & Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Bandelt, H. J., Forster, P. & Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036 (1999).
Suwanarusk, R. et al. Chloroquine resistant Plasmodium vivax: In vitro characterisation and association with molecular polymorphisms. PLoS ONE 2, e1089. https://doi.org/10.1371/journal.pone.0001089 (2007).
Marfurt, J. et al. Molecular markers of in vivo Plasmodium vivax resistance to amodiaquine plus sulfadoxine–pyrimethamine: Mutations in pvdhfr and pvmdr1. J Infect Dis 198, 409–417. https://doi.org/10.1086/589882 (2008).
Korsinczky, M. et al. Sulfadoxine resistance in Plasmodium vivax is associated with a specific amino acid in dihydropteroate synthase at the putative sulfadoxine-binding site. Antimicrob. Agents Chemother. 48, 2214–2222. https://doi.org/10.1128/AAC.48.6.2214-2222.2004 (2004).
Wellems, T. E. & Plowe, C. V. Chloroquine-resistant malaria. J. Infect. Dis. 184, 770–776. https://doi.org/10.1086/322858 (2001).
Foote, S. J. et al. Several alleles of the multidrug-resistance gene are closely linked to chloroquine resistance in Plasmodium falciparum. Nature 345, 255–258. https://doi.org/10.1038/345255a0 (1990).
Fidock, D. A. et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6, 861–871. https://doi.org/10.1016/s1097-2765(05)00077-8 (2000).
Picot, S. et al. A systematic review and meta-analysis of evidence for correlation between molecular markers of parasite resistance and treatment outcome in falciparum malaria. Malaria J. 8, 89. https://doi.org/10.1186/1475-2875-8-89 (2009).
Amato, R. et al. Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: A genotype-phenotype association study. Lancet Infect. Dis. 17, 164–173. https://doi.org/10.1016/S1473-3099(16)30409-1 (2017).
Ariey, F. et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505, 50–55. https://doi.org/10.1038/nature12876 (2014).
Miotto, O. et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 47, 226–234. https://doi.org/10.1038/ng.3189 (2015).
Manske, M. et al. Analysis of Plasmodium falciparum diversity in natural infections by deep sequencing. Nature 487, 375–379. https://doi.org/10.1038/nature11174 (2012).
de Roode, J. C. et al. Virulence and competitive ability in genetically diverse malaria infections. Proc. Natl. Acad. Sci. USA 102, 7624–7628. https://doi.org/10.1073/pnas.0500078102 (2005).
Lopez, A. C. et al. Genetic diversity of Plasmodium vivax and Plasmodium falciparum in Honduras. Malaria J. 11, 391. https://doi.org/10.1186/1475-2875-11-391 (2012).
Zelaya, V. G. N. et al. Escolar con malaria por Plasmodium falciparum de África: Riesgo para la salud. Rev. Med. Hondur 87, 20–26 (2019).
Jovel, I. T. et al. Drug resistance associated genetic polymorphisms in Plasmodium falciparum and Plasmodium vivax collected in Honduras, Central America. Malaria J. 10, 376. https://doi.org/10.1186/1475-2875-10-376 (2011).
Acknowledgements
This publication uses data from the MalariaGEN SpotMalaria Project as described online https://www.malariagen.net/projects/spotmalaria; the project is coordinated by the MalariaGEN Resource Centre with funding from Wellcome (206194, 090770). The authors would like to thank the staff of Wellcome Sanger Institute Sample Management, Genotyping, Sequencing and Informatics teams for their contribution.
Funding
This work was supported by the Armed Forces Health Surveillance Division (AFHSD) and its Global Emerging Infections Surveillance and Response (GEIS) Section (P0143_19_N6_02 – 2019/2020). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
H.O.V participated in study conception, design and data analysis, F.E.V and S.E.L participated in data analysis and manuscript writing, J.G and J.A participated in data collection and research design. D.K.B participated in research design. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Valdivia, H.O., Villena, F.E., Lizewski, S.E. et al. Genomic surveillance of Plasmodium falciparum and Plasmodium vivax cases at the University Hospital in Tegucigalpa, Honduras. Sci Rep 10, 20975 (2020). https://doi.org/10.1038/s41598-020-78103-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78103-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
