
Abstract
We have studied the diversity and specificity of interactions of amphibactin produced by Vibrio genus bacterium (Vibrio sp. HC0601C5) with iron and various metal ions in + 3 oxidation state in an octahedral (Oh) environment. To survive in the iron-deficient environment of their host, pathogenic bacteria have devised various efficient iron acquisition strategies. One such strategy involves the production of low molecular weight peptides called siderophores, which have a strong affinity and specificity to chelate Fe3+ and can thus facilitate uptake of this metal in order to ensure iron requirements. The Fe uptake by amphibactin and the release of iron inside the cell have been studied. Comparison of the interaction of different transition metal ions (M3+) with amphibactin has been studied and it reveals that Co and Ga form stable complexes with this siderophore. The competition of Co and Ga with Fe impedes iron uptake by bacteria, thereby preventing infection.
Similar content being viewed by others

Introduction
Metal ions are present in all microorganisms in many complexed forms, and they play key roles in numerous biological processes. Further studies of metal ion uptake by microorganisms are expected to shed more light on their life cycles and survival in different environmental conditions. The element Fe is essential for the growth of almost all microorganisms because it catalyzes several enzymatic processes, oxygen metabolism, electron transfer, and DNA and RNA syntheses, among others1. The Vibrio genus is curved-rod shaped gram-negative bacterium that is found in estuarine and marine environments worldwide. Several species of this genus can cause foodborne infection2,3,4,5,6,7,8,9. The seawater environments that are home to Vibrionaceae are slightly alkaline (pH 7.5–8.4), and the total concentration of soluble means Fe+++, FeOH++, Fe(OH)2+, Fe++, and FeOH+ in the absence of any chelating ligands is as low as 10–10 M, which is less than the amount required for bacterial growth (10−6–10−7 M)10,11,12. In biological systems, these Fe levels are further reduced (10−15–10−25 M) through sequestration by molecules such as transferrin and lactoferrin. For biological processes, Fe is required in a + 2 oxidation state. In aqueous-aerobic conditions and neutral pH, Fe is present mostly as insoluble Fe(III) hydroxide, which cannot be taken up by microorganisms13,14. In response to Fe scarcity and high biological demand, microorganisms have evolved strategies to acquire iron from the environment. In this regard, microorganisms secrete small-molecule peptides called siderophores, which chelate Fe3+, thereby ensuring their capacity to maintain biological processes15,16,17,18. Maintaining a dynamic equilibrium of Fe availability for the cell, i.e., Fe homeostasis is therefore critical for optimal cellular function, and indeed for life. Siderophores are metal-chelating agents with low molecular masses (200–2000 Da) produced by microorganisms and plants, especially under Fe-limiting conditions2,19. Several hundred siderophores, varying considerably in chemical composition, have been isolated and characterized. In this regard, they can be classified as hydroxycarboxylate-, catecholate-, or hydroxamate-type on the basis of the acidic moieties they hold in their metal-binding sites2.
The prime role of siderophores is to scavenge Fe. However, these small bioactive molecules also form complexes with other essential elements (i.e. Mo, Mn, Co and Ni), thereby mobilizing them and making them available for microorganisms20,21,22,23,24. The formation of Fe3+–siderophore complexes is affected by pH because of competition between free protons and Fe3+ ions for the free siderophore ligands25. In nature, Fe3+ competes for the siderophore binding sites not only with free protons but also with other metal ions, such as divalent cations Cd2+, Cu2+, Ni2+, Pb2+ and Zn2+, trivalent cations Mn3+, Co3+ and Al3+, and actinides Th4+, U4+ and Pu4+25,26,27. Several studies have shown that siderophores influence the mobility of these metal ions in the environment28.
In recent years, a novel family of cell-associated amphiphilic siderophores called amphibactin has been reported in marine gram-negative bacterium Vibrio sp. HC0601C5 (Fig. 1a). Containing a lipidic side chain attached to a polar head group, these siderophores are found only in waters with low Fe concentrations (< 0.3 nM)29,30,31,32,33,34,35,36,37,38. The amphibactin released from the bacteria specifically bind to Fe (Fe3+) from the environment, transport it into the cell, and then release it for cellular functions. Here we studied how amphibactin binds to Fe3+ and other + 3 oxidation state metal ions and whether this siderophore has greater affinity to bind metals other than Fe3+. Computational quantum chemistry tools can provide insights into the energetic parameters of metal complexation with amphibactin, the metal specificity of amphibactin, the metal–amphibactin interaction, the redox reaction, and the release of the Fe–amphibactin complex. Here we have computed different parameters to understand the binding of different M3+ ions with amphibactin and tried to deduce from these calculated parameters why certain M3+ ions form stable complexes with amphibactin. Fe is essential for many metabolic processes in microorganisms and developing an effective strategy to limit access to this metal will be a good defense against bacterial infections15,39. Several strategies to inhibit bacterial infections have been reported, and the research community faces the major challenge of antibiotic resistance of drugs40,41,42. In this context, the relative stability, interaction energy and affinity of the chelator amphibactin with various transition metal ions in + 3 oxidation state, other than Fe, are studied in order to examine the potential of using amphibactin–metal ion interaction as a tool to develop new drugs for antibiotic resistance. In this regard, here we explored the antibacterial activity of vibrio genus bacterium (Vibrio sp. HC0601C5) based on the chelation of metal ions Co3+ and Ga3+ which have similar ionic radii as Fe3+ with amphibactin. We demonstrate that the bacterial Fe uptake transport system can be exploited to deliver antimicrobials based on Co3+ and Ga3+ to the intracellular space of target bacteria with high specificity. This approach impedes Fe uptake and therefore blocks cellular processes.

(a) Amphibactin, (b) amphibactin–Fe(III) complex. Color scheme: grey-carbon, red-oxygen, blue-nitrogen, purple/blue-iron, gray-hydrogen. All the structures are drawn using BIOVIA, Dassault Systèmes, Discovery Studio Visualizer, version: 17.2.0.16349, 2017. https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/visualization/ and PerkinElmer Informatics, ChemBioDraw UltraVersion: 14.0.0.117. https://cambridgesoft.com/Ensemble_for_Chemistry/details/Default.aspx?fid=14&pid=666.
Results
Molecular structure and stability of the complexes
Fe3+ prefers a hexa-coordinate octahedral ligand coordination sphere. The amphibactin–Fe(III) is a high spin d5 complex. Amphibactin has three hydroxamate ends; three of these groups bind Fe3+ in an Oh geometry, forming a neutral 1:1 complex with a single unpaired electron centered on the Fe43. Figure 1 shows optimized minimum energy structures for free amphibactin and amphibactin–Fe(III) complex. The optimized bond lengths and bond angles are given in Tables S1 and S2, and Fig. S1 (Supplementary information).
The geometric parameters of the optimized local minimum structures of amphibactin–metal ion complexes are given in Fig. S1 (SI). We performed a comparative study of the stability and affinity of amphibactin for Fe3+/Fe2+ and various transition metal ions in + 3 oxidation states by calculating and analyzing the parameters discussed below. The 1st row transition metals (Sc–Co), the Fe group (Ru, Os) and Mo, all of these with a preference for + 3 oxidation states and octahedral (Oh) geometry, were examined.
Frontier molecular orbital analysis
The ground state reactivity was analyzed for amphibactin–transition metal ion complexes from the frontier molecular orbitals (FMOs): HOMO (highest occupied molecular orbitals) and LUMO (the lowest unoccupied molecular orbitals)44. The energy of the HOMO (EHOMO), energy of the LUMO (ELUMO) and the HOMO–LUMO gap (ΔEH–L) are used to describe the global reactivity and kinetic stability of a system44,45,46,47,48. The higher the value of ΔEH–L, the lower the reactivity of a molecule and vice versa. To evaluate the chemical reactivity or kinetic stability, the ΔEH-L of all twelve amphibactin–transition metal ion complexes was calculated at B3LYP and M06/DEF2TZVP level (Table 1). The order of decreasing ΔEH-L was as follows: Ga3+ > Sc3+ > Cr3+ > Mn3+ > Co3+ > V3+ > Fe3+ > Ti3+ > Mo3+ > Ru3+ > Fe2+ > Os3+ (B3LYP/def2tzvp level). At higher level calculation (M06/def2tzvp level) the order of decreasing ΔEH-L gap was as follows: Ga3+ > Sc3+ > Cr3+ > Co3+ > Mn3+ > V3+ > Fe3+ > Mo3+ > Ti3+ > Ru3+ > Os3+ > Fe2+. The amphibactin–Ga3+ complex had the highest ΔEH–L, and hence was the most kinetically stable complex, the least stable being the amphibactin–Fe2+ and amphibactin–Os3+ complex from B3LYP/M06/def2tzvp calculations. Ionic radius of the metal ion is an important factor for amphibactin chelation with the metal ion. Here Sc3+ with high ΔEH–L gap, but with large ionic radius compared to Fe3+ ion results in reduced preference for complexation with amphibactin (Table 2). The Sc3+ in d0 Oh geometry with high ΔEH-L gap showed low Eint, MIA, δ and ∆G (Tables 2 and 3). The Cr3+, Mn3+, Co3+ and V3+ have higher ΔEH–L than Fe3+. The high ΔEH–L for Cr3+ may be due to high LFSE (Ligand Field Splitting Energy) value (~ − 1.2Δ0, high spin t2g3) in an Oh geometry. The high spin d2 V3+ also showed high ΔEH–L than Fe3+ due to high LFSE it had in an Oh environment. The Mn3+ shows a higher ΔEH–L gap, but the d4 high spin t2g3 eg1 Mn3+ ion was unstable and readily decomposes to Mn2+ and Mn4+ in aqueous condition49. The Co3+ had large ΔEH-L gap value as it forms stable Oh complex with amphibactin. The optimized structure of the amphibactin–Mo complex was broken with the loss of metal–ligand six-coordinate Oh geometry. The results from Table 1 confirmed that out of the metal ions with similar ionic radii as Fe3+, amphibactin–Fe3+/Ga3+/Co3+/Cr3+/Mn3+ complexes were the most kinetically stable or were less reactive than other complexes.
Hardness
Chemical hardness (η), first put forward by Pearson, is a measure of the resistance of a chemical species to change its electronic configuration53,54. According to the Hard and Soft Acid and Bases (HSAB) principle, a hard acid prefers a hard base and a soft acid prefers a soft base. Here, free Fe3+ has an ionic radius of 67 pm and acts as a “hard” acid, preferring “hard” oxygen ligands such as phenolate (from Tyr) and carboxylate (from Asp or Glu). Free Fe2+ has an ionic radius of 83 pm and is borderline between a “hard” and “soft” acid, favoring coordination of nitrogen atoms (from His or pyrrole) and sulfur ligands (from Cys or Met) over oxygen ligands55. Since the parameter η indicates a preference of a hard/soft acid for a hard/soft base, for our purposes it provides a quantitative value of the preference of the chelator amphibactin for distinct metal ions.
The \(\eta\) values were calculated for all the amphibactin–metal complexes (Table 1). Since the ionic radius was an important factor deciding the complexation of amphibactin with metal ion, the Sc3+ with large ionic radius (Table 2) can be excluded even though it had large \(\eta\) value. The Fe3+, Ga3+, Co3+, Cr3+ and Mn3+ have similar ionic radii (Table 2).The \(\eta\) values were higher for amphibactin complexes with Fe3+, Ga3+, Cr3+, Mn3+ and Co3+, with the amphibactin–Ga complex showing the highest \(\eta\) value. The high \(\eta\) value for the amphibactin–Ga complex indicates that it was chemically the least reactive after complexation and hence it was kinetically stable or inert.
Interaction energy
Interaction energies (Eint) between amphibactin and the different transition metal ions were calculated using the following equation
where Eion–amphibactin is the energy of the ion–amphibactin complex, Eamphibactin is the energy of the isolated amphibactin, and Eion is the energy of the isolated metal ion. The larger the negative interaction energy, the stronger the intermolecular interaction between amphibactin and the transition metal ion. The Eint with ZPE and BSSE correction for all the amphibactin–metal complexes are reported in Table 2. An important factor for amphibactin metal complexation is the size of the metal ion as the amphibactin was produced especially to chelate Fe3+ ions (~ 64 pm). The Fe3+, Co3+, Ga3+, and Cr3+ have similar ionic radii around (62–64 pm) and the ionic radii of Mn3+ and V3+ were next closer to Fe3+. Among these six metal ions the order of Eint was Co3+ > Fe3+ ~ Mn3+ > Ga3+ > Cr3+ > V3+.The amphibactin–Fe3+/Co3+/Ga3+/Mn3+ complexes showed large negative Eint values, with Co3+ having maximum interaction energy (Table 2). Amphibactin showed a high preference to complex with Fe3+, Ga3+, Mn3+ and Co3+, and the Co3+ ion interacted with the chelator strongly to form the most stable complex. The amphibactin–Fe2+ complex showed the lowest Eint, making it the least stable complex and also the resulting optimized structure did not retain Oh geometry at B3LYP/DEF2TZVP level. At higher level calculation M06/DEF2TZVP, the amphibactin–Fe2+ complex showed a distorted Oh geometry with a large metal–ligand distance (5, Fig. S1, Table S1). It should be noted that amphibactin bound strongly to Fe3+, Co3+, Mn3+ and Ga3+, and all ions have almost the same ionic radius (Table 2). This observation reveals that amphibactin is specific and forms stable complexes with metal ions with this particular ionic radius. The amphibactin–Mn3+ complex was also found to be considerably stable, as reflected by the Eint value. The high spin d4 Mn3+ (~ 66–67 pm) preferred a tetragonal coordination compared to Oh due to Jahn–Teller effect (evident from bite angle and bond distances Tables S1 and S2). The Mn3+ ions shows a high tendency to disproportionate to Mn2+ or Mn4+ in neutral and acidic condition in water and was less stable56,57,58. The Cr3+ with an ionic radius of around 62–63 pm has Eint value lower than that of Fe3+, Ga3+ and Co3+ due to decreased metal–ligand interaction as the effective nuclear charge or penetration effect increases from left to right along the 1st row transition metals. The higher values for Eint for Ru3+ and Os3+ may be due to the high penetration effect or effective nuclear charge and hence stronger metal–ligand interactions. As we go down the Fe group, the penetration effect increases due to more diffuse orbitals and hence it was reflected in the higher values for Eint, MIA and ∆G. The remaining metal ions (Mo3+, Sc3+, Ti3+, V3+) showed lower Eint values and larger ionic radii.
Relaxation/deformation energy
Relaxation or deformation energies (Erelax) are derived by subtracting the complexation energies (unrelaxed single point energy, amphibactin without metal ion) from the interaction energy value (relaxed, optimized amphibactin). The Erelax values for the amphibactin–metal complexes are given in Table 2 and they provide a measure of how a metal ion can induce a specific conformation (Oh geometry) from the preferred conformation of amphibactin
The large the value of Erelax, indicates greater the capacity of metal ion to coordinate with amphibactin in six-coordinate Oh conformation and form a stable complex. The Fe3+, Co3+, Ga3+, and Cr3+ have similar ionic radii around (62–64 pm). The ionic radii of Mn3+ and V3+ were next closer to Fe3+. The order of relaxation energy (δ) is Co3+ > Mn3+ > Cr3+ > V3+ > Fe3+ > Ga3+ at M06/def2tzvp level. The metal ions Ru3+, Os3+, Sc3+, except (Mn3+, Ti3+ and V3+) had low values of Erelax. The inducing effects of the ions on the conformation of amphibactin also depended on the strength of the amphibactin–metal ion interactions. This observation may explain why Cr3+, Mn3+ and V3+ showed slightly higher. The high spin d4 Mn3+ (~ 66–67 pm) with a stable tetragonal geometry compared to Oh due to Jahn–Teller effect causes two of the bond move apart and other four Mn-ligand bonds to come closer (Table S1). This more inducing effect of Mn3+ on amphibactin conformation causes high Erelax and closer metal–ligand interaction. The high spin d3 Cr3+ with t2g3 levels has high preference for an Oh structure with a high LFSE gap resulting in a larger Erelax value. The d10 Ga3+ with a LFSE = 0 has equal preference for both an Oh and Td geometry. With large ligands, to avoid ligand–ligand repulsion and metal ions with zero LFSE (d0, d5 and d10), the tetrahedral geometry is preferred compared to Oh. For a metal ion to induce an Oh geometry causes more Erelax and hence this may be the reason for higher Erelax value for Cr3+ than Ga3+. The high spin d2 V3+ also has higher preference for Oh geometry than Ga3+ and therefore a higher Erelax value. Therefore from above the discussion, Co3+ metal ion had the largest Erelax values, thereby making it the most stable Oh complexes, Fe2+ being the least stable with lowest Erelax. The Erelax values decreased with the increase in the atomic radii of the ions upon complexation with amphibactin. The Mo3+ does not retain an Oh geometry after optimization (one of the bonds is broken) and the high δ value accounts for that broken bond.
Metal ion affinity
Metal ion affinity (MIA) is one of the methods to study the stability of metal–ligand interactions and was calculated as,
where Eel is the electronic energy obtained from the SCF (Self Consistent Field) computation and Evib include the Zero point energy and temperature corrections from 0 to 298 K obtained by the thermo-chemical analysis of vibrational frequencies59,60,61. MIA is considered as the negative of the enthalpy for the dissociation of the amphibactin–metal complex to free amphibactin and metal ion. The higher the MIA value, the greater the affinity of the metal ion for amphibactin. MIA values of the amphibactin–metal complexes showed a similar trend to the Eint values (Table 2). Among these six (Co3+, Fe3+, Mn3+, Ga3+, Cr3+, V3+) metal ions with almost same ionic radii, the order of MIA are: Co3+ > Fe3+ ~ Mn3+ > Ga3+ > Cr3+ > V3+. The Mn3+ with its d4 high spin tetragonal structure promotes better metal–ligand interaction and hence the large MIA value. The decreased metal–ligand interaction in case of Cr3+ and V3+ results in low MIA. The higher values for MIA for Ru3+ and Os3+ may be due to high (LFSE) expected in these complexes due to more interaction between the ligand and diffuse valence metal orbitals. These distorted Oh structures were less stable which is evident from their relaxation energies (Table 2 and Table S2). Out of the metal ions with similar ionic radii as Fe3+, MIA values for Fe3+, Ga3+ and Co3+ were larger than the other metal ions examined, Co3+ having the highest value. This observation further confirms the affinity of amphibactin for Co3+ followed by Fe3+ and Ga3+ and compared to other transition metals in the + 3 oxidation state.
Chelate angle and interatomic distances
The rigidity of the chelate ligand is measured by the bite angle (α) of the chelate ring and is sensitive to the metal–ligand distance. The bite angle α is defined as the L–M–L angle (Fig. 2). The longer the bond distance, the smaller the α. Octahedral and square planar complexes show a preference for a α of around 90°, while for tetrahedral complexes it is 110°. If the Ls are too far apart, thus resulting in a much bigger α, one end of the ligand will dissociate, indicative of low stability of the complex. For chelate rings with a α of less than 90°, the coordination sphere extends from Oh toward the trigonal prism with smaller α. As shown in Table S2, the α for Fe3+, Ga3+, Mn3+, Cr3+ and Co3+ were close to 90°, as required for an Oh coordination sphere, thus making these metal ions more stable than others with a smaller bite angle. The decreasing order of α: Co3+ > Mn3+ > Ga3+ ~ Cr3+ > Fe3+. The bite angles for remaining metal ions were farther away from 90°, thus indicating a more deviation from Oh geometry. If ligands were large; so as to avoid ligand-ligand repulsion, for metal ions (Sc3+, Fe3+, Ga3+, Ru3+, Os3+) with zero LFSE (d0, d5 and d10), the tetrahedral geometry was preferred over Oh. These metal ions have showed deviation from Oh geometry (Table S2). The Co3+ shows maximum value of 82.9/83.6° making amphibactin–Co3+ the most stable complex. In the case of the amphibactin–Mo complex, it had the largest deviation from α of 90° with most deviation from Oh geometry.

Chelate angle α = < L–M–L, where L ligand, M metal.
Thermodynamic properties
To further understand the stability of amphibactin–metal ion complexes and binding preference of amphibactin for a particular metal ion, the enthalpy, free energy, entropy and individual contributions (translational, rotational, and vibrational) of the transition metal ions complexed with amphibactin are reported in Table 3. The ∆G, ∆H, ∆S parameters explain the thermodynamic stability of the complex. A negative ∆G/∆H indicates the formation of a stable complex. Chelation or complex formation causes ∆S to be negative. As a result of negative S values, G values are less negative than their corresponding H values. The ∆H values recorded for the complexes were slightly less negative than their ∆G values (Table 3). Examination of the thermodynamic parameters revealed Fe3+ and Co3+ as the metal ions with the largest ∆G/∆H values, thereby allowing them to form strong complexes with amphibactin. In contrast, Fe2+ was the least stable metal ion, as reflected by the lowest ∆G/∆H values. The ∆G/∆H values depended on the value of ∆S. Large negative ∆S values were found for Ga3+, Co3+, Fe3+, Mn3+, Os3+ and Ru3+. We calculated the individual contributions to total ∆S. The ∆Strans and ∆Srot became more negative as the size of the complex grew and is purely size-dependent. This explains the reason for large negative ∆Strans values for Os3+ and Ru3+ complexes because as we go down the periodic table the size increases due to more diffuse orbitals. The ∆Srot depends on the symmetry and geometry of the complex formed. If the resulting complex has highly symmetric, like Oh geometry the ∆Srot will be more negative. From Table 3 it was clear that Co3+, Fe3+, Ga3+, Cr3+ and Mn3+ had larger negative ∆Srot values implying that the complexes formed by these metal ions have more symmetric Oh geometry and contributes to its stability. Also the bite angles calculated for above mentioned metal ions were closer to Oh geometry. The ∆Svib depended on the strength of the interaction between the metal ion and amphibactin. As the complex became stronger, the vibrations became stiffer, and the ∆Svib became more negative. In case of Ga3+, Co3+, Fe3+, Cr3+, Mn3+, the ∆Svib values were high, indicating strong amphibactin–metal interactions in these complexes. The ions Cr3+, Sc3+, Ti3+, Os3+ and Ru3+ had high ∆Svib but with low ∆G values. The high ∆Svib value for Cr3+ may be due to the high LFSE value exhibited by Cr3+ in Oh coordination, but the weak metal–ligand interaction results in low ∆G value. In the case of Sc3+ and Ti3+ as well, the high LFSE in Oh environment contribute to high ∆Svib values. The higher penetration effect in Os3+ and Ru3+ assist in better metal–ligand interaction and hence higher ∆Svib values for these ions. The Mn3+ also formed a stable complex with amphibactin. However, Mn3+ complexes are generally unstable in aqueous neutral and acidic pH environments and they decompose in water to produce Mn2+ and Mn4+56,57,58. Also Mn3+ prefers a tetragonal geometry in d4 state62. In tetragonal geometry either two ligands move apart or come closer to the central metal ion to reduce the degeneracy. In amphibactin–Mn(III) complex two of the bond distances showed considerable difference compared to other four Mn–ligand bonds (Table S1). The thermochemical analysis also reveals amphibactin–Co3+ forms the stable complex compared to other amphibactin–metal ions studied.
Redox reaction
A powerful strategy to fight microbial infections is to prevent pathogens from acquiring Fe. The reduction of Fe3+ to Fe2+ is an important step in the Fe uptake process. A greater understanding of this step may contribute to the development of molecules that inhibit Fe uptake. All the following calculations were carried out at B3LYP/6-31g(d,p)/LANL2DZ level in gaseous and aqueous condition (dielectric constant ε = 78.4) in G09 CPCM model.
The molecular weights of the ferric–siderophore complexes exceeded the cut off for porins and therefore require specific outer membrane receptors for their uptake into the cell through the periplasmic space. Once siderophores enter the cytoplasm, the Fe from the ferric-siderophore complex can be released as Fe2+ for metabolic processes in the following two ways2,15,63.
-
1.
The reduction of amphibactin–Fe(III) to amphibactin–Fe(II) complex, followed by protonation and hydrolysis to release Fe2+ in to the cell cytoplasm (see Fig. 3). The first step is the reduction of amphibactin–Fe3+ complex that has entered the cytoplasm. The free energy change for the reduction of amphibactin–Fe(III) to amphibactin–Fe(II) complex with zero point correction in aqueous solvent \(({\Delta G}_{s}^{0})\) was negative (\(-112.35\) (kcal/mol)). The Eq. (S1) (Supplementary) gives the standard reduction potential, \({E}^{0}\left(V|RE\right)=0.44 V\), for the reduction reaction of amphibactin–Fe3+ to amphibactin–Fe2+. This was compared with standard hydrogen electrode (SHE) using Eqs. (S2) and (S3) and was positive, thereby indicating that the complex reduces readily in an acidic pH42,64. Bacterial efflux pumps (EPs) are proteins that are localized and imbedded in the plasma membrane of the Gram-negative bacterium are responsible for the pH dependent binding and release of the substrate and for other reactions like reduction65. Many vibrio species use NADH with ferric reductase enzyme66,67,68,69,70,71,72. Inside the cell, NADH acts as a potential electron donor for the enzyme. Therefore we can couple the reduction reaction of amphibactin–Fe3+ to amphibactin–Fe2+ with NADH oxidation reaction. The overall redox reaction is the sum of the two half-cell reactions or couples: (a) reduction of amphibactin–Fe3+ to amphibactin–Fe2+ and (b) oxidation of NADH → NAD− + H+. We calculated the above mentioned two half-cell reactions. \({E}_{Cell}^{0}\) was found to be positive (0.12 V), thereby establishing that the reaction slightly spontaneous, see Eq. (S4). A negative value of \({E}_{Cell}^{0}\) indicates that the reaction will proceed spontaneously in the opposite direction. The Ehcell at pH = 7 was calculated from the Nernest equations (S8–S10). The Ehcell calculated was 0.12 V at pH = 7 (S16). The E°cell and Ehcell calculated at pH = 0 and pH = 7 respectively for the reduction of amphibactin–Fe(III) by NADH was the same 0.12 V (Eqs. S4 and S16). Even though with small Ecell value, the reduction was facilitated at pH = 7. The standard electrode potentials calculated for the reduction amphibactin–Fe(III) to amphibactin–Fe(II) at pH = 0, E0 = 0.44 and at pH = 7, Eh = 0.02588 respectively are different and Eh < E0 (Eqs. S2 and S10). The Eh for Fe3+ → Fe2+ at pH = 2 is 0.32 V (Eq. S12) indicating the reduction would be more spontaneous at an acidic pH. The ∆G°h at pH = 7 was calculated from the Eh value of Fe3+ → Fe2+ reduction potential (Eqs. S22–23). The ∆G°h = − 0.60 kcal/mol (Eq. S11) is slightly non-spontaneous at pH = 7 and ∆G°h = − 7.42 kcal/mol (Eq. S13) at pH = 2, indicating that the complex will reduce readily at an acidic pH. It is well known that the reduction Fe3+ to Fe2+ happens inside the cell where proton gradient generated to lower the extracellular pH to acidic pH. Using a milder reducing agent than NADH like FADH2 in the cell may increase the Ecell value. Usually, the steps which were coupled with NADH reduction had a free energy change of about ΔG = − 100 to − 150 kcal/mol. The reduction of amphibactin–Fe(III) to amphibactin–Fe(II) has a free energy value of 110 kcal/mol. Therefore it might not be feasible to couple with NADH reduction but it would be favourable (thermodynamically) to couple with an easy-to-reduce and less energetic molecule FADH2. With FADH2 (free) as reducing agent, the Ecell = 0.22 V (Eq. S19) and FADH2 (bound) Ecell = 0.41 V (Eq. S21) at a pH = 7 indicating the reduction of amphibactin–Fe(III) to amphibactin–Fe(II) is spontaneous and favoured.
Figure 3 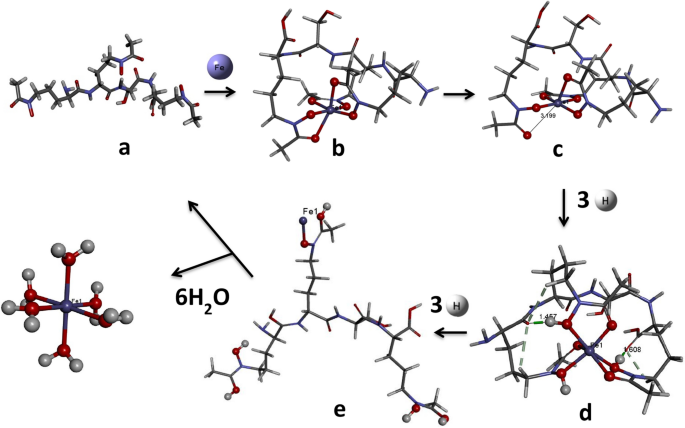
Mechanistic pathway showing Fe(III) acquisition followed by reduction to Fe(II) and further protonation steps to release Fe(II). Optimized minimum energy structures of (a) free amphibactin (b) amphibactin–Fe(III) complex (c) amphibactin–Fe(II) complex (d) three-hydrogen protonated amphibactin–Fe(II) complex (e) six-hydrogen protonated amphibactin–Fe(II) complex, respectively, at B3LYP/6-31g(d,p)/LANL2DZ level. All the structures are drawn using BIOVIA, Dassault Systèmes, Discovery Studio Visualizer, version: 17.2.0.16349, 2017. https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/visualization/.
Further protonation of the reduced amphibactin–Fe2+ complex by three hydrogen atoms in aqueous medium was calculated using Scheme 1 (see below) and the free energy change \({(\Delta {\varvec{G}}}_{{\varvec{t}}}^{\#})\) was found to be negative, typical of a spontaneous process. The CPCM model in Gaussian 09 was used to calculate the free energy for protonation from the optimized amphibactin–Fe2+ complex with three hydrogen atoms added to the (N–O)hydroxamate group coordinated Fe2+ metal ion in aqueous medium.
Scheme 1 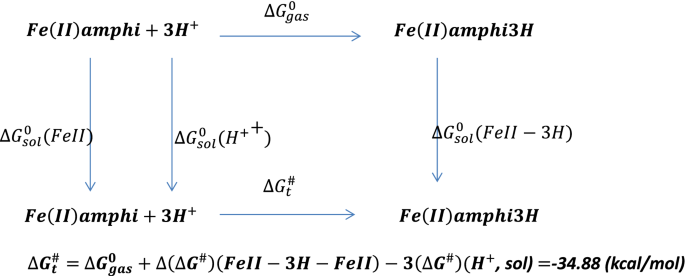
Protonation of amphibactin–Fe2+.
-
2.
The protonation of amphibactin–Fe3+ followed by reduction reaction. The free energy change for the protonation reaction of amphibactin–Fe3+ with three hydrogen atoms attached to the (N–O) hydroxamate group in aqueous medium was calculated using Scheme 2 (see below) and was found to be positive and thus a non-spontaneous reaction. Comparing the free energy changes obtained using Schemes 1 and 2, the former was more thermodynamically favorable than the latter. The former was a spontaneous process with a negative ∆G value whereas the latter was non-spontaneous with a positive ∆G.
At physiological pH = 7, the ∆G for Schemes 1 and 2 are − 6.26 kcal/mol and 39.07 kcal/mol respectively (Eq. S23). This implies that the Scheme 1 is spontaneous process and Scheme 2 non-spontaneous.
Scheme 2 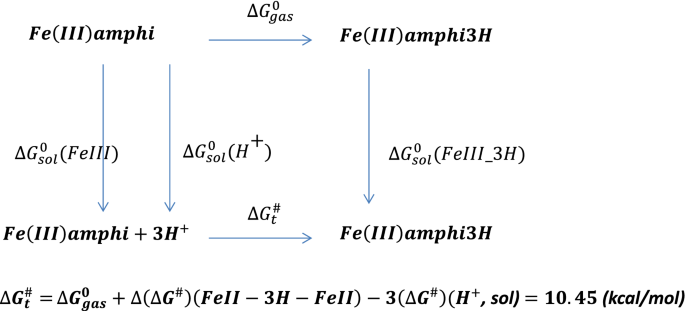
Protonation of amphibactin–Fe3+.



Protonation reaction
The ΔEH–L, chemical hardness (η), Eint and relaxation energy were calculated for amphibactin–Fe3+, amphibactin–Fe2+, 3H protonated amphibactin–Fe2+ and 6H protonated amphibactin–Fe2+ respectively (Table 4). The protonation of reduced amphibactin–Fe complex was done first by the addition of three hydrogen atoms to the three oxygen atoms of N–O (ligand) of amphibactin, followed by the addition of a further three hydrogen atoms to the three oxygen atoms of C=O chelating with Fe. Hydroxamates show pKa values from 8 to 9 in seawater environments that are home to Vibrionaceae with slightly alkaline (pH 7.5–8.4)73,74,75. The energy for protonation of O of CO 7.44 kcal/mol higher than protonation of O of NO of hydroxamate group implying NO protonation was more preferred compared to CO protonation. Here a proton (H+) was attached to one of the O of CO of hydroxamate group of the three hydrogen protonated at three O atoms of NO of hydroxamate group amphibactin–Fe(II)–3H complex to get amphibactin–Fe(II)–4H complex. Computationally from G09 the pKa for hydroxamate CO calculated from the Eq. (S26) was around 8.15. As we go from 1 to 4 the stability of the complex decrease from the Eint decreases and relaxation energy increases while its reactivity increases from the decrease in the ΔEH–L and η (Table 4). The higher ΔEH–L for 2 was due to additional hydrogen bonding interactions in the system due to conformational/geometry change on protonation (d, Fig. 3). This indicates that the reduction of Fe3+ to Fe2+ in the Fe–amphibactin complex, followed by 3H and 6H pronation reduced the thermodynamic stability and reactivity of the complexes. Fe3+ is a hard metal ion that has a preference for hard ligands, like oxygen. Fe2+ is a borderline metal ion that prefers tetrahedral coordination and softer/borderline ligands such as nitrogen (e.g., porphyrin, PhNH2) or sulphur (RSH, R2S). Therefore, the reduction of amphibactin–Fe3+ (1) to amphibactin–Fe2+ (2) drastically lower the affinity of the siderophore for the Fe2+ ion. The Eint also decreases down Table 4, with positive interaction energy for 4. This implies that Fe3+ (1) reduction to Fe2+ (2) followed by subsequent protonation (3 and 4) reduce the affinity of siderophore for Fe2+ ion, thereby releasing the Fe into the cell. The Fe-free siderophore is usually degraded or recycled by excretion through an efflux pump system (like H+/Na+ driven pump). Further hydrolysis of amphibactin–Fe2+–3H is a spontaneous process with a ΔG = − 517.16 kcal/mol.
The resulting aqua complex of Fe2+(Fe(H2O)62+) was stable, with a ΔEH–L value of 5.77 eV, and can therefore be further used for metabolic processes inside the cell.
Co (III) and Ga (III) complexes as antibacterial agents
An alternative approach to block Fe uptake to limit pathogen growth is to use the bacteria’s Fe uptake systems against them in a “Trojan-horse” strategy. Various Ga3+ complexes gained special attention for the treatment of infections associated with bacterial biofilms26. Ga3+ readily forms poly-hydroxo complexes in aqueous physiological conditions. Many chelated complexes of Ga3+ show antibacterial activity against Pseudomona aeruginosa and are stable in physiological conditions76,77,78,79,80,81,82,83. In addition to complexation, co-administration of Ga3+ salts with known antimicrobial agents, such as a combination of antimicrobial and non-anti-microbial agents, has been described to enhance antimicrobial potency84,85. A very small number of Co3+ complexes have biochemical roles. The Co3+ ion as such is unstable in water but can be stabilized against reduction to Co2+ by coordination to ligands or chelators. To date, only the N, O donor ligand type has been found to stabilize the Co3+ ion in aqueous conditions. The Co3+complexes derived from this ligand donor set have proved to be surprisingly efficient antibacterial or antiviral agents. Some of the most promising classes of Co3+ complexes containing N, O donor ligands, like Schiff base [Co(NH3)6]Cl3, are antibacterial and thermally and kinetically stable in aqueous solution, in addition to being easily synthesized86,87,88. The amphibactin is produced by bacteria to specifically chelate Fe3+ for biological process and hence the size of metal ion is an important factor. Our earlier analysis on the Eint, MIA, chemical hardness, ΔEH–L, and thermodynamic parameters (Tables 1 and 2) for metal ions with similar ionic radii as Fe3+ with amphibactin indicate that Ga3+ and Co3+ can replace and form better complexes than Fe3+ and the other metal ions examined. Ga3+ has similar ionic radius as Fe3+ and is more kinetically stable/inert compared to Fe3+ even though it is thermodynamically less stable than Fe3+. The Co3+ is both kinetically and thermodynamically stable than Fe3+. This means that once we replace the Fe3+ by Ga3+/Co3 + , the resulting amphibactin–Ga(III)/Co(III) complex is kinetically inert for further replacement reaction and the bacterial cell wall will allow its passage as the size is similar to Fe3+ ion. Since Fe is essential for the metabolic activities of bacteria and hence for their survival, the better complexation of amphibactin with Ga3+ and Co3+ than Fe3+ (with all three ions having the same ionic radii) will lead to competition for Fe acquisition by bacteria. So instead of Fe; Ga and Co form stable complexes with amphibactin, thereby impairing bacterial metabolic activities and causing cellular toxicity, since the entered Co3+ and Ga3+ cannot partake in the oxidation–reduction processes inside the cell. The free energy changes for the following reactions were studied
The ∆G with ZPE correction for Eqs. 1 and 2 at b3lyp/def2tzvp level were − 708.52 kcal/mol and − 785.83 kcal/mol, respectively, indicating spontaneity. The reactions (1) and (2) confirmed that the formation of amphibactin–Co3+ and amphibactin–Ga3+ complexes were thermodynamically feasible. The Co3+ was inert in aqueous condition but the replacement of monodentate ligand by multidentate was very favourable thermodynamically due to chelation effect. Entropy was largely responsible for the greater free energy change observed for chelates, compared to complexes of unidentate ligands with the same metal ion. The Co-hexamine was kinetically inert (high ΔEH–L = 6.625) in aqueous solution as it was a low spin d6 Oh complex with high LFSE. Instead of the Co-hexamine we can use [Co(CO3)3]3− complex (ΔEH–L = 5.14) as it is kinetically less stable or more labile making the forward replacement reaction kinetically feasible. The ∆G + ZPE = − 117.21 kcal/mol for Eq. (3) shows spontaneity89.
The Eqs. (1–3) confirm that Co3+ from the Co(NH3)6 complex and Ga3+ from the Ga(H2O)6 complex readily binds to amphibactin, forming thermodynamically stable amphibactin–Co and amphibactin–Ga complexes, which inhibit Fe uptake by the bacteria. Also, the different parameters calculated (Tables 1, 2, 3) confirms the Co3+ and Ga3+ forms stronger complexes with amphibactin. Thus Ga3+ and Co3+ emerge as good antibacterial agents and can be impressed as Trojan horses for marine bacteria Vibrio sp. HC0601C5, especially those producing amphibactin siderophores. This way of exploiting the nutritional pathways of bacteria to quell infection is an effective strategy to stop infection by introducing Co and Ga into the amphibactin–Fe3+ complex has potential therapeutic benefit and paves the way for the development of new antibiotics based on Co and Ga. Indeed, Co3+ and Ga3+ compounds coupled with known antibiotic agents emerge as potential tools to tackle sea food or waterborne infections.
Discussions
Here we studied the stability, affinity and energetics of amphibactin complexed with different transition metal ions in + 3 oxidation state in an Oh coordination sphere. The ΔEH–L, interaction energy (Eint), metal ion affinity (MIA), relaxation energy (δ) and thermodynamic parameters (∆G, ∆H, ∆S, ∆Strans, ∆Srot and ∆Svib) calculated indicate that out of the different metal ions studied (Tables 1, 2, 3) Fe3+, Ga3+ and Co3+ with similar ionic radii form stable complexes with amphibactin. The Mn3+ also forms strong complex with amphibactin, but the Mn3+ ion is unstable in aqueous condition. The free energy changes calculated for the studied process: reduction of Fe3+ → Fe2+, followed by protonation and subsequent hydrolysis, were found to be spontaneous, thereby making this the favored mechanism of Fe release from the chelated amphibactin–Fe3+ complex into the bacterial cell. We examined the energetics involved in the mechanistic pathway of Fe acquisition, followed by its reduction, protonation and subsequent release into the bacterial cell in order to understand the scope of blocking bacterial infection by hindering this process. Of the transition metal ions listed in Table 2, Co3+ and Ga3+ showed better chelation for amphibactin other than Fe3+. Here Ga3+ and Co3+, both with same ionic radius as Fe3+, competed with Fe3+ and formed better complexes, thereby impairing the capacity of bacteria to take up Fe and effectively starving the cells. These findings pave the way for Co3+ and Ga3+ as new anti-bacterial agents for bacteria-producing amphiphilic siderophores, amphibactin. The amphibactin–Co3+ forms strongest complex compared to other complexes studied thereby making it the best candidate as anti-bacterial agent. Our results also exemplify the potential to utilize Co3+ and Ga3+ akin to a Trojan horse to effectively block Fe acquisition by bacteria, enabling their specific and effective sterilization irrespective of their antibiotic-resistance type.
Methods
Density functional theory (DFT) calculations using the GAUSSIAN 09 suite of programs were carried out for all the amphibactin–metal complexes90. Geometry optimization of all the structures studied was performed using the hybrid density functional B3LYP91,92,93,94 and M0695,96,97 levels of theory with all atoms with DEF2TZVP basis set98,99, as implemented in the GAUSSIAN 09 quantum chemistry package. Calculations were done as unrestricted DFT calculation for ground state spin 2S + 1 > 1 and for 2S + 1 = 1 restricted DFT calculation respectively. Out of the twelve amphibactin–metal complexes studied five (Fe2+, Fe3+, Mn3+, Cr3+ and V3+) were high spin and remaining were low spin complexes. The input structure of amphibactin is made maintaining the (DDLL) configuration at asymmetric carbon centres as given in Fig. 1a. The lipidic side chain was replaced by hydrogen in amphibactin to reduce the computational expense. We did geometry optimization on this amphibactin structure followed by harmonic vibrational frequency analysis. To find the optimized minimum energy structure vibrational harmonic frequency analysis had been done. Vibrational frequency analysis did not show any imaginary frequencies indicating that the optimized minimum energy structure was reached. This optimized minimum energy amphibactin structure (DDLL) was complexed with metal ions and chosen as input structures for further optimization of the complexes. The gas phase contribution to the thermochemistry parameters Gibbs Free energy G, Enthalpy H, Entropy S, etc., were determined using DFT calculations. The HOMO–LUMO gaps, chemical hardness (η), interaction energy (Eint), metal ion affinity (MIA), and relaxation energies were further calculated using appropriate equations. The Eint, MIA, relaxation energies were ZPE corrected. The Eint was calculated after correcting the basis set super-position error (BSSE) by the counterpoise procedure (CP) of Boys and Bernardi100. Geometry optimization was done, followed by vibrational frequency analysis of amphibactin–Fe2+ and amphibactin–Fe3+ at 298 K and 1 atm., in aqueous medium (dielectric ε = 78.4) using the polarized continuum (overlapping spheres) model (CPCM), where a calculation was performed in the presence of a solvent by placing the solute in a cavity within the solvent reaction field101,102.
References
Richard, K. L., Kelley, B. R. & Johnson, J. G. Heme uptake and utilization by gram-negative bacterial pathogens. Front. Cell. Infect. Microbiol. 9, 81 (2019).
Payne, S. M., Mey, A. R. & Wyckoff, E. E. Vibrio iron transport: evolutionary adaptation to life in multiple environments. Microbiol. Mol. Biol. Rev. 80, 69–90 (2016).
Tan, D., Gram, L. & Middelboe, M. Vibriophages and their interactions with the fish pathogen Vibrio anguillarum. Appl. Environ. Microbiol. 80, 3128–3140 (2014).
Peng, E. D. & Payne, S. M. Vibrio cholerae VciB mediates iron reduction. J. Bacteriol. 199, e00874-e1816 (2017).
Plaza, N. et al. Bacteriophages in the control of pathogenic vibrios. Electron. J. Biotechnol. 31, 24–33 (2018).
Romalde, J. L., Diéguez, A. L., Lasa, A. & Balboa, S. New Vibrio species associated to molluscan microbiota: A review. Front. Microbiol. 4, 413 (2014).
Pernet, F. et al. Mass mortalities of Pacific oysters Crassostrea gigas reflect infectious diseases and vary with farming practices in the Mediterranean Thau lagoon, France. Aquacult. Environ. Interact. 2, 215–237 (2012).
Li, Y. & Ma, Q. Iron acquisition strategies of Vibrio anguillarum. Front. Cell. Infect. Microbiol. 7, 342 (2017).
Thode, S. K., Rojek, E., Kozlowski, M., Ahmad, R. & Haugen, P. Distribution of siderophore gene systems on a Vibrionaceae phylogeny: Database searches, phylogenetic analyses and evolutionary perspectives. PLoS ONE 13, e0191860 (2018).
Jurado, R. L. Iron, infections, and anemia of inflammation. Clin. Infect. Dis. 25, 888–895 (1997).
Boukhalfa, H. & Crumbliss, A. L. Chemical aspects of siderophore mediated iron transport. Biometals 15, 325–339 (2002).
Hem, J. D. & Cropper, W. H. Survey of Ferrous-Ferric Chemical Equilibria and Redox Potentials (USGPO, Washington, 1959).
Crichton, R., Crichton, R. R. & Boelaert, J. R. Inorganic Biochemistry of Iron Metabolism: From Molecular Mechanisms to Clinical Consequences (Wiley, New York, 2001).
Chu, B. C. et al. Siderophore uptake in bacteria and the battle for iron with the host; a bird’s eye view. Biometals 23, 601–611 (2010).
Caza, M. & Kronstad, J. Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front. Cell. Infect. Microbiol. 3, 80 (2013).
De Carvalho, C. C. & Fernandes, P. Production of metabolites as bacterial responses to the marine environment. Mar. Drugs 8, 705–727 (2010).
Sandy, M. & Butler, A. Microbial iron acquisition: Marine and terrestrial siderophores. Chem. Rev. 109, 4580–4595 (2009).
Hider, R. C. & Kong, X. Chemistry and biology of siderophores. Nat. Product Rep. 27, 637–657 (2010).
Aguado-Santacruz, G. A., Moreno-Gomez, B., Jimenez-Francisco, B., Garcia-Moya, E. & Preciado-Ortiz, R. E. Impact of the microbial siderophores and phytosiderophores on the iron assimilation by plants: A synthesis. Revista Fitotecnia Mexicana 35, 9–21 (2012).
Bellenger, J., Wichard, T., Kustka, A. & Kraepiel, A. Uptake of molybdenum and vanadium by a nitrogen-fixing soil bacterium using siderophores. Nat. Geosci. 1, 243 (2008).
Braud, A., Jézéquel, K., Bazot, S. & Lebeau, T. Enhanced phytoextraction of an agricultural Cr-and Pb-contaminated soil by bioaugmentation with siderophore-producing bacteria. Chemosphere 74, 280–286 (2009).
Braud, A., Hoegy, F., Jezequel, K., Lebeau, T. & Schalk, I. J. New insights into the metal specificity of the Pseudomonas aeruginosa pyoverdine–iron uptake pathway. Environ. Microbiol. 11, 1079–1091 (2009).
Zhang, X., Baars, O. & Morel, F. M. Genetic, structural, and functional diversity of low and high-affinity siderophores in strains of nitrogen fixing Azotobacter chroococcum. Metallomics 11, 201–212 (2019).
Vraspir, J. M. & Butler, A. Chemistry of marine ligands and siderophores. Annu. Rev. Mar. Sci. 1, 43–63 (2009).
Albrecht-Gary, A.-M. & Crumbliss, A. L. Coordination chemistry of siderophores: Thermodynamics and kinetics of iron chelation and release. Met. Ions Biol. Syst. 35, 239 (1998).
Dahlheimer, S. R., Neal, C. R. & Fein, J. B. Potential mobilization of platinum-group elements by siderophores in surface environments. Environ. Sci. Technol. 41, 870–875 (2007).
Behrends, T., Krawczyk-Bärsch, E. & Arnold, T. Implementation of microbial processes in the performance assessment of spent nuclear fuel repositories. Appl. Geochem. 27, 453–462 (2012).
Ahmed, E. & Holmström, S. J. Siderophores in environmental research: Roles and applications. Microb. Biotechnol. 7, 196–208 (2014).
Boiteau, R. M. et al. Patterns of iron and siderophore distributions across the California Current System. Limnol. Oceanogr. 64, 376–389 (2018).
Boiteau, R. M. et al. Siderophore-based microbial adaptations to iron scarcity across the eastern Pacific Ocean. Proc. Natl. Acad. Sci. 113, 14237–14242 (2016).
Cherkupally, P. et al. An efficient solid-phase strategy for total synthesis of naturally occurring amphiphilic marine siderophores: Amphibactin–T and moanachelin ala-B. Org. Biomol. Chem. 13, 4760–4768 (2015).
Vraspir, J. M., Holt, P. D. & Butler, A. Identification of new members within suites of amphiphilic marine siderophores. Biometals 24, 85–92 (2011).
Martinez, J. S. et al. Structure and membrane affinity of a suite of amphiphilic siderophores produced by a marine bacterium. Proc. Natl. Acad. Sci. 100, 3754–3759 (2003).
Gledhill, M. et al. Production of siderophore type chelates by mixed bacterioplankton populations in nutrient enriched seawater incubations. Mar. Chem. 88, 75–83 (2004).
Bossé, R., Patton, W. F. & Roby, P. (Google Patents, 2014).
Walker, L. R. et al. Unambiguous identification and discovery of bacterial siderophores by direct injection 21 Tesla Fourier transform ion cyclotron resonance mass spectrometry. Metallomics 9, 82–92 (2017).
Mawji, E. et al. Hydroxamate siderophores: Occurrence and importance in the Atlantic Ocean. Environ. Sci. Technol. 42, 8675–8680 (2008).
Bundy, R. M. et al. Distinct siderophores contribute to iron cycling in the mesopelagic at station ALOHA. Front. Mar. Sci. 5, 61 (2018).
Wilson, B. R., Bogdan, A. R., Miyazawa, M., Hashimoto, K. & Tsuji, Y. Siderophores in iron metabolism: from mechanism to therapy potential. Trends Mol. Med. 22, 1077–1090 (2016).
Schalk, I. J. Siderophore-antibiotic conjugates: Exploiting iron uptake to deliver drugs into bacteria. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Diseases 24, 801 (2018).
Tillotson, G. S. Trojan horse antibiotics—A novel way to circumvent gram-negative bacterial resistance?. Infect. Diseases Res. Treat. 9, IDRT.S31567 (2016).
Miethke, M. & Marahiel, M. A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 71, 413–451 (2007).
Butler, A. & Theisen, R. M. Iron (III)–siderophore coordination chemistry: Reactivity of marine siderophores. Coord. Chem. Rev. 254, 288–296 (2010).
Geerlings, P., De Proft, F. & Langenaeker, W. Conceptual density functional theory. Chem. Rev. 103, 1793–1874. https://doi.org/10.1021/cr990029p (2003).
Cohen, M. H. & Wasserman, A. On the foundations of chemical reactivity theory. J. Phys. Chem. A 111, 2229–2242. https://doi.org/10.1021/jp066449h (2007).
Fukui, K. The role of frontier orbitals in chemical reactions (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 21, 801–809 (1982).
Yang, W. & Parr, R. G. Hardness, softness, and the Fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci. 82, 6723–6726 (1985).
Fukui, K., Yonezawa, T. & Nagata, C. Frontier Orbitals and Reaction Paths: Selected Papers of Kenichi Fukui 43–53 (World Scientific, Singapore, 1997).
Weller, M., Overton, T., Armstrong, F. & Rourke, J. Inorganic Chemistry (Oxford University Press, Oxford, 2018).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A Crystal Phys. Diffract. Theoret. General Crystallogr. 32.5, 751–767 (1976).
Ahrens, L. H. The use of ionization potentials Part 1. Ionic radii of the elements. Geochim. Cosmochim. Acta 2.3, 155–169 (1952).
Choi, J., Park, H. & Hoffmann, M. R. Effects of single metal-ion doping on the visible-light photoreactivity of TiO2. J. Phys. Chem. C 114.2, 783–792 (2010).
Pearson, R. G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. 83, 8440–8441 (1986).
Shankar, R., Senthilkumar, K. & Kolandaivel, P. Calculation of ionization potential and chemical hardness: a comparative study of different methods. Int. J. Quantum Chem. 109, 764–771 (2009).
Pearson, R. G. Hard and soft acids and bases. J. Am. Chem. Soc. 85, 3533–3539 (1963).
Hem, J. D. Study and Interpretation of the Chemical Characteristics of Natural Water Vol. 2254 (Department of the Interior, US Geological Survey, Reston, 1985).
Klewicki, J. K. & Morgan, J. J. Kinetic behavior of Mn(III) complexes of pyrophosphate, EDTA, and citrate. Environ. Sci. Technol. 32, 2916–2922. https://doi.org/10.1021/es980308e (1998).
Takashima, T., Hashimoto, K. & Nakamura, R. Mechanisms of pH-dependent activity for water oxidation to molecular oxygen by MnO2 electrocatalysts. J. Am. Chem. Soc. 134, 1519–1527. https://doi.org/10.1021/ja206511w (2012).
Marino, T., Russo, N. & Toscano, M. Gas-phase metal ion (Li+, Na+, Cu+) affinities of glycine and alanine. J. Inorg. Biochem. 79, 179–185 (2000).
Belcastro, M., Marino, T., Russo, N. & Toscano, M. Interaction of cysteine with Cu2+ and group IIb (Zn2+, Cd2+, Hg2+) metal cations: a theoretical study. J. Mass Spectrom. 40, 300–306 (2005).
Mary, C. P. V., Vijayakumar, S. & Shankar, R. Metal chelating ability and antioxidant properties of Curcumin-metal complexes—A DFT approach. J. Mol. Graph. Model. 79, 1–14 (2018).
Cowan, J. A. Inorganic Biochemistry: An Introduction (Wiley, New York, 1997).
Ratledge, C. Iron metabolism and infection. Food Nutr. Bull. 28, S515–S523 (2007).
Amin, S. A., Green, D. H., Küpper, F. C. & Carrano, C. J. Vibrioferrin, an unusual marine siderophore: Iron binding, photochemistry, and biological implications. Inorg. Chem. 48, 11451–11458. https://doi.org/10.1021/ic9016883 (2009).
Amaral, L., Martins, A., Spengler, G. & Molnar, J. Efflux pumps of Gram-negative bacteria: What they do, how they do it, with what and how to deal with them. Front. Pharmacol. 4, 168 (2014).
Minato, Y. et al. Roles of the sodium-translocating NADH: Quinone oxidoreductase (Na+-NQR) on Vibrio cholerae metabolism, motility and osmotic stress resistance. PLoS ONE 9, e97083 (2014).
Hayashi, M., Nakayama, Y. & Unemoto, T. Recent progress in the Na+-translocating NADH-quinone reductase from the marine Vibrio alginolyticus. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1505, 37–44 (2001).
Schröder, I., Johnson, E. & De Vries, S. Microbial ferric iron reductases. FEMS Microbiol. Rev. 27, 427–447 (2003).
Kracke, F., Vassilev, I. & Krömer, J. O. Microbial electron transport and energy conservation—The foundation for optimizing bioelectrochemical systems. Front. Microbiol. 6, 575 (2015).
Ernst, J. & Winkelmann, G. Enzymatic release of iron from sideramines in fungi. NADH: Sideramine oxidoreductase in Neurospora crassa. Biochimica et Biophysica Acta (BBA)-General Subjects 500, 27–41 (1977).
Pierre, J., Fontecave, M. & Crichton, R. Chemistry for an essential biological process: The reduction of ferric iron. Biometals 15, 341–346 (2002).
Bird, L. J., Bonnefoy, V. & Newman, D. K. Bioenergetic challenges of microbial iron metabolisms. Trends Microbiol. 19, 330–340 (2011).
García, B. et al. Hydroxamic acids as weak base indicators: Protonation in strong acid media. J. Org. Chem. 66, 7986–7993 (2001).
Brown, D. A. et al. Conformational behaviour of hydroxamic acids: Ab initio and structural studies. J. Chem. Soc. Perkin Trans. 2(12), 2673–2679 (1996).
Beccia, M. R. Hydroxamic Acids Interactions with Metals in Aqueous and Micellar Media: A Mechanistic Study of Complexation Reactions and Metallacrown Formation. Ph.D Thesis, Università degli studi di Pisa (2012).
Bucalo, L., Schwendner, S., Wirtz, U. & Sreedharan, S. (Google Patents, 2007).
Goss, C. H. et al. Gallium disrupts bacterial iron metabolism and has therapeutic effects in mice and humans with lung infections. Sci. Transl. Med. 10, eaat7520. https://doi.org/10.1126/scitranslmed.aat7520 (2018).
DeLeon, K. et al. Gallium maltolate treatment eradicates Pseudomonas aeruginosa infection in thermally injured mice. Antimicrob. Agents Chemother. 53, 1331–1337 (2009).
Banin, E. et al. The potential of desferrioxamine-gallium as an anti-Pseudomonas therapeutic agent. Proc. Natl. Acad. Sci. 105, 16761–16766 (2008).
da Silva, J. G., Azzolini, L. S., Wardell, S. M., Wardell, J. L. & Beraldo, H. Increasing the antibacterial activity of gallium (III) against Pseudomonas aeruginosa upon coordination to pyridine-derived thiosemicarbazones. Polyhedron 28, 2301–2305 (2009).
Rzhepishevska, O. et al. The antibacterial activity of Ga3+ is influenced by ligand complexation as well as the bacterial carbon source. Antimicrob. Agents Chemother. 55, 5568–5580 (2011).
Mjos, K. D., Cawthray, J. F., Polishchuk, E., Abrams, M. J. & Orvig, C. Gallium (III) and iron (III) complexes of quinolone antimicrobials. Dalton Trans. 45, 13146–13160 (2016).
Coleman, M. et al. In vitro antimicrobial activity of gallium maltolate against virulent Rhodococcus equi. Vet. Microbiol. 146, 175–178 (2010).
Halwani, M. et al. Co-encapsulation of gallium with gentamicin in liposomes enhances antimicrobial activity of gentamicin against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 62, 1291–1297 (2008).
Ejim, L. et al. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat. Chem. Biol. 7, 348 (2011).
Miodragović, D. U. et al. Interesting coordination abilities of antiulcer drug famotidine and antimicrobial activity of drug and its cobalt (III) complex. J. Inorg. Biochem. 100, 1568–1574 (2006).
Chang, E. L., Simmers, C. & Knight, D. A. Cobalt complexes as antiviral and antibacterial agents. Pharmaceuticals 3, 1711–1728 (2010).
Veeralakshmi, S., Nehru, S., Arunachalam, S., Kumar, P. & Govindaraju, M. Study of single and double chain surfactant–cobalt (III) complexes and their hydrophobicity, micelle formation, interaction with serum albumins and antibacterial activities. Inorg. Chem. Front. 1, 393–404 (2014).
McAuley, A. Inorganic Reaction Mechanisms Vol. 6 (Royal Society of Chemistry, London, 2007).
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J. A., Jr., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ö., Foresman, J. B., Ortiz, J. V., Cioslowski, J. & Fox, D. J. Gaussian 09, revision E. 01. (2009).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098 (1988).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Physical review B 37, 785 (1988).
Perdew, J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 33, 8822 (1986).
Salomon, O., Reiher, M. & Hess, B. A. Assertion and validation of the performance of the B3LYP⋆ functional for the first transition metal row and the G2 test set. J. Chem. Phys. 117, 4729–4737 (2002).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoret. Chem. Acc. 120, 215–241 (2008).
Zhao, Y. & Truhlar, D. G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 125, 194101 (2006).
Cramer, C. J. & Truhlar, D. G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 11, 10757–10816 (2009).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Pritchard, B. P., Altarawy, D., Didier, B., Gibson, T. D. & Windus, T. L. New basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 59, 4814–4820 (2019).
Boys, S. F. & Bernardi, F. D. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 19, 553–566 (1970).
Tomasi, J., Mennucci, B. & Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094 (2005).
Takano, Y. & Houk, K. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 1, 70–77 (2005).
Acknowledgements
This work was funded in part by the following: the National Research Foundation (NRF) (Blue Skies 120386) and the University of KwaZulu-Natal (South Africa); Marató TV3 foundation 2018 (#20183530), MINECO, (RTI2018-093831-B-100), and the Generalitat de Catalunya (2017 SGR 1439) (Spain). The authors acknowledge the Centre for High Performance Computing (CHPC), South Africa, for providing computational resources to this research project. The authors thank the anonymous reviewer for the valuable comments which helped to improve the manuscript.
Author information
Authors and Affiliations
Contributions
V.K. conceived the project, performed the calculations and wrote the manuscript. A.S. performed a critical reading of the manuscript. F.A. and B.G.T. proposed the project, reviewed the manuscript and provided support.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaipanchery, V., Sharma, A., Albericio, F. et al. Insights into the chemistry of the amphibactin–metal (M3+) interaction and its role in antibiotic resistance. Sci Rep 10, 21049 (2020). https://doi.org/10.1038/s41598-020-77807-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-77807-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
