
Abstract
Cryptosporidiosis, caused by protozoan parasites of the genus Cryptosporidium, is estimated to rank as a leading cause in the global burden of neglected zoonotic parasitic diseases. This diarrheal disease is the second leading cause of death in children under 5 years of age. Based on the C. parvum transcriptome data, glutathione transferase (GST) has been suggested as a drug target against this pathogen. GSTs are diverse multifunctional proteins involved in cellular defense and detoxification in organisms and help pathogens to alleviate chemical and environmental stress. In this study, we performed genome-wide data mining, identification, classification and in silico structural analysis of GSTs in fifteen Cryptosporidium species. The study revealed the presence three GSTs in each of the Cryptosporidium species analyzed in the study. Based on the percentage identity and comprehensive comparative phylogenetic analysis, we assigned Cryptosporidium species GSTs to three new GST classes, named Vega (ϑ), Gamma (γ) and Psi (ψ). The study also revealed an atypical thioredoxin-like fold in the C. parvum GST1 of the Vega class, whereas C. parvum GST2 of the Gamma class and C. melagridis GST3 of the Psi class has a typical thioredoxin-like fold in the N-terminal region. This study reports the first comparative analysis of GSTs in Cryptosporidium species.
Similar content being viewed by others
Introduction
Cryptosporidiosis is a zoonotic parasitic disease that is caused by Cryptosporidium spp.1,2,3. This disease is estimated to be among the highest ranking causes in the global burdens of zoonotic parasitic disease, with an estimate of 8.37 million disability-adjusted life years2,4. Recently, large population studies revealed that cryptosporidiosis has become a fast-growing burden to children under the age of 5 years5,6. Moreover, the Global Enteric Multicenter Study (GEMS) showed that Cryptosporidium is significantly associated with diarrheal disease among children < 24 months of age in sub-Saharan Africa and South Asia5. Similar studies also found Cryptosporidium to be the second leading cause of moderate to severe diarrhea in infants after Rotavirus6. It is interesting to note that vaccines/treatment are already available or fast being developed for three of four diarrheal pathogens (Rotavirus, Shigella and heat-stable, enterotoxigenic Escherichia coli), the exception being Cryptosporidium, highlighting the need to address this disease7. Despite the global burden of cryptosporidiosis, to date nitazoxanide (NTZ) is the only treatment available for this disease. NTZ only appears to be effective in patients with a good immune response, whilst having limited efficacy in malnourished children and ineffective in immunocompromised people8,9,10. The lack of effective treatment for cryptosporidiosis, coupled with the fact that it is now considered the most common cause of human parasitic diarrhea in the world, highlights the need for more research on Cryptosporidium to identify new drug targets and thus develop new drugs11.
Cryptosporidiosis is typically characterized by nausea, profuse watery diarrhea, abdominal cramps, vomiting and low-grade fever, which manifest after 14 days and last up to 2.5 months in immune-competent patients12,13. These symptoms are usually self-limiting in immune-competent patients; however, in immunocompromised hosts they can be devastating, with the disease manifesting as life-threatening and often becoming extraintestinal13. The gastrointestinal infection can spread to other sites, such as the gall bladder, biliary tract, pancreas and pulmonary system. Cryptosporidiosis can be contracted through the fecal–oral route, through contact with infected animals or humans or contaminated food or water13.
Of the Cryptosporidium species that exist, C. hominis and C. parvum are responsible for the highest level of clinically relevant infections worldwide3. The remaining species have mild zoonotic properties causing moderate-to-severe diarrhea in humans3. Cryptosporidium species are reported to have an efficient defense mechanism that allows it to cope with a wide range of environmental stresses such as changes in temperature, drugs, free radicals, as well as the host’s immune responses at various life stages12. Genome analysis of C. parvum revealed that it contains various defense proteins such as glutathione transferase (GST), glutathione peroxidase and superoxide dismutase, which are known for detoxification, signal modulation and aromatic amino acid catabolism14. The existence of these enzymes may provide C. parvum with the abilities to maintain its parasitic lifecycle, enabling it to survive and persist in its host.
Among the above-mentioned enzymes, GST is found to be expressed in all stages of the C. parvum parasite’s life cycle15, thus making it a promising therapeutic target16. GSTs have been studied as drug targets against infectious agents and metabolic disorders17,18,19. GSTs are a diverse group of multifunctional proteins that are distributed ubiquitously in eukaryotes and prokaryotes20,21. These enzymes play an important role in cellular defense and detoxification20,22,23. They catalyze the nucleophilic conjugation of the reduced tripeptide glutathione (GSH) thiol group to the electrophilic substrates to convert them to less harmful, more soluble compounds. Based on the location, the GST superfamily is divided into three sub-families namely, soluble or cytosolic GSTs, mitochondrial GSTs and membrane-associated proteins involved in eicosanoid and gluthatione metabolism (MAPEG) with the cytosolic GSTs being the most characterized (Table S1). The GSTs are generally divided into classes based on amino acid sequence similarity, with GSTs within each class sharing similar immunological cross-reactivity and specificity towards the electrophilic substrate and sensitivity to inhibitors20,24,25. GSTs within each class typically share as little as 60% amino acid sequence identity; however, some classes can share from as little as 40%20,23,26,27,28. It is generally accepted that the assignment of different GSTs to specific classes must fall within these limits, with sequences sharing less than 25–30% designated to their own class20,23,26,27,28. Information on different GST classes found in organisms, their cellular localization and functions are listed in Table S1.
Typical GSTs are dimeric in structure and each monomer is divided into two domains20,23. The N-terminal domain of conical GSTs assumes a topology resembling the thioredoxin fold with a βαβ-ββα motif. This domain also houses an important conserved region of the active site where a catalytically active Tyr, Ser or Cys is found to interact with the GSH thiol group. The C-terminal domain of typical GSTs is all helical and connected by a short linker sequence called the cis-Pro loop with a highly-conserved proline residue in cis conformation23. The active site is comprised of the glutathione binding site (G-site) and the hydrophobic substrate binding site (H-site), located in the N- terminal and C-terminal domain respectively. The G-site exclusively binds glutathione and is highly conserved, whilst the H-site accepts more variability so to accommodate an extensive range of toxic electrophilic substances20,23.
Despite the importance of GSTs, especially as a potential drug target against Cryptosporidium16, to the best of our knowledge, no literature is available to date on Cryptosporidium GSTs with regards to their distribution, the GST classes and structural information. Thus, this study is aimed at addressing this research gap. In this study, genome data mining, identification, phylogenetic and structural analysis of GSTs in fifteen Cryptosporidium species has been carried out.
Methods
Species and database
Cryptosporidium species genomes that are available for public use at the Cryptosporidium database or CryptoDB29 (https://cryptodb.org/cryptodb/app; release 48 beta, 27 August 2020; accessed on 14 September 2020) and at National Center for Biotechnology information (NCBI)30 (https://www.ncbi.nlm.nih.gov/datasets/genomes/?txid=5806; accessed on 14 September 2020) were used in the study. The Cryptosporidium pathogens examined in this study include ones from both humans and other mammals (Table 1).
Genome data mining, identification and classification of GSTs
Cryptosporidium species genomes available at CryptoDB29 were mined for GSTs. Two different methods followed for GST mining. First, the genomes of Cryptosporidium species were mined using the term “glutathione transferase”. Second, the species genomes were blasted with GST proteins from Homo sapiens (protein ID: P08263)53 and C. parvum Iowa II (protein ID: EAK89476.1)14,38. The BLASTP mined proteins revealed a range of apicomplexan species which were filtered out to show only Cryptosporidium species. The hit proteins were then collected and subjected to protein family analysis using the Pfam54 and InterPro55 programs. The results were analyzed and the hit proteins that were classified as GST by Pfam (PF14497, PF13417 and, PF17172)54 and InterPro (IPR036282, IPR004045 and IPR010987)55 were selected.
For the collection of more hits, Cryptosporidium species genomes available at NCBI database30 was blasted with two GST proteins from C. andersoni 30847 (cand_012830 & cand_023790) and from C. meleagridis UKMEL1 (CmeUKMEL1_05845) that were collected from CryptoDB29. The hit proteins were screened for GSTs following the method described above.
A final total count was presented by deleting the duplicated GSTs. The selected GSTs were then grouped into different classes or groups based on their percentage identity, following the conventional criterion of less than 25–30% identity being a new class20,23,26,27,28.
Analysis of homology
The percentage identity between GSTs was deduced using Clustal Omega56. The full-length GSTs were subjected to Clustal analysis which produced the percentage identity amongst each of the proteins as matrix identity results. These results were laid out in an Excel spreadsheet where the results were analyzed to identify the percentage identity between GSTs.
Collection of different GST classes’ protein sequences
For comparative analysis, GST protein sequences belonging to different GST classes were collected using multiple methods to build a library for phylogenetic analysis. On the European Molecular Biology Laboratory (EMBL) site57, GSTs sequences that are placed under the GST superfamily (IPR040079) were retrieved. The GST classes namely CLIC (IPR002946), Alpha (IPR003080), Mu class (IPR003081), Pi (IPR003082), Omega (IPR005442), Zeta (IPR005955) and Sigma (IPR003083) were collected under EMBL. More sequences were obtained through text search using the UniProt protein knowledge base58. A specific GST class was searched on the site and the hits obtained were further verified using Pfam54 and InterPro55 to ensure uniformity with the GSTs collected from the EMBL site57. The remaining GSTs that were not in the databases were retrieved from published articles.
The Cryptosporidium species GST sequences along with protein sequences of different GST classes used in the phylogenetic analysis are presented in Supplementary Dataset 1.
Phylogenetic analysis
The GST sequences in supplementary dataset 1 were used to make a phylogenetic tree for inferring their evolutionary relationship. First, all the GST protein sequences were aligned by MAFFT v6.864 embedded on the Trex-online server59. Then, the alignment was automatically submitted to the server for inferring the tree with different models and the optimized tree was selected. Finally, the tree was submitted to iTOL for viewing and annotation60. Thioredoxin from Oryctolagus cuniculus (protein ID: P08628) was used as an outgroup.
For the construction of the phylogenetic tree of the Cryptosporidium GST proteins, the protein sequences were aligned using MUSCLE software61 embedded in MEGA762. The evolutionary history was inferred by using the maximum likelihood method with 100 bootstrap replication based on the JTT matrix-based model63. Evolutionary analyses were conducted in MEGA7.
Cellular localization and transmembrane helices prediction
Cellular localization of GSTs was predicted using the Bologna Unified Subcellular Component Annotator (BUSCA)64. BUSCA is the latest, accurate program available for the prediction of proteins’ subcellular localization; it integrates different computational tools such as identifying signal and transit peptides (DeepSig and TP-pred3), GPI-anchors (PredGPI) and transmembrane domains (ENSEMBLE3.0 and BetAware) with tools for discriminating subcellular localization of both globular and membrane proteins (BaCelLo, MemLociand SChloro)64. The outcomes of these different programs were processed and integrated to predict subcellular localization of both eukaryotic and bacterial proteins64. Prediction of transmembrane helices in GSTs was done using TMHMM Server v. 2.065. This program is well known for its high degree of accuracy in the prediction of transmembrane helices and discrimination between soluble and membrane proteins.
Template identification
To construct 3D models of proteins, reference protein structures previously solved by crystallization or Nuclear Magnetic Resonance are needed. These would serve to simulate not only the fold of a protein but also a full atom model to build. These proteins are referred to as templates. Either single or multiple templates can be used in constructing the 3D model of a protein66. In this study, three different web servers, namely NCBI BLAST (v2.10.1)67, i-TASSER (v5.1)68 and PHYRE (v2.0)69, were consulted to identify the most suitable templates for GST proteins. Based on the highest percentage identity and sequence coverage, the best templates were selected for modeling each GST protein. In cases where the templates had the same percentage identity and sequence coverage, we selected the template with the highest resolution for modelling.
Protein sequence alignment for modeling
T-COFFEE webserver70 was used for aligning the GST proteins and the template sequences. The aligned files were downloaded in FASTA format and modified to generate files to be used for protein modelling71.
Protein modeling, optimization and validation
The MODELLER v9.21 program71 was used to build GST models. Multiple structures were produced by Modeller 9.21. The model with the best DOPE assessment was selected as the output structure to be used. The structures modeled were viewed using PyMOL72. The model for each GST was then subjected to evaluation for stereochemistry and energetic quality at the Structural Analysis and Verification Server (http://servicesn.mbi.ucla.edu/SAVES/) and ProsaII (https://prosa.services.came.sbg.ac.at/)73. Based on the validation results, the protein models were then refined on the GalaxyWeb Refiner server74. After refinement, the models were again subjected to evaluation and validation using programs such as ERRAT75, Verify3D76, PROCHECK77,78, and RAMPAGE79 and ProsaII73.
Results and discussion
Two different sizes of GSTs present in Cryptosporidium species
Genome data mining of 15 Cryptosporidium species revealed the presence of 3 GST genes in each of the species genomes (Table 2). The presence of more than one GST gene is common in eukaryotic species23. Among 45 GSTs, 30 were found to have the characteristic GST motifs20,27, such as the N-terminal domain, which houses the G site, and C terminal domain, which determines the substrate specificity (H-site) (Table 2 and Fig. S1). The remaining 15 GSTs have one of the characteristics GST motifs indicating either these sequences are diverse or fragmented or not properly annotated (Table 2). These GSTs were considered incomplete and were not included for further analysis unless indicated. Future genome editing and better gene prediction programs will help in getting the complete sequences for these GSTs and possibly predicting characteristic N- and C-terminal motifs. In total, 30 GSTs were taken for further analysis. Analysis of GST protein sizes revealed the presence of two different lengths of GSTs in Cryptosporidium species (Table 2). One type of GST protein is shorter in size with amino acids ranging between 157 and 268, and another type of GST protein is longer in size, with amino acids ranging between 373 and 466 (Table 2). GSTs from Cryptosporidium species seem to be the longest in amino acid length, as most of the GSTs reported in other organisms to date are 200–250 amino acids in length23. Furthermore, it can be noted that the addition in length is found only on the outer N- and C-terminal regions, with the center of the protein containing the GST-superfamily domains (Table 2). In order to assess whether Cryptosporidium species GST proteins are indeed properly annotated gene products, we further analyzed the gene structure. Interestingly, all the longer GSTs had a single exon, thus no introns, but shorter GSTs were the products of 1–4 exons (Table 2). This could be indicative of shorter GSTs being prone to having multiple isoforms owing to gene shuffling. Due to presence of these multiple introns, the production of more diverse short GSTs can be expected compared to longer GSTs80.
Cryptosporidium species GSTs are cytosolic in nature
Most of the GSTs identified in organisms are cytosolic in nature, with the exception of GSTs belonging to the classes MAPEG and Kappa (mitochondrial) (Table S1). In order to identify the cellular localization, we subjected Cryptosporidium species GST protein sequences to the TMHMM Server v. 2.0 for the prediction of transmembrane helices in their structure65 and the BUSCA server64 for identifying possible localization in a cell. TMHMM prediction revealed that none of the Cryptosporidium species GSTs had transmembrane helices, indicating they were soluble and thus possibly cytosolic (Table S2). To authenticate our results, we also subjected 395 GSTs belonging to 17 different classes to TMHMM prediction (Table S3). The TMHMM predicted the presence of no transmembrane helices in previously designated cytosolic GSTs, whereas transmembrane helices were predicted for previously designated microsomal GSTs (Table S3). This indicated that the TMHMM results on the prediction of no transmembrane helices in Cryptosporidium species GSTs were in agreement with previous annotations. Furthermore, BUSCA indicated that all 30 Cryptosporidium species GSTs were cytosolic (Table S4). Based on these in silico results, we concluded that the 30 Cryptosporidium species GSTs were cytosolic in nature.
Cryptosporidium species GSTs belongs to new classes
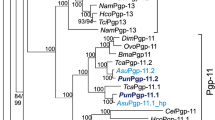
Phylogenetic analysis of Cryptosporidium species GSTs revealed that the 30 GSTs could be grouped into three different groups (Fig. 1). The shorter GSTs were grouped together (Group 1) and so were the longer GSTs (group 2). Interestingly, despite the short amino acid length, four GSTs diverged from these two groups (Group 3) (Fig. 1). Analysis of the amino acid percentage identity among Cryptosporidium species GSTs further confirmed that they indeed belonged to three different groups. Group 1 GSTs shared an amino acid percentage identity of 54–100%, whereas groups 2 and 3 shared identities of 48–100% and 42–71%, respectively. Group 3 GSTs had 13–21% identity with Group 2 GSTs and 14–22% identity to Group 1 GSTs. The percentage identity between Groups 1 and 2 was 17–25%. This indicates that all three groups of Cryptosporidium species GSTs indeed belonged to three different classes as the percentage identity between these groups was below 25–30%, qualifying them to be their own class20,23,26,27,28.

Phylogenetic analysis of glutathione transferase (GST) proteins from Cryptosporidium species. The evolutionary history was inferred by using the maximum likelihood method based on the JTT matrix-based model63. Evolutionary analyses were conducted in MEGA762. The percentage of trees (bootstrap value) in which the associated taxa clustered together is shown next to the branches.
Although the above results clearly indicated that Cryptosporidium species GSTs belong to three different groups, it was still not clear whether they fell under one of the GST classes described in the literature (Table S1). Thus, the comprehensive phylogenetic analysis of proteins belonging to 17 known GST classes and Cryptosporidium species GSTs was carried out (Fig. 2). Phylogenetic analysis revealed that Cryptosporidium species GSTs did not align with any of the 17 pre-existing GST classes and formed three new groups (Fig. 2). This clearly indicates that Cryptosporidium species GSTs belong to three different new GST classes. Thus, we named groups 1, 2 and 3 of Cryptosporidium GSTs Vega (ϑ), Gamma (γ) and Psi (ψ), respectively. A point to be noted is that all the GST proteins aligned together as per their GST class on the phylogenetic tree, indicating our phylogenetic analysis is correct and thus we conclude that Cryptosporidium species GSTs indeed belong to new GST classes.

Phylogenetic tree of the glutathione transferases (GSTs) protein sequences of Cryptosporidium species with GSTs from 17 different GST classes. Thioredoxin from Oryctolagus cuniculus (protein ID: P08628) is used as an outgroup. Three new GST classes reported in this study from Cryptosporidium species named Vega, Gamma and Psi are also shown in the tree. A high-resolution phylogenetic tree is provided in Supplementary Dataset 2.
Cryptosporidium parvum GST1 of Vega class has atypical thioredoxin-like fold
Identification of three new GST classes in Cryptosporidium species in this study necessitated examination of the structural aspects of these new classes to see if any deviations or novel folds might be present, compared to the canonical structure of GSTs20,27. Analysis of the primary structure revealed that all Cryptosporidium species GSTs have N- and C-terminal regions characteristic of GSTs that usually contain a G-site and H-site20,27, respectively (Table 2 and Fig. S1). All GSTs have the highly conserved proline amino acid residue (Fig. S1) that is part of the cis-Pro loop responsible for connecting the N- and C-terminal regions in order to maintain the GST structural integrity81. It was observed from Fig. S1 that Psi class GSTs have a Tyr residue in the N-terminal domain in close proximity to the expected active site Tyr. The same was observed with the Vega class GSTs with the expectation of C. muris and C. baileyi. Vega and Psi GSTs have a few tyrosine residues in the N-terminal region, but they are not at a position that is considered part of an active site20,27 (Fig. S1). Similarly, the majority of the Gamma class GSTs consist of an active site Tyr residue with the exception C. andersoni, C. baileyi, C. ryanae and C. bovis species. In these species, Phe replaces the active site Tyr residue. Mutagenesis studies have shown that the presence of Phe at the supposed position of the active site Tyr significantly reduces the catalytic activity. This highlights the critical role played by the active site Tyr in the catalytic activity of GST82,83. The effect of these mutations in the context of Cryptosporidium GSTs is yet to be studied.
Multiple sequence alignments of Vega and Gamma GSTs revealed that amino acids in the N- and C-terminal regions of these GSTs are highly conserved (Fig. S1). For this reason, we selected C. parvum GSTs 1 and 2 (CpGST1 and CpGST2) as representative of the Vega and Gamma GST classes for structural analysis along with C. meleagridis UKMEL1 GST3 (CmGST3) for the Psi class. Structural analysis of the three GSTs was carried out using in silico homology modeling. The structural analysis was aimed at assessing only the secondary structural elements that are characteristic of GST proteins20,27. These GST models are not aimed to assess the binding affinities or the residues involved in binding to different ligands. In order to build 3D models we performed a template search at three different webpages, namely NCBI67, PHYRE69 and I-TASSER68. The templates found were of low sequence identity but had relatively good coverage (Table S5). This was expected, since these GSTs are new. We then proceeded to build 3D models using a multiple template method, as this approach is known to improve the quality of homology models84. We built 3D models for all three GSTs, attempting single and multiple templates, while also using different combinations of the available templates listed in Table S5. The best 3D models with good quality closest to the templates were chosen for the structural analysis.
Here, we present the combination of templates that gave CpGST1, CpGST2 and CmGST3 models. The templates used to model CpGST1 were a Bombyx mori Sigma class GST (3VPQ-A)85 that had 94% coverage and 26% identity and a Penaeus vannamei Mu class (5AN1-A)86 with 98% coverage and 23% identity (Fig. 3 and Table S5). For CpGST2 the templates were both from Homo sapiens Alpha class (1K3Y-B)87 and Pi class (19GS-A)88, with sequence identity at 21%, coverage at 94% and 22% identity and 84% coverage (Fig. 4 Table S5), respectively. The CmGST3 templates used were from Caenorhabditis elegans Pi class GST (1ZL9-A) (https://www.rcsb.org/structure/1ZL9) with 94% coverage and 21% identity and a Homo sapiens Alpha class (1K3Y-B)87 with 98% coverage and 22% identity (Fig. 5 and Table S5).

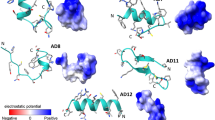
In silico structural analysis of Vega class representative Cryptosporidium parvum glutathione transferase 1 (CpGST1). 3D model of CpGST1 (A) and its amino acid sequence alignment with templates (B). Secondary structural annotations were done as per modeled structure where α-helices and corresponding amino acids are colored in red while the β-sheets and their corresponding amino acids are colored in yellow. The active-site tyrosine and the cis-proline residues are boxed in purple and grey respectively. The template Protein Data Bank codes, 3VPQ-A and 5AN1-A, represents GST protein crystal structures from Bombyx mori (Sigma class GST) and Penaeus vannamei (Mu class GST).

In silico structural analysis of Gamma class representative Cryptosporidium parvum glutathione transferase 2 (CpGST2). 3D model of CpGST2 (A) and its amino acid sequence alignment with templates (B). Secondary structural annotations were done as per modeled structure where α-helices and corresponding amino acids are colored in red while the β-sheets and their corresponding amino acids are colored in yellow. The active-site tyrosine and the cis-proline residues are boxed in purple and grey respectively. The template Protein Data Bank codes, 1K3Y-B and 19GS-A, represents GST protein crystal structures of Alpha class (1K3Y-B) and Pi class (19GS-A) GSTs from humans.

In silico structural analysis of Psi class representative Cryptosporidium meleagridis strain UKMEL1 GST3 glutathione transferase 3 (CpGST3). 3D model of CmGST3 (A) and its amino acid sequence alignment with templates (B). Secondary structural annotations were done as per modeled structure where α-helices and corresponding amino acids are colored in red while the β-sheets and their corresponding amino acids are colored in yellow. The active-site tyrosine and the cis-proline residues are boxed in purple and grey respectively. The template Protein Data Bank codes, 1K3Y-B and 1ZL9-A, represents GST protein crystal structures from Human (Alpha class GST) and Caenorhabditis elegans (Pi class GST).
For each GST, 20 models were built using the MODELLER v9.21 program71. The best model evaluated by DOPE score was selected and subjected to structural quality analysis. The selected model for each GST was then refined on the GalaxyWeb Refiner server74 and further subjected to structural quality evaluation using different programs such as ERRAT75, Verify3D76, PROCHECK77,78, RAMPAGE79 and ProsaII73. The overall quality of the models was assessed by the combination of these programs’ values and by comparing these with the templates’ structural evaluation scores (Tables S6 and S7). The models generated for CpGST1 and CpGST2 were found to be of good quality, as different structural validation programs indicated that the quality of the model structures was close to the quality of the template structures (Tables S6 and S7). The model generated for CmGST3 had all parameters in acceptable range including Z-score of − 3.68 indicating the model is of good quality with the exception of Verify3D where 26% residues had an average 3D-1D score > 0.2 (Tables S6 and S7). The three GST models generated in the study, along with their corresponding sequence alignments with their templates, are presented in Figs. 3, 4 and 5.
Structural analysis revealed the presence of 2β-sheets and 3α-helices in the N-terminal region and 6α-helices in the C-terminal region of CpGST1 (Fig. 3). The overall structure of CpGST1 at the N-terminal domain seems completely different compared to the canonical GST N-terminal domain20,27. The N-terminal region of CpGST1 did not have the typical thioredoxin-like fold, nor did it follow the βαβ-α-ββα motif; it was rather composed of two antiparallel β-sheets and 3α-helices (Fig. 3). It is rare to find GSTs that do not possess the conventional thioredoxin βαβ-α-ββα motif. Kappa class GSTs, which are mitochondrial GSTs, are the closest GSTs that do not follow the traditional thioredoxin fold but have still been found to carry out a similar molecular function as conical GSTs89,90,91. This is also common for MAPEG GST and the mPGES-1 (microsomal ProstaGlandin E-Synthase type 1) subfamily of proteins, as they too are a group of structurally unrelated proteins with GSH transferase activities23,91. Because the GST superfamily shares such vast variations in terms of their structural conformation, this ααββα conformation of CpGST1 can be considered a unique Vega class feature.
In contrast to the CpGST1 model, the CpGST2 and CmGST3 models N-terminal domain follows the thioredoxin-like fold, which is characteristic of cytosolic enzymes in the GST superfamily20,22,27. The N-terminal domain was complete with 4β-sheets and 3α-helices following a βαβ and ββα arrangement with the two motifs linked by an α2 (Figs. 4 and 5). The C-terminal domain contains helices with each model CpGST2 and CmGST3 having a varying number of helices (Figs. 4 and 5). It has been suggested that an increase in the number of helices in the C-terminal domain, may allow for a broader substrate range and/or offer a deeper catalytic pocket that facilitates the conjugation of larger substrates92,93.
Conclusions
In this genomic era, in silico based comparative studies at genome level or at protein family level have become an important tool to uncover novel aspects in organisms. This study is such an example, where genomes of Cryptosporidium species were mined for glutathione transferases (GSTs), enzymes playing a key role in cellular defense and detoxification that are also a potential drug target against pathogens and metabolic disorders. Analysis revealed an interesting feature, namely the presence of two different sizes of GSTs (short and long) in these species. The longer GST proteins were found to be longer than the GSTs found in other organisms, with the size attributed to C- and N-terminal extensions. One of the major findings of the study is the identification of GSTs belonging to three new GST classes in Cryptosporidium species. In addition, Cryptosporidium parvum GST1 had an atypical thioredoxin fold in the N-terminal region with an αα-ββ-α motif rather than the typical thioredoxin-like fold with a βαβ-α-ββα motif. Future study includes functional and structural (X-ray or NMR) characterization of Cryptosporidium species GSTs. The study results serve as reference for future mining and annotation of GSTs Cryptosporidium species.
References
Savioli, L., Smith, H. & Thompson, A. Giardia and Cryptosporidium join the ‘neglected diseases initiative’. Trends Parasitol. 22, 203–208 (2006).
Pisarski, K. The global burden of disease of zoonotic parasitic diseases: Top 5 contenders for priority consideration. Trop. Med. Infect. Dis. 4, 44 (2019).
Pumipuntu, N. & Piratae, S. Cryptosporidiosis: A zoonotic disease concern. Vet. World 11, 681 (2018).
Hotez, P. J. et al. The global burden of disease study 2010: Interpretation and implications for the neglected tropical diseases. PLoS Negl. Trop. Dis. 8, e2865 (2014).
Sow, S. O. et al. The burden of Cryptosporidium diarrheal disease among children < 24 months of age in moderate/high mortality regions of sub-Saharan Africa and South Asia, utilizing data from the Global Enteric Multicenter Study (GEMS). PLoS Negl. Trop. Dis. 10, e000472 (2016).
Kotloff, K. L. et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 382, 209–222 (2013).
Striepen, B. Parasitic infections: Time to tackle cryptosporidiosis. Nat. News 503, 189 (2013).
Bhalchandra, S., Cardenas, D. & Ward, H. D. Recent breakthroughs and ongoing limitations in Cryptosporidium research. F1000Research 7, F1000 Faculty Rev-1380 (2018).
Amadi, B. et al. High dose prolonged treatment with nitazoxanide is not effective for cryptosporidiosis in HIV positive Zambian children: A randomised controlled trial. BMC Infect. Dis. 9, 195 (2009).
Cabada, M. M. & White, A. C. Jr. Treatment of cryptosporidiosis: Do we know what we think we know?. Curr. Opin. Infect. Dis. 23, 494–499 (2010).
Widmer, G. et al. Update on Cryptosporidium spp.: highlights from the Seventh International Giardia and Cryptosporidium Conference. Parasite 27, 14 (2020).
Certad, G., Viscogliosi, E., Chabé, M. & Cacciò, S. M. Pathogenic mechanisms of Cryptosporidium and Giardia. Trends Parasitol. 33, 561–576 (2017).
Leitch, G. J. & He, Q. Cryptosporidiosis-an overvie. J. Biomed. Res. 25, 1–16 (2011).
Abrahamsen, M. S. et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science 304, 441–445 (2004).
Mauzy, M. J., Enomoto, S., Lancto, C. A., Abrahamsen, M. S. & Rutherford, M. S. The Cryptosporidium parvum transcriptome during in vitro development. PLoS ONE 7, e31715 (2012).
Khoza, T., Mfeka, S. M. & Achilonu, I. Targeting Cryptosporidium GST for rational drug discovery against parasitic infectious diseases. Biochem. Mol. Biol. J. https://doi.org/10.21767/2471-8084-C4-018 (2018).
Pljesa-Ercegovac, M. et al. Glutathione transferases: Potential targets to overcome chemoresistance in solid tumors. Int. J. Mol. Sci. 19, 3785 (2018).
Harwaldt, P., Rahlfs, S. & Becker, K. Glutathione S-transferase of the malarial parasite Plasmodium falciparum: Characterization of a potential drug target. Biol. Chem. 383, 821–830 (2002).
Rao, U., Salinas, G., Mehta, K. & Klei, T. R. Identification and localization of glutathione S-transferase as a potential target enzyme in Brugia species. Parasitol. Res. 86, 908–915 (2000).
Sheehan, D., Meade, G. & Foley, V. M. Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 360, 1–16 (2001).
Allocati, N., Federici, L., Masulli, M. & Di Ilio, C. Distribution of glutathione transferases in Gram-positive bacteria and Archaea. Biochimie 94, 588–596 (2012).
Wilce, M. C. & Parker, M. W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1205, 1–18 (1994).
Frova, C. Glutathione transferases in the genomics era: New insights and perspectives. Biomol. Eng. 23, 149–169 (2006).
Salinas, A. E. & Wong, M. G. Glutathione S-transferases—A review. Curr. Med. Chem. 6, 279–310 (1999).
Glisic, B. et al. Characterization of glutathione-S-transferases in zebrafish (Danio rerio). Aquat. Toxicol. 158, 50–62 (2015).
Allocati, N., Federici, L., Masulli, M. & Di Ilio, C. Glutathione transferases in bacteria. FEBS J. 276, 58–75 (2009).
Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 43, 138–151 (2011).
Soranzo, N., Gorla, M. S., Mizzi, L., De Toma, G. & Frova, C. Organisation and structural evolution of the rice glutathione S-transferase gene family. Mol. Genet. Genomics 271, 511–521 (2004).
Heiges, M. et al. CryptoDB: A Cryptosporidium bioinformatics resource update. Nucleic Acids Res. 34, D419–D422 (2006).
Agarwala, R. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 46, D8–D13 (2018).
Liu, S. et al. Evolution of mitosome metabolism and invasion-related proteins in Cryptosporidium. BMC Genomics 17, 1006 (2016).
Ifeonu, O. O. et al. Annotated draft genome sequences of three species of Cryptosporidium: Cryptosporidium meleagridis isolate UKMEL1, C. baileyi isolate TAMU-09Q1 and C. hominis isolates TU502_2012 and UKH1. FEMS Pathog. Dis. 74, ftw080 (2016).
Widmer, G., Köster, P. C. & Carmena, D. Cryptosporidium hominis infections in non-human animal species: revisiting the concept of host specificity. Int. J. Parasitol. 50, 253–262 (2020).
Guo, Y. et al. Comparative genomic analysis reveals occurrence of genetic recombination in virulent Cryptosporidium hominis subtypes and telomeric gene duplications in Cryptosporidium parvum. BMC Genomics 16, 320 (2015).
Xu, P. et al. The genome of Cryptosporidium hominis. Nature 431, 1107–1112 (2004).
Isaza, J. P. et al. Revisiting the reference genomes of human pathogenic Cryptosporidium species: Reannotation of C. parvum Iowa and a new C. hominis reference. Sci. Rep. 5, 16324 (2015).
Xiao, L., Fayer, R., Ryan, U. & Upton, S. J. Cryptosporidium taxonomy: Recent advances and implications for public health. Clin. Microbiol. Rev. 17, 72–97 (2004).
Bankier, A. T. et al. Integrated mapping, chromosomal sequencing and sequence analysis of Cryptosporidium parvum. Genome Res. 13, 1787–1799 (2003).
Ebeid, M., Mathis, A., Pospischil, A. & Deplazes, P. Infectivity of Cryptosporidium parvum genotype I in conventionally reared piglets and lambs. Parasitol. Res. 90, 232–235 (2003).
Sateriale, A. et al. Protective immunity in a genetically tractable natural mouse model of cryptosporidiosis. Available at SSRN 3362259.
Prediger, J. et al. Native and introduced squirrels in Italy host different Cryptosporidium spp. Eur. J. Protistol. 61, 64–75 (2017).
McDONALD, V., Deer, R., Uni, S., Iseki, M. & Bancroft, G. Immune responses to Cryptosporidium muris and Cryptosporidium parvum in adult immunocompetent or immunocompromised (nude and SCID) mice. Infect. Immun. 60, 3325–3331 (1992).
Uni, S., Iseki, M., Maekawa, T., Moriya, K. & Takada, S. Ultrastructure of Cryptosporidium muris (strain RN 66) parasitizing the murine stomach. Parasitol. Res. 74, 123–132 (1987).
Ng, J., Pavlasek, I. & Ryan, U. Identification of novel Cryptosporidium genotypes from avian hosts. Appl. Environ. Microbiol. 72, 7548–7553 (2006).
Pavlásek, I. The black-headed gull (Larus ridibundus L.), a new host for Cryptosporidium baileyi (Apicomplexa: Cryptosporidiidae). Vet. Med. 38, 629–638 (1993).
Elwin, K., Hadfield, S. J., Robinson, G., Crouch, N. D. & Chalmers, R. M. Cryptosporidium viatorum n. sp. (Apicomplexa: Cryptosporidiidae) among travellers returning to Great Britain from the Indian subcontinent, 2007–2011. Int. J. Parasitol. 42, 675–682 (2012).
Koehler, A. V., Wang, T., Haydon, S. R. & Gasser, R. B. Cryptosporidium viatorum from the native Australian swamp rat Rattus lutreolus—An emerging zoonotic pathogen?. Int. J. Parasitol. Parasites Wildlife 7, 18–26 (2018).
Guo, Y. et al. Subtyping novel zoonotic pathogen Cryptosporidium chipmunk genotype I. J. Clin. Microbiol. 53, 1648–1654 (2015).
Stenger, B. L. et al. North American tree squirrels and ground squirrels with overlapping ranges host different Cryptosporidium species and genotypes. Infect. Genet. Evol. 36, 287–293 (2015).
Fayer, R., Santín, M. & Trout, J. M. Cryptosporidium ryanae n. sp. (Apicomplexa: Cryptosporidiidae) in cattle (Bos taurus). Vet. Parasitol. 156, 191–198 (2008).
Fayer, R., Santín, M. & Xiao, L. Cryptosporidium bovis n. sp. (Apicomplexa: Cryptosporidiidae) in cattle (Bos taurus). J. Parasitol. 91, 624–629 (2005).
Elwin, K. & Chalmers, R. M. Contemporary identification of previously reported novel Cryptosporidium isolates reveals Cryptosporidium bovis and the cervine genotype in sheep (Ovis aries). Parasitol. Res. 102, 1103–1105 (2008).
Board, P. G. & Webb, G. C. Isolation of a cDNA clone and localization of human glutathione S-transferase 2 genes to chromosome band 6p12. Proc. Natl. Acad. Sci. 84, 2377–2381 (1987).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Mitchell, A. L. et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360 (2019).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 (2019).
Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
Boc, A., Diallo, A. B. & Makarenkov, V. T-REX: A web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res. 40, W573-579. https://doi.org/10.1093/nar/gks485 (2012).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. https://doi.org/10.1093/nar/gkh340 (2004).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Jones, D. T., Taylor, W. R. & Thornton, J. M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 (1992).
Savojardo, C., Martelli, P. L., Fariselli, P., Profiti, G. & Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 46, W459–W466 (2018).
Möller, S., Croning, M. D. & Apweiler, R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17, 646–653 (2001).
Fiser, A. In Computational Biology 73–94 (Springer, Berlin, 2010).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. https://doi.org/10.1016/s0022-2836(05)80360-2 (1990).
Yang, J. & Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181 (2015).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845 (2015).
Di Tommaso, P. et al. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39, W13-17. https://doi.org/10.1093/nar/gkr245 (2011).
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protocols Bioinform. 54, 5.6.1-5.6.37. https://doi.org/10.1002/cpbi.3 (2016).
72Schrodinger, L. The PyMOL molecular graphics system. Version 1, 0 (2010).
Wiederstein, M. & Sippl, M. J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410 (2007).
Ko, J., Park, H., Heo, L. & Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 40, W294–W297. https://doi.org/10.1093/nar/gks493 (2012).
Colovos, C. & Yeates, T. O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 2, 1511–1519. https://doi.org/10.1002/pro.5560020916 (1993).
Eisenberg, D., Luthy, R. & Bowie, J. U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 277, 396–404 (1997).
Laskowski, R., MacArthur, M. & Thornton, J. PROCHECK: Validation of protein-structure coordinates. (2006).
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 (1993).
Wang, W. et al. Data set for phylogenetic tree and RAMPAGE Ramachandran plot analysis of SODs in Gossypium raimondii and G. arboreum. Data Brief 9, 345–348. https://doi.org/10.1016/j.dib.2016.05.025 (2016).
Xu, G., Guo, C., Shan, H. & Kong, H. Divergence of duplicate genes in exon–intron structure. Proc. Natl. Acad. Sci. 109, 1187–1192 (2012).
Allocati, N. et al. Functional analysis of the evolutionarily conserved proline 53 residue in Proteus mirabilis glutathione transferase B1–1. FEBS Lett. 445, 347–350 (1999).
Liu, S., Stoesz, S. P. & Pickett, C. B. Identification of a novel human glutathioneS-transferase using bioinformatics. Arch. Biochem. Biophys. 352, 306–313 (1998).
Stenberg, G., Board, P. G. & Mannervik, B. Mutation of an evolutionarily conserved tyrosine residue in the active site of a human class alpha glutathione transferase. FEBS Lett. 293, 153–155 (1991).
Larsson, P., Wallner, B., Lindahl, E. & Elofsson, A. Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci. 17, 990–1002 (2008).
Yamamoto, K. et al. Crystal structure of a Bombyx mori sigma-class glutathione transferase exhibiting prostaglandin E synthase activity. Biochim. Biophys. Acta Gen. Subjects 1830, 3711–3718 (2013).
Juárez-Martínez, A. B., Sotelo-Mundo, R. R. & Rudiño-Piñera, E. Crystal structure of a class-mu glutathione S-transferase from whiteleg shrimp Litopenaeus vannamei: Structural changes in the xenobiotic binding H-site may alter the spectra of molecules bound. J. Biochem. Mol. Toxicol. 31, e21838 (2017).
Le Trong, I., Stenkamp, R. E., Ibarra, C., Atkins, W. M. & Adman, E. T. 1.3-Å resolution structure of human glutathione S-transferase with S-hexyl glutathione bound reveals possible extended ligandin binding site. Proteins Struct. Funct. Bioinform. 48, 618–627 (2002).
Oakley, A. J., Bello, M. L., Nuccetelli, M., Mazzetti, A. P. & Parker, M. W. The ligandin (non-substrate) binding site of human Pi class glutathione transferase is located in the electrophile binding site (H-site). J. Mol. Biol. 291, 913–926 (1999).
Atkinson, H. J. & Babbitt, P. C. Glutathione transferases are structural and functional outliers in the thioredoxin fold. Biochemistry 48, 11108–11116 (2009).
Ladner, J. E., Parsons, J. F., Rife, C. L., Gilliland, G. L. & Armstrong, R. N. Parallel evolutionary pathways for glutathione transferases: Structure and mechanism of the mitochondrial class kappa enzyme rGSTK1-1. Biochemistry 43, 352–361 (2004).
Lallement, P.-A., Brouwer, B., Keech, O., Hecker, A. & Rouhier, N. The still mysterious roles of cysteine-containing glutathione transferases in plants. Front. Pharmacol. 5, 192 (2014).
Fritz-Wolf, K. et al. X-ray structure of glutathione S-transferase from the malarial parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. 100, 13821–13826 (2003).
Ji, X., Zhang, P., Armstrong, R. N. & Gilliland, G. L. The three-dimensional structure of a glutathione S-transferase from the Mu gene class. Structural analysis of the binary complex of isoenzyme 3-3 and glutathione at 2.2-. ANG. resolution. Biochemistry 31, 10169–10184 (1992).
Acknowledgements
Mbalenhle Sizamile Mfeka thanks the Department of Science and Technology—National Research Foundation (DST-NRF), South Africa for Innovation Master’s Scholarship (Grant Numbers 112242). Khajamohiddin Syed expresses sincere gratitude to the NRF, South Africa for a research grant (Grant Number 114159) and University of Zululand (Grant number C686). Thandeka Khoza also thanks the NRF (Grant number 121275); South African Medical Research Council (SAMRC) and University of KwaZulu-Natal for research grants. The authors want to thank Barbara Bradley, Pretoria, South Africa for English language editing.
Author information
Authors and Affiliations
Contributions
K.S., I.A. and T.K. designed and conceptualized the study. K.S. and T.K. provided funding for the study. All authors are involved in generation, analysis and interpretation of data. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mfeka, M.S., Martínez-Oyanedel, J., Chen, W. et al. Comparative analyses and structural insights of new class glutathione transferases in Cryptosporidium species. Sci Rep 10, 20370 (2020). https://doi.org/10.1038/s41598-020-77233-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-77233-5
This article is cited by
-
Analysis of codon usage bias of thioredoxin in apicomplexan protozoa
Parasites & Vectors (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
