
Abstract
Taxonomy is the science of defining and naming groups of biological organisms based on shared characteristics and, more recently, on evolutionary relationships. With the birth of novel genomics/bioinformatics techniques and the increasing interest in microbiome studies, a further advance of taxonomic discipline appears not only possible but highly desirable. The present work proposes a new approach to modern taxonomy, consisting in the inclusion of novel descriptors in the organism characterization: (1) the presence of associated microorganisms (e.g.: symbionts, microbiome), (2) the mitochondrial genome of the host, (3) the symbiont genome. This approach aims to provide a deeper comprehension of the evolutionary/ecological dimensions of organisms since their very first description. Particularly interesting, are those complexes formed by the host plus associated microorganisms, that in the present study we refer to as “holobionts”. We illustrate this approach through the description of the ciliate Euplotes vanleeuwenhoeki sp. nov. and its bacterial endosymbiont “Candidatus Pinguicoccus supinus” gen. nov., sp. nov. The endosymbiont possesses an extremely reduced genome (~ 163 kbp); intriguingly, this suggests a high integration between host and symbiont.
Similar content being viewed by others

Introduction
Taxonomy is the science of defining and naming groups of biological organisms based on shared characteristics and, more recently, based on evolutionary relationships. Classical taxonomy was exclusively based on morphological-comparative techniques requiring a very high specialization on specific taxa. For this reason, and due to the development and rise of modern molecular tools, in the last decades this discipline has faced a significant period of crisis1,2. Lately, traditional taxonomy has been renewed in the so-called integrative taxonomy, which also includes ultrastructural and phylogenetic-molecular analysis1,3.
Now, in our opinion, new concepts and tools would allow taxonomy to make another step forward: to consider the host associated symbiont/microbiome, pursuing a further multidisciplinary integration with modern available technologies, such as bioinformatics and genomic analyses.
Indeed, although at present not mandatory for the description of an organism, the characterization of obligatory or occasional symbionts sensu de Bary, i.e. according to the definition of symbiosis as “the living together of two differently named organisms”4, and of its associated microbial consortium (= microbiome), could be indeed considered an important descriptor of the state of an organism, potentially influencing its development, physiology, and morphology. This opinion is in agreement with the findings of several previous studies5,6,7,8,9,10,11,12,13,14.
Based on the above reasons we are prone to consider the complex formed by the host plus associated microorganisms as a “holobiont”: Lynn Margulis defined the term “holobiont”15 as the assemblage of “two or more organisms, members of different species” (bionts), which remain associate “throughout a significant portion of the life history”16. Nowadays, the term holobiont is ambiguously defined; indeed, it can span from including only obligate mutualistic symbionts17 to somehow associated microbial consortia18,19,20,21,22,23. In our opinion, it would be appropriate to apply the concept of holobiont each time the association between different organisms gives rise to a functional unit in which emerging characteristics and properties are not typical of the different parts separately taken (e.g. symbiosis, mutualistic, neutral, or parasitic phenomena).
Therefore, the updated approach we are proposing implies the possibility to describe each of the organisms potentially associated to a host and, consequently, the need to build networks of complementary skills, able to combine bio-taxonomy tools, classical morphology, ultrastructure, molecular phylogeny, genomics, and bioinformatics. Hence, we propound to define this updated approach as “next generation taxonomy” (see “Discussion” section).
To exemplify the feasibility of the proposed approach we herein present the taxonomic description of the ciliate protist Euplotes vanleeuwenhoeki sp. nov. (Euplotia, Ciliophora) and its bacterial endosymbiont “Candidatus (Ca.) Pinguicoccus supinus” gen. nov., sp. nov. (Opitutae, Verrucomicrobia). Ciliates are known to form stable associations with eukaryotic24,25,26,27,28,29 and prokaryotic6, 30,31,32,33,34,35,36,37,38 organisms, and thus represent an ideal case of study for the proposed “next generation taxonomy”. Moreover, to the best of our knowledge, this is the first study addressing the concept of holobionts of protists in general.
In detail, we used the proposed approach combining the requirements of an integrative taxonomic description39, and some state of the art analyses, such as the host mitochondrial genome characterization, with the genomic study on the endosymbiont. Interestingly, we found that the endosymbiont “Ca. Pinguicoccus supinus” has an extremely small genome (~ 163 kbp), comparable in size to extremely reduced genomes of insect symbionts, making this bacterium the first of this category to be found in a unicellular host40,41. The extremely small genome size is suggestive of a high level of integration with the host, further indicating the appropriateness of the use of a unifying approach to ensure a suitable functional and taxonomical description of all the partners involved in such kind of symbioses.
Results
Description of Euplotes vanleeuwenhoeki sp. nov. (Figs. 1, 2, 3, 4 and 5, Table 1)

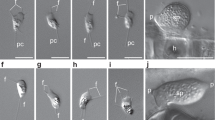
Morphology of Euplotes vanleeuwenhoeki sp. nov. (A) Schematic drawings of the dorsal side (left), ventral side (right), and nuclear apparatus (middle); (B) Live picture, ventral side. (C) Live picture, dorsal side; (D) Feulgen staining, showing macronucleus (Ma) and micronucleus (Mi); (E) Silver staining, dorsal side; (F) Silver staining, ventral side; (G–J) SEM pictures of dorsal side (G), ventral side (H), and lateral views (I,J); Arrow: paroral membrane; Arrowhead: dorsal furrow; Double arrowhead: food vacuole containing algae; AZM adoral zone of membranelles, Br bristle, CA cortical ampules, CC caudal cirri, CV contractile vacuole, FVC fronto-ventral cirri, Ma macronucleus, MC marginal cirri, Mi micronucleus, MK mid-dorsal kinety, TC transverse cirri. Bars stand for 10 µm.

TEM picture of Euplotes vanleeuwenhoeki sp. nov. (A–C) Cortex region; (D–F) Oral region; (A) Ciliary pit (CP) with dikinetid (Dk); (B) Detail of bristle pit containing filamentous material (arrowhead); cortical ampules (CA) are visible; (C) Transverse section of dikinetid (Dk) surrounded by cortical ampules; sections of the macronucleus (Ma), two mitochondria (Mt) and an endosymbiotic bacterium (arrow) are visible; (D) Detail of oral membranelles, composed of two longer rows (1, 2) and one shorter row (3) of cilia; membranelles are separated by ridges (R); (E) Section of oral region, showing paraoral membrane (Pa) in front of oral membranelles and pharingeal disks (PD); (F) Closer view of paraoral membrane and transverse section of macronucleus; (G) Endosymbiotic bacterial cells (arrow) inside Euplotes cytoplasm, nearby macronucleus and micronucleus (Mi); rosettes of glycogen (G) and lipid droplets (L) are present; (H) Detail of contractile vacuole (CV) and transverse cirrus (TC); (I) Section of cirrus (double arrowhead), close to mitochondria; (J) Two phagosomes (Ph). CA cortical ampules, CP ciliary pit, CV contractile vacuole, Dk dikinetid, G rosette of glycogen, L lipid droplet, Ma macronucleus, Mi micronucleus, Mt mitochondrion, Pa paraoral membrane, PD pharingeal disk, Ph phagosome, R ridge, TC transverse cirrus, Arrowhead: filamentous material; Arrow: endosymbiotic bacterium; Double arrowhead: transverse section of cirrus. Bars stand for 1 µm.

Phylogenetic tree of genus Euplotes based on the 18S rRNA gene. Numbers associated to nodes represent posterior probabilities and bootstrap values, respectively (only values above 0.80–75 are shown). Sequence obtained in the present work is in bold.

Mitochondrial gene map of Euplotes vanleeuwenhoeki sp. nov. The gene map of the mitochondrial genome of E. vanleeuwenhoeki in comparison with those belonging to Euplotes minuta and Oxytricha trifallax is represented. Homologous regions among the three genomes are indicated by pale coloured areas. Names of split genes are suffixed by a letter or a lowercase Roman numeral. The direction of transcription is indicated by an arrow at each end of the mitochondrial map.

TEM pictures of "Candidatus Pinguicoccus supinus". (A) Endosymbiont cells (arrow) in host cytoplasm, lying underneath the cortex, aggregated in clusters; macronucleus (Ma) and some mitochondria (Mt), also in proximity or in apparent close contact with endosymbionts, are visible; (B) Closer view of “Ca. Pinguicoccus supinus” showing inner membrane (IM) and outer membrane (OM); an invagination of the inner membrane (IM) is present (arrowhead); (C) Different cell shapes of three “Ca. Pinguicoccus supinus” specimens (arrow), from rounded to ovoid; the IM is invaginated (arrowhead); (D) Endosymbiont cell with an irregular shape, with an evident nucleoid (Nu) and an invagination of the IM is present (arrowhead); (E,F) “Ca. Pinguicoccus supinus” cells showing evagination of the outer membrane (double arrowhead); (F) Endosymbiont cell in proximity to a mitochondrion; (G) “Ca. Pinguicoccus supinus” showing emphasized folding (asterisk) of membrane area; (H) Endosymbiont cells (arrow) appear to be in close contact with lipid droplets (L); (I) “Ca. Pinguicoccus supinus” (arrow) during binary fission; the division septum is well visible. IM inner membrane, L lipid droplet, Ma macronucleus, Mt mitochondrion, Nu nucleoid, OM outer membrane. Arrow: “Ca. Pinguicoccus supinus” cell; Arrowhead: evagination of the outer membrane; Asterisk: folding of membrane area; Double arrowhead: section of bacterial cell folding. Bars stand for 1 µm (A–F,H,I) and 0.5 µm (G).
Phylum Ciliophora Doflein, 1901.
Class Spirotrichea Bütschli, 1889.
Subclass Euplotia Jankowski, 1979.
Order Euplotida Small and Lynn, 1985.
Family Euplotidae Ehrenberg, 1838.
Genus Euplotes Ehrenberg, 1831.
Diagnosis
Size in vivo (X ± SD) 49.1 ± 4.7 × 32.7 ± 3.8 μm. Dorso-ventrally flattened, with an oval to ellipsoidal shape. “C–shaped” or “3-shaped” macronucleus and a single micronucleus. Dargyrome of double-eurystomus type, 7–8 dorso-lateral ridges, with 13–14 dikinetids in the mid–dorsal row. About 22–29 adoral membranelles. Cirri pattern: ten fronto-ventral, five transverse, two marginal, and two caudal cirri. Freshwater.
Type locality
Freshwater emissary of Kolleru Lake, in the proximity of the Allapadu-Kolletikota road (16°36′05.0″N 81°18′47.8″E), West Godavari District of Andhra Pradesh, India. This species inhabits freshwater sites covered by Eichhornia sp. (water hyacinth).
Etymology
We dedicated this new species of Euplotes to Antoni Philips van Leeuwenhoek (1632–1723), Dutch optician and naturalist. Van Leeuwenhoek is best known for his pioneering work in microscopy and for his contributions toward the establishment of microbiology as a scientific discipline. For this reason, he is also known as "the father of microbiology", being one of the first microscopists and microbiologists.
Type material
The slide with the silver-stained holotype specimen (indicated with a black circle of ink on the coverslip) and some paratype specimens has been deposited in the collection of the “Museo di Storia Naturale dell’Università di Pisa” (Calci, Pisa, Italy) with registration number "2019-1". Two slides with silver-stained paratype specimens (indicated with a black circle of ink on the coverslip) were deposited in the collection of the Natural History Museum of London (registration number: NHMUK 2019.3.16.1), and in the collection of the Unit of Zoology-Anthropology of the Department of Biology at Pisa University (registration number: UNIPI_2019-1), respectively.
Morphological description
Size (X ± SD) in vivo 49.1 ± 4.7 × 32.7 ± 3.8 μm. Size after silver staining 45.6 ± 3.1 × 29.1 ± 4.1 μm. Cell reduction after fixation: 8%. Cells dorso-ventrally flattened, with an oval to ellipsoidal shape (Fig. 1). Right margin usually straight or slightly convex, left margin tapered in the anterior, becoming convex in the mid–body, and both ends are rounded (Fig. 1A). Ciliates can crawl on the substrate and swim freely in the medium. Cytoplasm transparent with some roundish, yellow granules; few food vacuoles containing green algae and bacteria (Fig. 1B,C). Single contractile vacuole located at the level of transverse cirri (Fig. 1B). On dorsal side, cortical ampules arranged around each bristle form conspicuous rosettes with their cortical insertions (Fig. 1C). Macronucleus (Ma) “C-shaped” or “3-shaped” (size: 36.6 ± 4.5 × 5.7 ± 1.0 μm) with irregularly dense chromatin, and a single, roundish micronucleus (Mi) (diameter: 2.0 ± 0.2 μm), usually located in a small depression close to Ma (Fig. 1D).
Dargyrome of the double–eurystomus type, with two rows of polygonal alveoli between each pair of dorsolateral kineties (Fig. 1E,G,I). Dorsal surface crossed by three longitudinal furrows (i.e. right marginal, median, and left marginal), reaching the posterior region of the cell (Fig. 1G). Six dorsal kineties plus 1–2 lateral, three in correspondence of dorsal furrows, carrying short bristle-like cilia (Fig. 1G); the leftmost kinety is placed in a slightly ventrolateral position (Fig. 1J). Mid–dorsal row containing up to 13–14 dikinetids, while other five dorsal rows contain 11–14 dikinetids (in detail, from the left, row1: 11; row2: 12; row3: 13–14; row5: 12–13; row6: 11–13) (Fig. 1E).
On the ventral side, invariably ten frontoventral cirri (FVC), five transverse cirri (TC), two well developed caudal cirri (CC), and two marginal cirri (MC) on the left side, in the posterior end of the cell (Fig. 1F,H,J). Argyrome is highly irregular (Fig. 1H,J) and the ventral surface presents five longitudinal ridges hosting cirral insertions; the three ridges on the left are more prominent (Fig. 1H). The first and the fifth ridges reach the posterior part of the cell at the level of the CC, while the other three ridges terminate beyond the TC (Fig. 1H).
Narrow peristome, extending for about 63% of the body length, on the ventral side. Adoral zone of membranelles (AZM) comprises 22–29 membranelles, starting at the top of the cell, travelling down along the left side and reaching the first ventral ridge, with a slight curve towards the centre of the body, at the level of transverse cirri (Fig. 1H). Length of AZM after silver staining: 43.1 ± 1.9 μm. The paroral membrane (4.7 ± 0.4 μm in length) appreciable in silver stained specimens (Fig. 1F), and in SEM-processed specimens (Fig. 1J), although carrying cilia shorter than those forming the AZM. Morphometric data are shown in Table 1.
Fine structure
The fine structure of E. vanleeuwenhoeki (Fig. 2) matches that of the other previously described Euplotes species, in general showing typical features42,43,44,45,46,47. Under the cell cortex flat alveoli are present (Fig. 2A–F,H–J). On the dorsal side, somatic cilia consisting of dikinetids (Fig. 2A,C) are deeply inserted into the cytoplasm (~ 1.4 µm); from kinetosomes only a single bristle-like cilium emerges (Fig. 1C,G). In the bristle pit, some filamentous material is sometimes visible (Fig. 2B). This is likely released by cortical ampules, the typical exocytotic organelles associated with both Euplotes dorsal bristle and compound ciliary organelles of the ventral surface; these organelles probably represent specialized compartments of the cell in which materials that need to be excreted are accumulated, stored, and released according to the requirements of the different Euplotes species48. Ampules associated with dorsal bristles of E. vanleeuwenhoeki appear elongated (size: ~ 1.6 × 0.3 µm) and usually empty possibly also due to the fixation procedure (Fig. 2C). Membranelles bordering the upper and left side of the oral cavity are separated from each other by ridges (Fig. 2D). Each membranelle of AZM consists of three rows of cilia: two equally long plus a shorter one (Fig. 2D). Axonemes contain many electron dense granules (Fig. 2D). Kinetosomes of membranelles are linked at their base. (Fig. 2E). A polystichomonad paroral membranelle is inserted on the right margin of the terminal oral cavity; its cilia appear linked to each other at the kinetosome level (Fig. 2D,E). Many flat, electron lucid pharyngeal disks are associated to the base of the cytostome, in correspondence of AZM bases (Fig. 2E,F). Macronucleus contains large pieces of chromatin and large nucleoli (Fig. 2F,G). Micronucleus consists of fine chromatin (Fig. 2G). A single contractile vacuole with an irregular silhouette is observed near a transverse cirrus (Fig. 2H). On the ventral side, kinetosomes of cilia forming cirri contain large electron dense granules (Fig. 2I). Mitochondria show variable shape and size and typical tubular cristae (Fig. 2B,C,E,F). Lipidic reserve substances consist of large granules; polysaccharidic reserve substances are represented by rosettes of glycogen abundantly and sparsely distributed throughout the cytoplasm (Fig. 2G,H). Large, irregular phagosomes are also present, with various content in different digestion stages (Fig. 2J).
Numerous, morphologically similar endosymbiotic bacteria, presenting variable shape and size, are located in the cytoplasm (Fig. 2): a detailed morphological description is presented below.
Gene sequence
The 18S rRNA gene sequence of E. vanleeuwenhoeki (strain KKR18_Esm) obtained from PCR resulted 1849 bp long, and it has been deposited in NCBI GenBank database with the accession number KY855568. The 18S rRNA gene sequence of E. vanleeuwenhoeki showed the highest identity with sequences of Euplotes cf. antarcticus (FJ998023) and E. trisulcatus, (EF690810): 99.0% (3 gaps, 16 mismatches) and 98.7% (13 gaps, 19 mismatches), respectively (Supplementary Table 1).
Phylogeny
The 18S rRNA gene-based phylogeny placed E. vanleeuwenhoeki in the so-called “clade A” of genus Euplotes49, 50, clustering together with Euplotes cf. antarcticus (FJ998023; Gao and Song unpublished) and with E. trisulcatus, (EF690810; Schwarz and Stoeck unpublished), with high statistical support (1.00/100%) (Fig. 3). This clade resulted sister to a clade comprising sequences attributed to E. charon (AF492705), E. magnicirratus (AJ549210), and E. euryhalinus (EF094968, JF903799) group (Fig. 3) (see later discussion on species attribution).
Mitochondrial genome
The assembly resulted in a single linear contig 41,682 bp long with a GC content of ~ 25%, representing the complete mitochondrial genome of E. vanleeuwenhoeki. It has been deposited in NCBI GenBank database with the accession number MK889230. It contains 36 protein coding genes and 16 tRNA genes. The genome presents the 16S rRNA and 23S rRNA genes split in two loci, with the 23S rRNA further divided into two genes, separated by a short interposing region of approximately 350 nucleotides (Fig. 4). The predicted direction of the transcription is away from a central region constituted of low-complexity repeated units (Fig. 4). The splitting of the rRNA genes and the presence of a central repeat region is a common feature shared by the two so far investigated Euplotes mitochondrial genomes (Euplotes minuta and Euplotes crassus)51 and by that of Oxytricha trifallax52. The novel genome shows an overall synteny with the mitochondrion of of Euplotes minuta, Euplotes crassus51, and Oxytricha trifallax52, with the exception of the two terminal regions, which show a different structure with respect to the other three genomes (Fig. 4).
Microbial consortium
To investigate the presence of possible bacteria related to E. vanleeuwenhoeki the whole sequenced DNA material was checked for the presence of 16S rRNA gene sequences (for detail see “Endosymbiont genome assembly and annotation” in “Experimental procedures” section). The screening of the preliminary assembly for bacterial 16S rRNA genes allowed to identify the presence of a single microorganism associated to E. vanleeuwenhoeki. Further analyses proved that this bacterium was localized in the cytoplasm of the ciliate, and that it was a novel endosymbiont we named “Ca. Pinguicoccus supinus” (see “Endosymbiont characterization” section). No other bacterial 16S rRNA gene sequence was detected in the sequencing reads. Moreover, most of the other contigs that were preliminary flagged as bacterial from the best megablast hit in the Blobology pipeline actually belonged to the mitochondrial or nuclear genome of Euplotes. In addition, contigs that were short (< 600 bp) or at a very low coverage (< 30×) were considered as from undetermined origin, possibly representing only minor contaminations. In both cases these contigs were discarded from the analysis (Supplementary Table 2).
Endosymbiont characterization: “Candidatus Pinguicoccus supinus” gen. nov. sp. nov. (Figs. 5, 6, 7, 8, 9 and 10)
Morphological description
“Ca. Pinguicoccus supinus” gen. nov. sp. nov. is a roundish-ovoid bacterium detected in the cytoplasm of E. vanleeuwenhoeki (Fig. 5) with a diameter of 1.3–2.3 µm (on average (X ± SD): 1.9 ± 0.3 µm). It usually lies beneath the ciliate cortex, often in clusters of several individuals (Fig. 5A). Although the most common bacterial shape observed is rounded (Fig. 5B), sometimes ovoid (Fig. 5C) and irregular (Fig. 5D,E) individuals can be detected as well. This cell shape plasticity might possibly be due to the pressure exerted by the host cytoplasm on the ductile body of the bacterium. No symbiosome is observed to isolate the endosymbiont from ciliate cytoplasm (Fig. 5). “Ca. Pinguicoccus supinus” is delimited by a double membrane with a thin space between the two layers possibly corresponding to the paryphoplasm (Fig. 5B), the intracellular space defined for the first time by Linsday and colleagues53. In several individuals, the increase of membrane area is visible: in some cases, a slight invagination of the inner membrane occurs (Fig. 5B–D), while in others the evagination of the external membrane can be observed (Fig. 5E,F). In the latter case, different inclusions of unknown origin have also been observed in the space between the inner and outer membranes (Fig. 5E). The bacterial cytoplasm (possibly corresponding to the pirellulosome53) generally appears homogeneous and a compact, more electrondense region, likely corresponding to bacterial nucleoid, is visible in some bacteria with an eccentric localization (Fig. 5D,E,H,I). Occasionally, specimens show a very emphasized folding of the membrane area, making it difficult to recognize whether the folding comes from the inner or the outer membrane (Fig. 5G). Out of a total of ~ 80 endosymbionts observed in the thin section, roughly one third are in proximity (i.e. at a distance of ~ 0.25 µm or less) of mitochondria (Fig. 5A–C,F,I) and one fifth are in proximity to lipid droplets (Fig. 5E,G,H). Intriguingly, in these cases, bacterial double membrane is even seen somehow in direct contact with mitochondrial external membrane (Fig. 5C) and lipid droplets (Fig. 5H). “Ca. Pinguicoccus supinus” reproduces in the host cytoplasm by binary fission and has, apparently, a typical symmetrical division (Fig. 5I).

COG analysis of “Candidatus Pinguicoccus supinus” and other bacteria with highly reduced genome. (A) Principal Component Analysis of numerosity in COG classes; explained variance by Component one 32%; explained variance by Component two 24%; (B) Distribution of COGs in each analysed genome; (C) Distribution of COGs, showing: i. percentage of COGs shared by all the analysed genomes, COGs unique for each genome, and COGs shared by at least two genomes; ii. Number of organisms sharing COGs. Each set groups together the COGs shared by a given number of organisms (i.e. 1, 2, 3…7), regardless of their identity; iii. Percentage of unique COGs in analyzed genomes.

Phylogenetic tree of Phylum Verrucomicrobia, based on the 16S rRNA gene. The phylogenetic position of "Candidatus Pinguicoccus supinus" is shown. Numbers associated to nodes represent posterior probability and bootstrap values, respectively (only values above 0.80–75 are shown). Black circles indicate organisms also employed in phylogenomic analysis (Fig. 8). Asterisks indicate sequences employed in the 16S rRNA gene screening on IMNGS (Fig. 10; Supplementary Table 6). Numbers in square brackets, associated to collapsed branches, indicate how many sequences are not shown (for list of hidden sequences see Supplementary Table 9). Sequence obtained in the present work is in bold.

Phylogenomic tree of Verrucomicrobia, showing evolutionary relationships of "Candidatus Pinguicoccus supinus". Numbers associated to nodes represent bootstrap values (only values above 75 are shown). Black circles indicate organisms also employed in phylogenetic analysis (Fig. 7). Genome of “Ca. Pinguicoccus supinus” obtained in the present work is in bold (accession number: CP039370).

Fluorescence in situ hybridization experiments on Euplotes vanleeuwenhoeki sp. nov. (A–C) Specimen hybridized with probe EUB338 VII. (D–F) Two specimens hybridized with probe Pingui_1174. (A,D) Pictures at DIC microscope of fixed cells; (B,E) DAPI staining, showing position of nuclear apparatus; (C) Cell positive to probe EUB338 VII, fluorophore emitting in red (Cyanine-3); (F) Cell positive to probe Pingui_1174, fluorophore emitting in red (Cy-3). Bars stand for 10 µm.

Diversity of "Candidatus Pinguicoccus supinus" based on 16S rRNA gene amplicon search in IMNGS. 16S rRNA gene hypervariable regions phylogenetic trees. OTUs were clustered with 99% identity and were longer than 300 bp. A total number of 90 OTUs were separated for each hypervariable region taken into analysis: 29 for V1–V2, 60 for V4–V6, 2 for V7–V8. Complete 16S rRNA gene was employed in the analysis to enlighten the diversity of “Ca. Pinguicoccus supinus”. White triangle indicates the “Ca. Pinguicoccus supinus” full-length 16S rRNA gene; black triangle indicates the full-length 16S rRNA gene.
Gene sequence
The 16S rRNA gene sequence of “Ca. Pinguicoccus supinus” resulted 1517 bp long and has been deposited in NCBI GenBank database with the accession number MK569697. Best BLAST hits of the obtained 16S rRNA gene against NCBI database were sequences from uncultured bacteria: JQ993517 and MNWT01000005, AB826705, and AY571501 (identity values: 78.8%, 78.3%, and 78.2%, respectively). The best hit with a cultured bacterium was 76.8% with Ruficoccus amylovorans (KT751307) (Verrucomicrobia, Opitutae, Puniceicoccaceae). In general, identity values with the closest relatives resulted quite low (75.7–78.8%) (Supplementary Table 3).
Genome assembly
The assembly of the symbiont’s genome resulted in a single circular chromosome, 163,218 bp long with a GC content of 25.1%. The complete genome sequence of “Ca. Pinguicoccus supinus” has been deposited in NCBI GenBank database with the accession number CP039370. It contains 168 protein coding sequences, 34 tRNA genes and a single rRNA operon composed of a 16S rRNA gene, a 23S rRNA gene and a 5S rRNA gene. The overall coding percentage is 92.3%. The ORFs were subjected to clusters of orthologous groups (COGs) classification, and 131 COGs were identified, most of which were related to the general cellular function categories: J (translation, ribosomal structure, and biogenesis), O (post-translational modification, protein turnover, chaperons), M (cell wall/membrane/envelope biogenesis), and I (lipid transport and metabolism), for a total of 112 COGs (Supplementary Table 4). This repertoire is a subset of the previously characterized Verrucomicrobia, i.e. without any exclusive metabolic pathway or gene (Supplementary Table 5). Considering the drastic genome reduction of “Ca. Pinguicoccus supinus”, we decided to perform comparative analyses using as reference the few other symbiotic bacteria with highly reduced genome although belonging to unrelated lineages (i.e. Bacteroidetes, Alphaproteobacteria, Gammaproteobacteria, Betaproteobacteria).
A Principal Component Analysis (PCA) based on retrieved COGs was able to capture almost 56% of the whole variance (Fig. 6A) in the COG dataset, and thus capturing the functional diversity among the analyzed genomes. While almost all the bacteria with highly reduced genome cluster together (Fig. 6A), the variance explained by the first component positions “Ca. Pinguicoccus supinus” is remarkably distant. This component is mainly correlated with the COG classes Q—Secondary metabolite biosynthesis, transport and catabolism (eigenvalue: 0.36), I—Lipid transport and metabolism (0.34), and K—Transcription (0.33). The variance explained by the second component separates “Ca. Zinderia insecticola” from the other bacteria with highly reduced genome. This component is mainly correlated with COG classes P—Inorganic ion transport and metabolism (0.42), H—Coenzyme transport and metabolism (0.39), and C—Energy production and conversion (0.33) classes.
COG analysis also showed the presence of a set of 33 genes shared by all the small genome bacteria in analysis (Fig. 6B,C), mostly related to DNA replication, transcription, and translation. Although the metabolic capability of “Ca. Pinguicoccus supinus” is, in general, similar to that of other highly reduced genomes40,54 (88% of COGs shared with at least another analysed genome; Fig. 6B,C), the novel genome does not code for the synthesis of any amino acid. Moreover, no catalytic subunit of the DNA polymerase was identified. “Ca. Pinguicoccus supinus” possesses 30 genes (12% of the total retrieved COGs in this bacterial genome) that are absent in all the other tiny genomes, mostly related to COG classes I (lipid transport and metabolism) and M (cell wall/membrane/envelope biogenesis). In general, with respect to the other analyzed bacteria, this endosymbiont includes a richer set of genes involved in fatty acid biosynthesis, in glycosylation and glycan modification (Supplementary Table 4).
Phylogeny and phylogenomics
The 16S rRNA gene-based phylogeny showed “Ca. Pinguicoccus supinus” as a member of the family Puniceicoccaceae (Verrucomicrobia, Opitutae) (Fig. 7). It clustered with sequences from uncultured organisms, forming a clade related to the genera Coraliomargarita, “Fucophilus”, Cerasicoccus, and Ruficoccus (Fig. 7). The long branch of “Ca. Pinguicoccus supinus” suggests a higher evolutionary rate with respect to related Verrucomicrobia. Moreover, we observed that the inclusion of the “Ca. Pinguicoccus supinus” sequence in the Verrucomicrobia tree reduces the values of statistical supports for the nodes of the Puniceicoccaceae clade (data not shown). This is consistent with the occurrence of a long branch attraction phenomenon between “Ca. Pinguicoccus supinus” and the outgroup, which destabilizes Puniceicoccaceae despite high taxon sampling55.
The result of the phylogenomic analysis is fully consistent with the phylogeny of the 16S rRNA gene, confirming the position of the endosymbiont inside the phylum Verrucomicrobia and in the class Opitutae (Fig. 8). “Ca. Pinguicoccus supinus” clustered together with uncultured organisms (GB_GCA_001872735, GB_GCA_002309885, GB_GCA_002336265), in a clade related to Coraliomargarita akajimensis (RS_GCF_000025905), and thus inside the family Puniceicoccaceae (Fig. 8).
Localization inside host cell
FISH experiments showed 100% of E. vanleeuwenhoeki cells positive to specifically designed probes for “Ca. Pinguicoccus supinus” (Fig. 9). The number of endosymbionts per host cell ranged from 12 to 36 bacteria (X ± SD: 25.1 ± 7.3; n = 16). The symbionts showed a peculiar pattern of distribution in almost all the observed Euplotes cells: beneath the cortex of the host, in clusters of several bacteria grouped at one side of the cell (Fig. 9C), or more commonly at the two poles (Fig. 9F). These observations support data from TEM analysis, showing “Ca. Pinguicoccus supinus” generally lying close to the cortex and forming clusters with other conspecifics.
Diversity and environmental screening of the endosymbiont
The Integrated Microbial Next Generation Sequencing (IMNGS) platform56, was used to investigate the molecular diversity and environmental distribution of sequences showing high identity with the novel endosymbiont of Euplotes. The 16S rRNA gene of the endosymbiont was queried at 95% similarity threshold, and a total number of 4572 sequences were obtained and subsequently used to investigate the molecular diversity of 16S rRNA gene hypervariable regions. Sequences were clustered in 90 OTUs with 99% threshold identity, then OTUs were grouped according to their hypervariable region in V1–V2, V4–V6 and V7–V8, and phylogenetic trees were inferred accordingly (Fig. 10). Phylogenetic trees showed that six diverse groups can be distinguished for all hypervariable regions considered, and their phylogenetic relations are in agreement with those shown by the full-length 16S rRNA gene phylogenetic reconstruction (Fig. 10). The first group (red) includes the novel endosymbiont and related sequences (AB614893, JQ993517; Fig. 7), the second one (violet) is formed by sequences from uncultured bacteria (JQ993599, AB826705), the third one (purple) consists of a single uncultured bacterium (EU462461), the fourth one (green) includes Coraliomargarita akajimensis (CP001998), “Fucophilus fucodainolyticus” (AB073978), and Puniceicoccus vermicola (DQ539046), the fifth one (yellow) has Cerasicoccus frondis (AB372850) and Ruficoccus amylovorans (KT751307), and the last one (blue) includes just epixenosomes from Euplotidium arenarium (Y19169) (Fig. 10).
The IMNGS environmental screening also showed that metagenomic samples positive to “Ca. Pinguicoccus” were very limited in number, namely 0.01% of total samples. Indeed, the related sequences were present in just 32 out of a total of 303,362 samples (Supplementary Table 6). The environmental origin of positive samples is reported in Supplementary Table 6. Positive hits originated from very diverse samples, such as seawater, wastewater, microbial mats, but also potential host organisms, such as shrimps and plants (Supplementary Table 6). However, the overall abundance of positive hits was extremely low and always below 1% within each sample. The highest abundance was found in samples originating from a study on wastewater57, where “Ca. Pinguicoccus” abundance ranged between 0.005 and 0.752% (Supplementary Table 6).
Discussion
First biont: Euplotes vanleeuwenhoeki
Comparison with congeners
Euplotes vanleeuwenhoeki showed morphological and molecular affinity with other members of the genus, such as E. antarcticus58,59, E. trisulcatus60, E. euryhalinus61, E. charon62,63, and E. magnicirratus60. Indeed, they shared the same dargyrome and frontoventral cirri patterns (for details see Table 2), and clustered together in our molecular phylogeny (Fig. 3). As for its morphology, E. vanleeuwenhoeki was particularly similar to E. trisulcatus60. Nevertheless, the combination of several characters (i.e. posterior end rounded versus pointed; peristome length, number of membranelles in the AZM, number of dikinetids in mid-dorsal row—for details see Table 2), the overall body shape, the type of habitat (freshwater vs marine), and the 18S rRNA gene sequence supported its attribution to a novel species.
Unfortunately, the sequences closest to that of E. vanleeuwenhoeki in our phylogeny reconstruction (i.e. E. magnicirratus—AJ54921064; E. charon—AF49270565; E. euryhalinus—EF09496866; JF903799 (Keerthi and Reddy unpublished); E. trisulcatus EF690810 (Schwarz and Stoeck unpublished), Euplotes cf. antarcticus—FJ998023 (Gao and Song unpublished) are not accompanied by any morphological description. Consequently, data used in our morphometric comparison inevitably derive from previous, exclusively morphological studies59,60,61,62,63. Moreover, there is no certainty that the above-mentioned Euplotes-derived sequences were properly attributed to the correct ciliate species; thus, a certain cautiousness is recommended until a comprehensive redescription of such organisms, joining morphology and molecular data, has been performed. In this somehow unclear landscape, for the sake of completeness, we decided to include also E. charon in our comparison among different Euplotes species (Table 2), because it shares some basic features with E. vanleeuwenhoeki, (i.e. dargyrome, and frontoventral cirri pattern), although the phylogenetic position of this species is far from being resolved67. Indeed, the sequence AF492705, under the name of E. charon, is not linked to any morphological description65; this sequence was used by Shao and colleagues63 as a molecular reference in their redescription of E. charon, for which, unfortunately, they used a different Euplotes strain for morphological analysis. However, there are several other available sequences that are named after E. charon, but none are supported by any morphological data [i.e. AJ30524964; JF69404368; FJ87077-80 (Huang and Song unpublished)] and these sequences cluster far away from E. vanleeuwenhoeki. A critical revision of the species E. charon using at least integrative taxonomy is consequently highly recommended.
As a general remark, E. vanleeuwenhoeki shares with many other species the plesiomorphic condition of the genus, i.e. the occurrence of ten FVC and a double dargyrome of eurystomus-type (e.g.: E. alatus, E. antarcticus, E. balteatus, E. charon, E. crenosus, E. curdsi, E. enigma, E. euryhalinus, E. focardii, E. harpa, E. inkystans, E. magnicirratus, E. neapolitanus, E. octocirratus, E. palustris, E. parabalteatus, E. platysoma, E. plicatum, E. polycarinatus, E. qatarensis, E. quinquecarinatus, E. rariseta, E. trisulcatus, and E. tuyraui). Although these two features have always been reported in morphological description, due to their plesiomorphic status they are of limited use for taxonomic identification67. As an example, some authors69 even proposed to synonymise almost all species possessing ten FVC and a double dargyrome under the name of E. charon. Obviously, nowadays, this proposal is not acceptable on the basis of molecular data50,67,70 and because of other morphological features characterizing some of the organisms (e.g. dorsal furrows/ridges, and body shape and proportions).
It has been proved that, in many cases, this ancestral condition underwent severe, independent modifications during the evolutionary history of the genus49,64,67. Indeed, the double dargyrome evolved in other different patterns, such as “single”, “double patella-type”, “multiple”60, and “complex”, in many distinctive events49,67. The dargyrome modification was explained either by increasing in number or by unification of alveoli49,67,71. It is not by chance that some Euplotes species present a great phenotypic plasticity, determined by particular physiological and/or ecological conditions, such as the presence of a predator and/or food availability (i.e. E. focardii72, E. balteatus73, E. octocarinatus74,75, and E. variabilis76. Similarly, one or more FVC were independently lost many times, as also evidenced by the vestiges still detectable in some Euplotes species49,64,67,77,78.
Mitochondrial genome
Ciliates possess very peculiar mitochondrial genomes, being among the first to be identified as linear79,80 and showing several split rRNA genes81 and protein genes82. To our best knowledge, in ciliates, the set of potentially split genes in the mitochondrial genome includes nad1, nad2, rps3, rnl and rns. This list now includes the cox1 gene, which is split in E. vanleeuwenhoeki. Considering that the splits in nad1, nad2, rps3, rnl and rns genes occur approximately at the same positions in the mitochondrial genomes of Spirotrichea, it has been hypothesized they could be present in the last common ancestor of Euplotes and Oxytricha52. However, since the cox1 gene is split only in the E. vanleeuwenhoeki genome, it is likely that this represents a more recent event. The occurrence of multiple and evolutionary independent split genes in ciliates could be considered indicative of a possible inherently higher tolerance to split events in mitochondrial genes in these organisms.
The mitochondrial genome of E. vanleeuwenhoeki shows a high level of colinearity with the other two available Euplotes mitochondrial genomes. In addition, all Euplotes share a good colinearity with O. trifallax (two single-gene inversions and one single-gene transposition). Since the amino acid substitution rates in ciliate mitochondrial genomes appear to be high52, this result enforces the idea that mitochondrial genome colinearity can be employed in phylogenetic analysis as a valid supporting feature to define the systematics of ciliates, or to distinguish different taxonomic levels among close related species using the non-colinear terminal regions83,84. Indeed, mitochondrial genes, such as cox1, cox2 and rns, are being increasingly used in taxonomy, both for phylogenetic analysis85, and for discriminating among species with similar morphological features. For example, mtDNA genes have been employed to reveal several cryptic species in Ciliophora86,87 and metazoan such as annelids88 and insects89, or to discriminate between subspecies of the bee Apis mellifera90. The limited number of available mitochondrial genome sequences of ciliates currently does not permit phylogenomic analyses based either on sequences or on colinearity. Nevertheless, we predict that such analyses will be extremely helpful, in the future, to resolve phylogenetic and taxonomic issues as already occurred in other taxa (e.g. metazoa) in which more sequences are already available91,92. We expect the same for ciliates as soon as enough novel mitochondrial genome sequences are available.
Symbiosis with a Verrucomicrobia member
Members of genus Euplotes are known to host symbiotic eukaryotes, mutualistic such as endocellular algae46,93 or opportunistic such as microsporidia94 and trypanosomes27. Moreover, Euplotes species display a certain propension to harbor one or even multiple bacterial symbionts95,96,97,98,99. This second condition appears more common if the ciliate presents the obligate symbiosis with Polynucleobacter necessarius or “Ca. Protistibacter heckmannii”99,100,101,102,103,104,105,106. In those cases, the secondary or tertiary symbionts, if present, frequently belong to Alphaproteobacteria, e.g. Rickettsiaceae99,103,105,107 or “Ca. Midichloriaceae”99,102,104,108. Also, Gammaproteobacteria were retrieved in different Euplotes species99, 101,106,109,110. For a detailed and comprehensive review on Euplotes endosymbionts see Ref.99.
To date, “Ca. Pinguicoccus supinus” as endosymbiont of E. vanleeuwenhoeki constitutes a unicum, being the first member of Verrucomicrobia with a highly reduced genome, and also the first found as an endosymbiont of a protist. Indeed, bacteria of the phylum Verrucomicrobia are mostly free-living organisms, nearly ubiquitous, present both in the soil and in wet environments111,112, and often retrieved in extreme habitats113,114. Little is known about Verrucomicrobia living as obligate symbionts, which at present were described in nematodes115, echinoderms116, squids117, and in the gut of different metazoans118,119, including humans120. The only other case of association of Verrucomicrobia with protists is the symbiosis between the ciliate Euplotidium and the so-called epixenosomes33,121,122 which are verrucomicrobial ectosymbionts able to extrude and defend their host against predators6.
Second biont: “Candidatus Pinguicoccus supinus”
Endosymbiont morphology
The morphology of some PVC members is renowned to be peculiar. Indeed, some of them present a certain degree of cellular compartmentalization, even if some aspects of the subject are still under debate53,123,124. Cell compartmentalization has been proposed for some members of Verrucomicrobia, such as Prostechobacter dejongeii (Verrucomicrobiaceae) and Coraliomargarita akajimensis (Puniceicoccaceae) based on molecular and ultrastructural studies125,126. In those two bacteria, the presence of two different cellular compartments, delimited by an intracytoplasmic membrane, has been described. These two compartments were recognizable as a ribosome-free region, the paryphoplasma, and a region containing ribosomes and nucleoid, the pirellulosome. In this framework, some morphological traits of “Ca. Pinguicoccus supinus”, such as membrane invaginations and folding, could be considered homologous to those observed in other Verrucomicrobia and PVC members in general, although our data, based solely on TEM observation, definitely do not provide clear evidence of cell compartmentalization. A possible future development could be the use of cryo-electron tomography127.
Endosymbiont genome
The genome of “Ca. Pinguicoccus supinus” was found to be extremely small (163,218 Kbp), being, to our best knowledge, the fourth smallest bacterial genome sequenced to date (smallest being “Ca. Nasuia deltocephalinicola”: 112,031 bp)128. The novel genome is highly reduced and lacks many genes, especially compared with the relative free living Verrucomicrobia (range 2.2–7.3 Mbp)114,129 but also with other Verrucomicrobia symbionts, such as the ones belonging to the genus “Ca. Xiphinematobacter” (~ 916 Kbp)130. The tiniest genomes (< 500 Kb) are found in putatively ancient mutualistic symbioses, where symbionts are beneficial to their hosts’ metabolism and hosts have in turn evolved to support and control the symbiosis40,54,128,131. Such levels of co-evolution often also imply phenomena of severe genome reduction, with gene loss and often a subsequent modification of the nucleotide base composition54. This framework gives us some clues for understanding the peculiar characteristic of the novel symbiont “Ca. Pinguicoccus supinus”. All other symbionts with highly reduced genomes provide their hosts with nutrients that are absent in their diet. In those cases, the nutritional support role is clear, due to the fact that they retain some metabolic pathways for the biosynthesis of amino acids essential for their hosts132 (for a review see Ref.54). Moreover, several of these symbionts are co-obligate with other bacteria complementing each other for the enzymatic repertoire necessary to provide nutritional support to the host. This seems not to be the case of “Ca. Pinguicoccus supinus”, which is devoid of genes related to amino acids, co-factors or vitamin biosynthesis, and is the only symbiont retrieved in the E. vanleeuwenhoeki holobiont. The extensive gene loss and genome modification suggest that this symbiosis may have an ancient origin, and that it may play an important role for the host, although its function is yet to be understood. This case could be partly similar to that of the well-ascertained mutualist bacterium, Polynucleobacter necessarius in other Euplotes spp.133,134. Indeed, this bacterium is necessary for the survival of its hosts, but no precise clues on the metabolic nature of the interaction have been provided by genomic analyses133,134. This has been linked to the peculiar ecology of ciliates, which normally feed on free-living bacteria, and are thus unlikely to require dietary compensations from bacterial endosymbionts. It has been hypothesized that they may require help in catabolism or other undefined functions, and such a contribution would not be simple to detect by analyses of the symbionts’ genomes only133.
The comparison between “Ca. Pinguicoccus supinus” and the other symbionts with highly reduced genomes shows the presence of a core set of 33 genes retained by all these evolutionary unrelated bacteria. Those genes are mostly involved in DNA replication, transcription, and translation, as well as in protein folding and stability. Clearly, these core cellular functions are required even in bacteria with such extremely reduced genomes. Interestingly, in “Ca. Pinguicoccus supinus”, it was not possible to find a gene homologous to a DNA polymerase II catalytic subunit (dnaE), or, in general, to any other protein with predicted DNA polymerase catalytic activity. This is a striking difference with respect to the other analysed symbionts, and, to our best knowledge, it is a unique case among all bacteria. A Blast analysis on the entire preliminary assembly of the whole holobiont did not reveal the presence of any dnaE gene, suggesting that there is no such gene integrated in the host genome. This finding strongly suggests that “Ca. Pinguicoccus supinus” relies on proteins obtained from the host to replicate its own genome, analogously to what has been proposed for other essential cellular functions for other symbionts with highly reduced genomes54.
A common feature of the previously characterized symbiotic bacteria with highly reduced genomes is the complete lack of genes related to the production of components of the cellular envelope such as phospholipids, lipopolysaccharide, peptidoglycan and related membrane proteins54. Unique in this regard, “Ca. Pinguicoccus supinus” presents the pathway for the initiation and elongation of fatty acids, and several glycosyl transferases, although the phospholipid synthesis pathway is missing. Obligate small genome symbionts of insects are found in specialized cells, the bacteriocytes and some symbionts of unicellular eukaryotes are located in a specific cellular structure, the symbiosome, while “Ca. Pinguicoccus supinus” resides free in the host cytoplasm. This could imply the requirement for a more complex regulation of the membrane structure (e.g. variation of fatty acid length in phospholipids) to face a “less stable” environment, thus possibly explaining the unique genes found in the genome of “Ca. Pinguicoccus supinus”. Interestingly, “Ca. Pinguicoccus supinus” has often been observed in close contact with the host’s lipid droplets. It is not yet elucidated whether the interaction with the host’s lipids could be another reason for the retention of genes devoted to fatty acid biosynthesis.
The genome of “Ca. Pinguicoccus supinus” presents a non-standard genetic code (NCBI genetic code “4”), in which the canonical stop triplet UGA is recoded for tryptophan, consistently with the absence of release factor 2, implied in the normal recognition of UGA as stop. This is consistent with other highly reduced genomes135 and has been tentatively linked to a directional mutation pressure related to the AT/GC composition136, or directly linked to genome reduction and energetic constrains135.
Endosymbiont phylogeny
Both the phylogenetic and phylogenomic analyses, the first including more taxa and the second more genes, support the position of “Ca. Pinguicoccus supinus” in the family Puniceicoccaceae (Opitutae, Verrucomicrobia). We suggest that the long branch of the novel species is due to an overall high evolutionary rate, and consequently may decrease support for some nodes inside the family-clade.
It is worth noticing that our phylogenetic and phylogenomic analyses are among the most taxonomically exhaustive up to date for Verrucomicrobia and resulted coherent with those from previous studies137,138,139,140. They also highlight the necessity for systematic revision for some species and taxa (i.e. Brevifollis gellanilyticus clustering within genus Prostechobacter; Subdivision 3 and Subdivision 6 still waiting for a proper naming).
Endosymbiont diversity and environmental distribution
In order to evaluate how widespread and abundant the novel taxon is, we evaluated the presence of sequences related to 16S rRNA gene of E. vanleeuwenhoeki endosymbiont in IMNGS, which resulted to be very limited. The main possible reason of the scarcity of “Ca. Pinguicoccus supinus” and related sequences in online repositories resides in the fact that 16S rRNA gene primers employed in metabarcoding and metagenomic studies do not often detect certain bacterial groups, such as Verrucomicrobia141,142. Indeed, the choice of primers used in such NGS analysis should be carefully pondered as some bacterial taxa might be undetected, thus not depicting the real diversity in the environment. Our analysis shows that the 16S rRNA sequences related to the endosymbiont were present in just 0.01% of total samples, and never reached 1% abundance in positive samples, a number of which resulted to be microbiome studies of multicellular organisms (Supplementary Table 6). The hypervariable region diversity analysis confirmed the limited presence of sequences from these bacteria sequences in metabarcoding samples. Extracting from the databases sequences similar to “Ca. Pinguicoccus supinus” and performing phylogenies on the hypervariable regions of the 16S rRNA gene, six diverse groups could be identified, and their phylogenetic relationships retraced those of the complete 16S rRNA gene phylogeny (Figs. 7 and 10). Moreover, hypervariable region diversity analysis pointed out that “Ca. Pinguicoccus supinus” was usually a stand-alone sequence within its group, with the only exception of the V4–V6 phylogenetic tree having a single OTU associated to the endosymbiont sequence (Fig. 10). On the contrary, the other related Puniceicoccaceae, which were isolated and characterized from the environment (mostly marine), clustered in groups encompassing numerous OTUs (Fig. 10). The same stand-alone phenomenon occurred also for the epixenosomes, the aforementioned ectosymbionts of the ciliate Euploditium arenarium, that always formed a defined and separated lineage in all our analyses (Fig. 10). The limited presence of OTUs associated to these microorganisms could be also explained with the symbiotic nature of these bacteria, which occupy a peculiar and circumscribed ecological niche, possibly making their detection more complicated. It is also worth noticing that the number of OTUs per clade is significantly different when region V1–V2 is compared to region V4–V6 (Fig. 10). Indeed, in region V1–V2 “Ca. Pinguicoccus supinus” and related clades (in red) are relatively richer in OTUs if compared to region V4–V6, whereas the opposite is true for the Coraliomargarita-“Fucophilus”-Puniceicoccus clade (in green). This observation suggests that commonly used primers for region V1–V2 and V4–V6 could recognize the two clades with a different efficiency, producing artefactual results in terms of abundance and biodiversity.
Proposal of an updated approach in taxonomy: the “next generation taxonomy”
The present work aims to propose and introduce a more comprehensive approach to the characterization of living beings, which integrates the holobiont concept according to Margulis16 into traditional and integrative taxonomy, leveraging the power of high-throughput DNA sequencing.
Starting from de Bary’s definition of symbiosis4, Lynn Margulis defined a holobiont as the compound of different species that together form a single ecological unit16. In recent times, an increasing interest regarding holobionts has arisen, leading to different interpretations of the concept143,144 and to different theories on the underlying evolutionary driving forces143,144,145,146,147,148,149. Nevertheless, the applicability of the “holobiont” definition, the evolutionary implications of the concept and, in particular, the ’boundaries’ of the holobiont, i.e. which associations should be considered within this definition, are still matters of debate.
Holobionts have been described among many and far-related groups of living beings, such as plants (for review see150,151), algae152, insects (for review see Refs.153,154,155), corals156 (for review see Refs.157,158), and even humans159. In this landscape, protists are under-represented, even though they are highly diverse and environmentally widespread160,161,162, and their association with a wide range of other microorganisms is well known163,164,165,166 (for review see Refs.167,168,169,170,171).
In our opinion, the application of the holobiont concept could be very useful for the characterization of living beings, introducing a new parameter for their taxonomic description, i.e. the presence of stably associated organisms, whenever present. Indeed, the presence of these organisms can influence the host, its physiology, and its adaptability to the environment6,12,13; moreover, in some cases it has been shown to influence also its morphology5,14, the fundamental parameter of traditional taxonomy.
The proposed approach aims to combine all the associated species characterizations into the holobiont description, providing not only morphological characters and molecular markers, but also functional and associative parameters. According to this line of thought, such a multidisciplinary approach represents a considerable improvement, allowed by the development of more sophisticated and powerful tools in molecular biology and genomics.
The whole mitochondrial genome, that we are proposing as a novel taxonomic descriptor for its amount of genetic information and the relative ease in sequencing and annotation, is becoming widely used in phylogenomics and evolutionary studies172,173,174. Indeed, the resolutive power of the mitogenome was employed to disentangle systematic issues175,176,177,178.
Although the number of articles treating mitogenomes are increasing, also leading to the birth of several dedicated online databases, such as ParameciumDB179, these analyses are still rarely used for a more comprehensive description of living beings, from the beginning (i.e. in their taxonomic description).
The aim of our proposal is, indeed, to fill this gap and, in parallel, to further promote the use of mitogenomes. This would increase the number of annotated mitogenomes of ciliates and other protists in online repositories, and therefore allow the development of dedicated databases.
To sum up, we suggest the inclusion of two additional descriptive parameters, i.e. the host's mitogenome and the characterization of the possibly present associated bionts, integrating novel useful features to the description of organisms. Proposed approach could be referred to as “next generation taxonomy”, to differenziate it from traditional “integrative taxonomy” that is not using Next Generation Sequencig data. It is to be emphasized that, applying the new approach by no mean implies that the traditional taxonomic tools are to be considered obsolete, as they are indisputably the base of species description.
To exemplify the concept of “next generation taxonomy” applied to holobionts, we present the ciliate E. vanleeuwenhoeki sp. nov. and its symbiont, “Ca. Pinguicoccus supinus” gen. nov., sp. nov., as a case study. In E. vanleeuwenhoeki sp. nov. a sole, stably associated bacterium was detected, namely the endosymbiont “Ca. Pinguicoccus supinus”, which makes the application of the holobiont concept to species description unambiguous. As detailed above, the peculiar features of this symbiont, especially the genomic ones, are strongly indicative of a long coevolutionary and adaptation history with its host, so that it might even be hypothesized that this bacterium might possibly represent an autapomorphy of the host species. Further study on different strain of the same species and on other related species will be necessary to assess this possibility.
Not by chance, it has already been demonstrated that some phylogenetically related Euplotes species showed the obligate symbiosis with certain bacteria (e.g. P. necessarius, “Ca. Protistibacter heckmannii”, “Ca. Devosia” spp.) as a peculiar evolutionary inherited trait49,67,98,99,180,181. Given the possible relevance of symbiotic events in Euplotes evolution and the extremely reduced genome of “Ca. Pinguicoccus supinus”, it seemed opportune to treat the sum of the different parts (E. vanleeuwenhoeki plus its symbiont) in a unitary and integrated way, starting from the very beginning, i.e. the taxonomic description”.
In conclusion, in our view, the proposed “next generation taxonomy” approach can constitute an updated and valuable standard for unicellular eukaryote descriptions and redescriptions. Indeed, integrating standard taxonomical analyses with the most modern tools in genomics and bioinformatics and the concept of holobiont, provides additional descriptors convenient not only to fully and unambiguously define a species but also to infer its interaction with other organisms and, at least partially, its biology. At a practical level, the suitability of the proposed approach for unicellular eukaryotic organisms, in particular free-living forms, associated with bacterial endobionts, is herein documented with the description of E. vanleeuwenhoeki, and we are rather confident that it should be similarly straightforward if applied to other kinds of holobiontic protists.
Description of “Candidatus Pinguicoccus” gen. nov.
Pinguicoccus [Pin.gui.coc’cus. L. adj. pinguis fat; N.L. n. coccus (from Gr. masc. n. kokkos grain, seed) coccus: N.L. masc. n. Pinguicoccus, a fat coccus, because of its rounded body shape and because it was often found associated with host’s lipid droplets].
The genus description at present is the same as the description of the type species “Ca. Pinguicoccus supinus”.
Description of “Candidatus Pinguicoccus supinus” sp. nov.
Pinguicoccus supinus (su.pi'nus. L. adj. supinus lying on its back, supine, indolent, lazy, because it is not a motile microorganism and because it lacks several fundamental metabolic pathways).
Type species of the genus. Non motile Verrucomicrobia bacterium of family Puniceicoccaceae (class Opitutae). Roundish-ovoid in shape, detected in the cytoplasm of the ciliate Euplotes vanleeuwenhoeki, with a diameter of 1.3–2.3 µm. It usually lies beneath the ciliate cortex, often in clusters of several individuals. Cells are delimited by a double membrane with a thin space between the two layers; no symbiosome has ever been observed. In several individuals, an invagination of the inner membrane and folding of the outer membrane are observed. Homogeneous bacterial cytoplasm. Sometimes shows presence of nucleoids. Reproduce by binary fission in the host cytoplasm, by means of an apparently typical symmetrical division. Its genome size is 163,218 bp and G + C content is 25%. The genetic code for protein translation is “translation table 4”.
Base of attribution: 16S rRNA gene sequence, MK569697; complete genome sequence CP039370; recognized by oligonucleotidic probes EUB338 VII and Pingui_1174.
Experimental procedures
Sample collection and cell culturing
The strain KKR18_Esm was collected on August 8th 2014, in one emissary of the freshwater Lake of Kolleru (16°36′05.0″N 81°18′47.8″E). Kolleru Lake is the largest freshwater body of India, and a protected Ramsar area due to seasonal migratory birds. It is situated between the deltas of two major rivers, Godavari and Krishna, 15 km south-east from the city of Eluru, in Andhra Pradesh state. The depth of Kolleru Lake is usually around 150–300 cm, but at the time of sampling, the lake was almost dried up and the water level had dropped to the depth of 60–90 cm181. Samples were collected at a depth of 15–30 cm near the shore of the lake using sterile 50 ml Falcon tubes: both water and sediment were collected at the same time181.
The original sample was screened by pouring about 20 ml of water in a Petri dish. Single cells were collected using a micropipette, washed several times in mineral water, put in a depression slide and enriched with a few drops of the monoclonal culture of Dunaliella tertiolecta (original salinity 5‰, diluted to freshwater) as food to obtain monoclonal cultures. These were maintained in an incubator at a temperature of 19 ± 1 °C and on a 12:12 h irradiance of 300 µmo photons/m2/s, and progressively adapted to 2.5‰ salinity by means of regular feeding (i.e. once a week) on D. tertiolecta.
Live observations
Live ciliates were observed for morphological identification using a differential interference contrast (DIC) microscope with a Leitz Orthoplan microscope (Weitzlar, Germany), with the help of a compression device182 in order to distort them as little as possible. For examination of the swimming behaviour, ciliates were observed in a Petri dish, under a stereomicroscope (WILD HEERBRUGG, Switzerland)181.
Silver and Feulgen stainings
Ciliates were treated for silver staining analysis with Champy’s solution and then with silver nitrate according to Ref.183, to stain the ciliary pattern. Feulgen staining procedure was performed to reveal the nuclear apparatus, using a protocol modified from Ref.184, after cell immobilization with celloidin-diethyl ether-alcohol solution181.
Scanning electron microscopy (SEM)
The specimens were fixed in 2% OsO4 for 40 min and glued on small coverslips (snipped from 1 × 1 cm size to the size of stub) previously coated with Poly-L-Lysine and subjected to consecutive dehydration in an ethanol series. Samples were critical point dried according to Ref.181. Later, these coverslips were fixed onto SEM stubs with carbon conductive tape. Finally, the samples were sputter-coated with gold (Edwards sputter coater S 150B) and analyzed with JSM-5410 scanning electron microscope181.
Transmission electron microscopy (TEM)
Euplotes cells were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer for 45 min, rinsed in 0.1 M cacodylate buffer and post-fixed in 1.5% aqueous osmium tetroxide in distilled water for 45 min at room temperature. Then cells were dehydrated and embedded in an Epon-araldite mixture as elsewhere described32. The blocks were sectioned with an RMC PowerTome X ultra-microtome. Sections were placed on copper grids and stained with uranyl acetate and lead citrate. Samples were visualized using a JEOL JEM-100SX electron microscope181.
Measurements and recordings
Morphometric data of properly oriented cells were taken by using both live and stained specimen preparations (i.e. Feulgen, silver staining, SEM). Optical microscopy pictures were captured with a digital camera (Canon Power Shot S45) and used to obtain dimensions of living and stained ciliates. Morphometric measurements were analyzed with ImageJ 1.46r software185.
Based on micrographs of living and stained cells, accurate schematic line drawings were produced with a procedure described by Ref.186, which employed bitmap graphics with the GNU Image Manipulation Program (GIMP)181.
Terminology and systematics are mainly according to Ref.187,188,189.
DNA extraction and 18S rRNA gene sequencing
Approximately 100–150 cells of KKR18_Esm strain were individually washed 3–5 times in sterile distilled water and fixed in 70% ethanol. Total genomic DNA extraction was performed using the NucleoSpin Plant II DNA extraction kit (Macherey–Nagel GmbH and Co., Düren NRW, Germany)181.
Polymerase chain reaction (PCR) was performed in a C1000 Thermal Cycler (Bio-Rad, Hercules, CA). The almost full-length of the 18S rRNA gene of Euplotes was amplified using the primer combination listed in Supplementary Table 7. High-fidelity Takara Ex Taq PCR reagents were employed (Takara Bio Inc., Otsu, Japan) according to the manufacturer’s instructions. PCR cycles were set as follows: 3 min 94 °C, 35 × [30 s 94 °C, 30 s 55 °C, 2 min 72 °C], 6 min 72 °C. PCR products were purified with the Eurogold Cycle-Pure Kit (EuroClone, Milan, Italy) and subsequently sent for direct sequencing to an external sequencing company (GATC Biotech AG, European Custom Sequencing Centre, Germany) by adding appropriate internal primers181 (see Supplementary Table 7).
Whole genome amplification and assembly
Starting from around 5–10 cells, the total DNA material was amplified via the whole-genome amplification (WGA) method, using REPLI-g Single Cell Kit (QIAGEN, Hilden, Germany). The cells of KKR18_Esm strain were washed in distilled water for three times and the last time in PBS buffer. Then, they were transferred to a 0.2 ml eppendorf together with 4 µl of PBS. The WGA protocol was completed following the manufacturer’s instructions. This DNA material was processed with a Nextera XT library and sequenced at Admera Health (South Plainfield, USA), using Illumina HiSeq X technology to generate 75,510,798 reads (paired-ends 2 × 150 bp). Preliminary assembly of resulting reads was performed using SPAdes software (v 3.6.0)190.
Mitochondrial genome assembly and annotation
Contigs representing the mitochondrial genome were identified using the Blobology pipeline191, and by tblastn searches using as queries proteins from reference genomes downloaded from NCBI, namely Oxytricha trifallax (JN383842) and Euplotes minuta (GQ903130). Contigs with a GC content comprised between 19 and 30%, and a coverage higher than 1000X were selected and a subset of the extracted reads were assembled with SPAdes. We decided to use approximately 10% of the extracted reads to artifically reduce the reads coverage, as coverage above 100 × generally produces worse assemblies, due to an increasing number of exactly replicated sequencing errors, which create false branches in the deBruijn graph. The assembled genome was annotated using PROKKA 1.10192, setting the DNA translation codon table “4” and then manually checked.
Endosymbiont genome assembly and annotation
The presence of symbionts and host related microbial consortium was inspected in the preliminary assembly, using Barrnap193 to detect 16S rRNA gene sequences, and manually checking all the contigs annotated as bacterial. This, in conjunction with the use of the Blobology pipeline as indicated above, allowed contigs selection with a GC content lower than 25% and a coverage higher than 1000X for the assembly of the symbiont genome, and a subset (about 10% of the extracted reads. See mitochondrial genome assembly and annotation in “Experimental procedures” section) of the extracted reads were re-assembled with SPAdes. Closure of the circular genome was confirmed via PCR. Specific primers were designed on the basis of genome assembly and used for PCR amplification [Pingui_F162297 (5′—GTT GTA GCT CTC GGA TCG—3′), Pingui_R436 (5′—GTA GAG CAT CTT CGA CTC G—3′)]; the following internal primers were used for sequencing PCR products [Pingui_F163091 (5′—CTC AGA GCA CTC TGA GAT AG—3′); Pingui_R199 (5′—GTT TAG CTC TTC CGA GAT CG—3′)]. PCR cycles were set as follows: 3 min 94 °C, 35 × [30 s 94 °C, 30 s 55 °C, 2 min 72 °C], 6 min 72 °C. We relied on the same reagents, instruments and sequencing company cited above.
The assembled genome was annotated using PROKKA 1.10192, setting the DNA translation codon table “4” and then manually checking the results. The predicted protein-coding genes were also classified using NCBI COGs194, and compared with selected small genomes (Supplementary Table 8) and previously characterized Verrucomicrobia genomes (Supplementary Table 5). The COGs thus obtained were used to carry out a Principal Component Analysis (PCA), using SciPy packages in Python, taking into account the numerosity of each COGs class.
Phylogenetic and phylogenomic analyses
The 18S rRNA gene of the host and the 16S rRNA gene of the newly characterized symbiont were aligned with the automatic aligner of the ARB software package version 5.5195 on the SSU ref NR99 SILVA database196. For the analysis on the host, 53 18S rRNA sequences of other members of the Euplotes genus, plus 7 sequences of other Spirotrichea as outgroup, were selected (dataset 1).
For the analysis on the symbiont, 98 16S rRNA sequences of other members of Verrucomicrobia were selected, plus 12 other sequences belonging to the superphylum Planctomycetes, Verrucomicrobia, Chlamydiae (PVC)197 as outgroup (dataset 2). Sequences not shown in the tree are listed in (Supplementary Table 9).
After manual editing to optimize base pairing in the predicted rRNA stem regions in each dataset, the two alignments were trimmed at both ends to the length of the shortest sequence. A positional filter was applied to dataset 1, to keep only those columns where the most conserved base was present in at least 10% of the sequences. Resulting matrices contained respectively 1843 (dataset 1) and 1456 (dataset 2) nucleotide columns, which were used for phylogeny and for the identity matrix calculation.
For each phylogenetic dataset, the optimal substitution model was selected with jModelTest 2.1198 according to the Akaike Information Criterion (AIC). Maximum likelihood (ML) trees were calculated with the PHYML software version 2.4199 from the ARB package, performing 1000 pseudo-replicates. Bayesian inference (BI) trees were inferred with MrBayes 3.2200, using three runs each with one cold and three heated Monte Carlo Markov chains, with a burn-in of 25%, iterating for 1,000,000 generations181.
For the phylogenomic analysis, 67 Verrucomicrobia (the endosymbiont plus 66 complete genomes from the genome taxonomy database (GTDB)140, and four other members of the PVC superphylum as outgroup, were used. A set of pre-aligned 120 single copy markers were employed140, and the “Ca. Pinguicoccus supinus” orthologs were identified by blastp search, then added to the existing alignments with MAFFT v7.123b201 and concatenated. A positional filter was applied to the concatenated genes according to Ref.140, to obtain a total of 34,747 sites. The best substitution model was estimated with ProtTest202. RaxML203 was used to estimate ML phylogeny with 1000 bootstraps.
Fluorescence microscopy
Euplotes specimens were fixed for Fluorescence In Situ Hybridization (FISH) experiments in 2% OsO4 and dehydrated after fixation with an increasing ethanol series for 10 min each. Specimens were processed for hybridization experiments according to previous publications181,204.
Preliminary FISH experiments were carried out using the generic probe for Verrucomicrobia EUB338 III205 and the generic probe for Bacteria EUB338206. Only the EUB338 III probe showed a slightly positive signal.
After 16S rRNA sequencing, we designed two probes on the 16S rRNA gene of the putative endosymbiont of Euplotes, EUB338 VII (5′—CTG CTG CCA TCC GTA GAT GT—3′) and Pingui_1174 (5′—ACT GAC TTG ACG TCA TCC TCA—3′). Both probes were tested in silico on the Ribosomal Database Project (RDP)207 and SILVA database using TestProbe 3.0196, allowing 0 mismatches. The sequence of probe EUB338 VII matched 475 bacterial sequences in the RDP database, 19 of them registered as Verrucomicrobia and 139 registered as “unclassified bacteria”. While probe Pingui_1174 matched 220 bacterial sequences, 35 of them registered as Verrucomicrobia and 180 registered as “unclassified bacteria”. Sequences of these two new probes were deposited into probeBase database208. Those probes were used to perform additional FISH experiments to confirm the presence of that particular species of Verrucomicrobia inside the host.
Hybridized slide preparations were observed under a Zeiss AxioPlan fluorescence microscope (Carl Zeiss, Oberkochen, Germany) equipped with an HBO 100 W/2 mercuric vapor lamp at the following UV wavelengths: ~ 495 nm, ~ 550 nm. Digital images were captured at different magnifications (40X and 100X) by means of a dedicated software (ACT2U, version 1.0)181.
Endosymbiont 16S rRNA gene screening on IMNGS
Diversity and environmental distribution of the bacterial endosymbiont was estimated using the IMNGS on-line platform56, which screens most of 16S rRNA gene amplicon datasets available. A query at 95% similarity was performed using the endosymbiont’s full-length 16S rRNA gene and related Puniceicoccaceae sequences (accession numbers: AB073978, AB372850, AB614893, AB826705, CP001998, DQ539046, EU462461, KT751307, JQ993599, JQ993517, Y19169, for their 16S rRNA full-length gene phylogenetic position see Fig. 7). The obtained sequences were longer than 300 bp and were divided into three different groups to perform phylogenetic analysis according to the 16S rRNA gene hypervariable regions V1–V2, V4–V6, and V7–V8. Sequences were clustered in OTUs with a 99% threshold similarity using UCLUST209, then they were aligned with MUSCLE210, and FastTree211 was employed to infer phylogenetic analyses. Thereafter, environmental distribution was investigated using the endosymbiont sequence as query and their abundances were calculated.
Nomenclature acts
The present work has been registered in ZooBank (code: LSID:urn:lsid:zoobank.org:pub:231DF703-DBA8-4839-9755-5C71D6872406), as well as the new Euplotes species, Euplotes vanleeuwenhoeki (code: LSID:urn:lsid:zoobank.org:act:19C2249B-BF2D-4E81-9391-A271D38CD7B6). The correspondent web pages are available at the following addresses: https://www.zoobank.org/References/231DF703-DBA8-4839-9755-5C71D6872406 and https://www.zoobank.org/NomenclaturalActs/19C2249B-BF2D-4E81-9391-A271D38CD7B6 , respectively.
Data availability
All data generated or analysed during this study are included in this published article. All the 18S rRNA sequences, mitogenome sequence, “Ca. Pinguicoccus supinus” genome have been deposited in GenBank. Permanent slides are available from museums (indicated in the text) and from the corresponding author on reasonable request.
References
Mallet, J. & Willmott, K. Taxonomy: Renaissance or Tower of Babel? Trends Ecol. Evol. 18, 57–59 (2003).
Agnarsson, I. & Kuntner, M. Taxonomy in a changing world: seeking solutions for a science in crisis. Syst. Biol. 56, 531–539 (2007).
Walter, D. E. & Winterton, S. Keys and the crisis in taxonomy: Extinction or reinvention?. Annu. Rev. Entomol. 52, 193–208 (2007).
de Bary, A. Die Erscheinung der Symbiose (Verlag von Karl J. Trübner, Strassburg, 1879).
Giambelluca, M. A. & Rosati, G. Behavior of epixenosomes and the epixenosomal band during divisional morphogenesis in Euplotidium itoi (Ciliata, Hypotrichida). Eur. J. Protistol. 32, 77–80 (1996).
Rosati, G., Petroni, G., Quochi, S., Modeo, L. & Verni, F. Epixenosomes: peculiar epibionts of the hypotrich ciliate Euplotidium itoi defend their host against predators. J. Eukaryot. Microbiol. 46, 278–282 (1999).
McFall-Ngai, M. Unseen forces: the influence of bacteria on animal development. Dev. Biol. 242, 1–14. https://doi.org/10.1006/dbio.2001.0522 (2002).
Gilbert, S. F. et al. Symbiosis as a source of selectable epigenetic variation: taking the heat for the big guy. Philos. Trans. R. Soc. B 365, 671–678 (2010).
Gilbert, S. F., Bosch, T. C. G. & Ledón-Rettig, C. Eco-Evo-Devo: developmental symbiosis and developmental plasticity as evolutionary agents. Nat. Rev. Genet. 16, 611. https://doi.org/10.1038/nrg3982 (2015).
Pradeu, T. A mixed self: the role of symbiosis in development. Biol. Theory 6, 80–88. https://doi.org/10.1007/s13752-011-0011-5 (2011).
Webster, N. S., Botté, E. S., Soo, R. M. & Whalan, S. The larval sponge holobiont exhibits high thermal tolerance. Environ. Microbiol. Rep. 3, 756–762. https://doi.org/10.1111/j.1758-2229.2011.00296.x (2011).
Bella, C. et al. Fitness impact of obligate intranuclear bacterial symbionts depends on host growth phase. Front. Microbiol. 7, 2084 (2016).
Grosser, K. et al. More than the “killer trait”: infection with the bacterial endosymbiont caedibacter taeniospiralis causes transcriptomic modulation in Paramecium host. Genome Biol. Evol. 10, 646–656 (2018).
Bright, M. et al. Thiotrophic bacterial symbiont induces polyphenism in giant ciliate host Zoothamnium niveum. Sci. Rep. 9, 1–1 (2019).
Meyer-Abich, A. Beiträge zur Theorie der Evolution der Organismen: Typensynthese Durch Holobiose (E.J Brill Archive, Leiden, 1950).
Margulis, L. Symbiogenesis and symbionticism. In Symbiosis as a Source of Evolutionary Innovation (eds Margulis, L. & Fester, R.) 1–14 (MIT Press, Cambridge, 1991).
Mindell,. Phylogenetic consequences of symbioses: Eukarya and Eubacteria are not monophyletic taxa. BioSystems. 27, 53–62 (1992).
Casiraghi, M. Being an individual (or a species) in a symbiotic world. Paradigmi 3, 59–69 (2012).
Moran, N. A. & Sloan, D. B. The hologenome concept: Helpful or hollow?. PLoS Biol. 13, e1002311 (2015).
Douglas, A. E. & Werren, J. H. Holes in the hologenome: why host-microbe symbioses are not holobionts. mBio 7, e02099-e2115 (2016).
Hester, E. R., Barott, K. L., Nulton, J., Vermeij, M. J. & Rohwer, F. L. Stable and sporadic symbiotic communities of coral and algal holobionts. ISME J. 10, 1157–1169 (2016).
Queller, D. C. & Strassmann, J. E. Problems of multi-species organisms: endosymbionts to holobionts. Biol. Philos. 31, 855–873 (2016).
Skillings, D. Holobionts and the ecology of organisms: Multi-species communities or integrated individuals?. Biol. Philos. 31, 875–892 (2016).
Graham, L. E. & Graham, J. M. Endosymbiotic Chlorella (Chlorophyta) in a species of Vorticella (Ciliophora). Trans. Am. Microsc. Soc. 99, 160–166 (1980).
Finlay, B. J., Berninger, U. G., Stewart, L. J., Hindle, R. M. & Davison, W. Some factors controlling the distribution of two pond-dwelling ciliates with algal symbionts (Frontonia vernalis and Euplotes daidaleos). J. Protozool. 34, 349–356 (1987).
Kodama, Y. & Fujishima, M. Cell division and density of symbiotic Chlorella variabilis of the ciliate Paramecium bursaria is controlled by the host’s nutritional conditions during early infection process. Environ. Microbiol. 14, 2800–2811 (2012).
Fokin, S. I., Schrallhammer, M., Chiellini, C., Verni, F. & Petroni, G. Free-living ciliates as potential reservoirs for eukaryotic parasites: occurrence of a trypanosomatid in the macronucleus of Euplotes encysticus. Parasite. Vector 7, 203 (2014).
Lanzoni, O. et al. Rare freshwater ciliate Paramecium chlorelligerum Kahl, 1935 and its macronuclear symbiotic bacterium “Candidatus Holospora parva”. PLoS ONE 11(12), e0167928. https://doi.org/10.1371/journal.pone.0167928 (2016).
He, M. et al. Genetic basis for the establishment of endosymbiosis in Paramecium. ISME J. https://doi.org/10.1038/s41396-018-0341-4 (2019).
van Hoek, A. H. et al. Multiple acquisition of methanogenic archaeal symbionts by anaerobic ciliates. Mol. Biol. Evol. 17, 251–258 (2000).
Ferrantini, F. et al. “Candidatus Cryptoprodotis polytropus”, a novel Rickettsia-like organism in the ciliated protist Pseudomicrothorax dubius (Ciliophora, Nassophorea). J. Eukaryot. Microbiol. 56, 119–129 (2009).
Modeo, L. et al. Morphology, ultrastructure and molecular phylogeny of the ciliate Sonderia vorax with insights into the systematics of order Plagiopylida. BMC Microbiol. 13, 40. https://doi.org/10.1186/1471-2180-13-40 (2013).
Modeo, L., Petroni, G., Lobban, C. S., Verni, F. & Vannini, C. Morphological, ultrastructural, and molecular characterization of Euplotidium rosati n. sp. (Ciliophora, Euplotida) from Guam. J. Eukaryot. Microbiol. 60, 25–36 (2013).
Gong, J., Qing, Y., Guo, X. & Warren, A. “Candidatus Sonnebornia yantaiensis”, a member of candidate division OD1, as intracellular bacteria of the ciliated protist Paramecium bursaria (Ciliophora, Oligohymenophorea). Syst. Appl. Microbiol. 37, 35–41 (2014).
Serra, V. et al. “Candidatus Gortzia shahrazadis”, a novelendosymb iont of Paramecium multimicronucleatum and revision of the biogeographical distribution of Holospora-like bacteria. Front. Microbiol. 7, 1704. https://doi.org/10.3389/fmicb.2016.01704 (2016).
Szokoli, F. et al. Disentangling the taxonomy of Rickettsiales and description of two novel symbionts (“Candidatus Bealeia paramacronuclearis” and “Candidatus Fokinia cryptica”) sharing the cytoplasm of the ciliate protist Paramecium biaurelia. Appl. Environ. Microbiol. 82, 7236–7247 (2016).
Castelli, M. et al. The hidden world of Rickettsiales symbionts: “Candidatus Spectririckettsia obscura”, a novel bacterium found in Brazilian and Indian Paramecium caudatum. Microb. Ecol. https://doi.org/10.1007/s00248-018-1243-8 (2019).
Fokin, S. I., Serra, V., Ferrantini, F., Modeo, L. & Petroni, G. “Candidatus Hafkinia simulans” gen. nov., sp. nov., a novel Holospora-like bacterium from the macronucleus of the rare brackish water ciliate Frontonia salmastra (Oligohymenophorea, Ciliophora): multidisciplinary characterization of the new endosymbiont and its host. Microb. Ecol. https://doi.org/10.1007/s00248-018-1311-0 (2019).
Warren, A. et al. Beyond the ‘Code’: a guide to the description and documentation of biodiversity in ciliated protists (Alveolata, Ciliophora). J. Eukaryot. Microbiol. 64, 539–554 (2017).
McCutcheon, J. P. & Moran, N. A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13 (2012).
Bennett, G. M., Abbà, S., Kube, M. & Marzachì, C. Complete genome sequences of the obligate symbionts “Candidatus Sulcia muelleri” and “Ca. Nasuia deltocephalinicola” from the pestiferous Leafhopper Macrosteles quadripunctulatus (Hemiptera: Cicadellidae). Genome Announc. 4, e01604-e1615 (2016).
Faure´-Fremiet E., Andre´ J. ,. Structure fine de l’ Euplotes eurystomus (WRZ). Arch. Anat. Micro. Morph. Ex. 57, 53–78 (1968).
Nobili, R. & Rosati, R. G. The fine structure of the oral apparatus of some ciliate Hypotrichs. Mon. Zool. Ital. 5, 65–80 (1971).
Kloetzel, J. A. Feeding in ciliated Protozoa. Pharyngeal disks in Euplotes: a source of membrane for food vacuole formation. J. Cell. Sci. 15, 379–401 (1974).
Ruffolo, J. J. Fine structure of the dorsal bristle complex and pellicle of Euplotes eurystomus, a ciliate protozoon. J. Morphol. 148, 469–488 (1976).
Lobban, C. S., Modeo, L., Verni, F. & Rosati, G. Euplotes uncinatus (Ciliophora, Hypotrichia), a new species with zooxanthellae. Mar. Biol. 147, 1055–1061 (2005).
Schwarz, M. V., Zuendorf, A. & Stoeck, T. Morphology, ultrastructure, molecular phylogeny, and autecology of Euplotes elegans Kahl, 1932 (Hypotrichida; Euplotidae) isolated from the anoxic Mariager Fjord, Denmark. J. Eukaryot. Microbiol. 54, 125–136 (2007).
Rosati, G. & Modeo, L. Extrusomes in ciliates: diversification distribution, and phylogenetic implications. J. Eukaryot. Microbiol. 50, 383–400 (2003).
Syberg-Olsen, M. J. et al. Biogeography and character evolution of the ciliate genus Euplotes (Spirotrichea, Euplotia), with description of Euplotes curdsi sp. nov. PLoS ONE 11, e0165442 (2016).
Boscaro, V., Syberg-Olsen, M. J., Irwin, N. A., del Campo, J. & Keeling, P. J. What can environmental sequences tell us about the distribution of low-rank taxa? The case of Euplotes (Ciliophora, Spirotrichea), including a description of Euplotes enigma sp. nov. J. Euk. Microbiol. 66, 281–293. https://doi.org/10.1111/jeu.12669 (2019).
de Graaf, R. M. et al. The mitochondrial genomes of the ciliates Euplotes minuta and Euplotes crassus. BMC Genom. 10, 514. https://doi.org/10.1186/1471-2164-10-514 (2009).
Swart, E. C. et al. The Oxytricha trifallax mitochondrial genome. Genome Biol. Evol. 4, 136–154. https://doi.org/10.1093/gbe/evr136 (2011).
Lindsay, M. R. et al. Cell compartmentalisation in planctomycetes: novel types of structural organisation for the bacterial cell. Arc. Microbiol. 175, 413–429 (2001).
Moran, N. A. & Bennett, G. M. The tiniest tiny genomes. Annu. Rev. Microbiol. 68, 195–215 (2014).
Bergsten, J. A review of long-branch attraction. Cladistics 21, 163–193 (2005).
Lagkouvardos, I. et al. IMNGS: a comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 6, 33721 (2016).
Al-Jassim, N., Ansari, M. I., Harb, M. & Hong, P. Y. Removal of bacterial contaminants and antibiotic resistance genes by conventional wastewater treatment processes in Saudi Arabia: Is the treated wastewater safe to reuse for agricultural irrigation?. Water Res. 73, 277–290 (2015).
Fenchel, T. & Lee, C. C. Studies on ciliates associated with sea ice from Antarctica. I. The nature of the fauna. Arch. Protistenk 114, 231–236 (1972).
Petz, W., Song, W. & Wilbert, N. Taxonomy and Ecology of the Ciliate Fauna (Protozoa, Ciliophora) in the Endopagial and Pelagial of the Weddell Sea, Antarctica 40 (OÖ Landesmuseum, Linz, 1995).
Carter, H. P. Infraciliature of eleven species of the genus Euplotes. Trans. Am. Microsc. Soc. 91, 466–492 (1972).
Valbonesi, A. & Luporini, P. Description of two new species of Euplotes and Euplotes rariseta from Antarctica. Polar Biol. 11, 47–53 (1990).
Song, W. & Packroff, G. Taxonomische Untersuchungen an marinen Ciliaten aus China mit Beschreibungen von zwei neuen Arten, Strombidium globosaneum nov. spec. and S. platum nov. spec. (Protozoa, Ciliophora). Arch. Protstenk. 147, 331–360 (1997).
Shao, C., Ma, H., Gao, S., Khaled, A. R. A. & Song, W. Reevaluation of cortical developmental patterns in Euplotes (sl), including a morphogenetic redescription of E. charon (Protozoa, Ciliophora, Euplotida). Chin. J. Ocean. Limnol. 28, 593–602. https://doi.org/10.1007/s00343-010-9128-9 (2010).
Petroni, G., Dini, F., Verni, F. & Rosati, G. A molecular approach to the tangled intrageneric relationships underlying phylogeny in Euplotes (Ciliophora, Spirotrichea). Mol. Phylogenet. Evol. 22, 118–130 (2002).
Li, L. & Song, W. Phylogenetic position of the marine ciliate, Certesia quadrinucleata (Ciliophora; Hypotrichia; Hypotrichida) inferred from the complete small subunit ribosomal RNA gene sequence. Eur. J. Protistol. 42, 55–61 (2006).
Vallesi, A., Di Giuseppe, G., Dini, F. & Luporini, P. Pheromone evolution in the protozoan ciliate, Euplotes: the ability to synthesize diffusible forms is ancestral and secondarily lost. Mol. Phylogenet. Evol. 47, 439–442 (2008).
Zhao, Y., Yi, Z., Warren, A. & Song, W. B. Species delimitation for the molecular taxonomy and ecology of the widely distributed microbial eukaryote genus Euplotes (Alveolata, Ciliophora). Proc. R. Soc. B Biol. Sci. 285, 20172159 (2018).
Huang, J., Dunthorn, M. & Song, W. Expanding character sampling for the molecular phylogeny of euplotid ciliates (Protozoa, Ciliophora) using three markers, with a focus on the family Uronychiidae. Mol. Phylogenet. Evol. 63, 598–605 (2012).
Jones, T. C. & Gates, M. A. A morphometric analysis of the Euplotes charon morphotype (Ciliophora: Euplotida. J. Eukaryot. Microbiol. 41, 441–450 (1994).
Lian, C. et al. Morphological and molecular redefinition of Euplotes platystoma Dragesco and Dragesco-Kernéis, 1986 and Aspidisca lynceus (Müller, 1773) Ehrenberg, 1859, with reconsideration of a “Well-known” Euplotes ciliate, Euplotes harpa Stein, 1859 (Ciliophora, Euplotida). J. Eukaryot. Microbiol. 65, 531–543 (2018).
Valbonesi, A. & Luporini, P. Euplotes bisulcatus, a key species for a better understanding of the organization and evolution of Euplotes argyrome patterns. Eur. J. Protistol. 31, 32–37 (1995).
Valbonesi, A. & Luporini, P. A new marine species of Euplotes (Ciliophora, Hypotrichida) from Antarctica. Bull. Br. Mus. Nat. Hist. (Zool.) 56, 57–61 (1990).
Tuffrau, M. Le maintien des caractères specifiques à travers le polymorphisme d’Euplotes balteatus. Archs. Zool. 104, 143–151 (1964).
Kuhlmann, H.-W. & Heckmann, K. Predation risk of typical ovoid and winged morphs of Euplotes (Protozoa, Ciliophora). Hydrobiologia 284, 219–227 (1994).
Kopp, M. & Tollrian, R. Reciprocal phenotypic plasticity in a predator–prey system: Inducible offences against inducible defences?. Ecol. Lett. 6, 742–748 (2003).
Tuffrau, M., Fryd-Versavel, G., Tuffrau, H. & Génermont, J. Description of Euplotes versatilis n. sp., a marine tropical ciliate exhibiting an unusually extensive phenotypic plasticity. Eur. J. Protistol. 36, 355–366 (2000).
Washburn, E. S. & Borror, A. C. Euplotes raikovi Agamaliev, 1966 (Ciliophora, Hypotrichida) from New Hampshire: description and morphogenesis. J. Protozool. 19, 604–608 (1972).
Jiang, J., Zhang, Q., Warren, A., Al-Rasheid, K. A. S. & Song, W. Morphology and SSU rRNA gene-based phylogeny of two marine Euplotes species, E. orientalis spec. nov. and E. raikovi Agamaliev, 1966 (Ciliophora, Euplotida). Eur. J. Protistol. 46, 121–132. https://doi.org/10.1016/j.ejop.2009.11.003 (2010) (PMID:20172700).
Suyama, Y. & Miura, K. Size and structural variations of mitochondrial DNA. Proc. Natl. Acad. Sci. U.S.A. 60, 235–242 (1968).
Morin, G. B. & Cech, T. R. The telomeres of the linear mitochondrial DNA of Tetrahymena thermophila consist of 53 bp tandem repeats. Cell 46, 873–883 (1986).
Schnare, M. N., Heinonen, T. Y., Young, P. G. & Gray, M. W. A discontinuous small subunit ribosomal RNA in Tetrahymena pyriformis mitochondria. J. Biol. Chem. 261, 5187–5193 (1986).
Burger, G. et al. Complete sequence of the mitochondrial genome of Tetrahymena pyriformis and comparison with Paramecium aurelia mitochondrial DNA1. J. Mol. Biol. 297, 365–380 (2000).
Blanchette, M., Kunisawa, T. & Sankoff, D. Parametric genome rearrangement. Gene 172, GV11–GV17 (1996).
Boore, J. L. & Brown, W. M. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 8, 668–674 (1998).
Dumilag, R. V. et al. Phenotypic and mtDNA variation in Philippine Kappaphycus cottonii (Gigartinales, Rhodophyta). Mitochondrial DNA 29, 951–963 (2018).
Barth, D., Krenek, S., Fokin, S. I. & Berendonk, T. U. Intraspecific genetic variation in Paramecium revealed by mitochondrial cytochrome c oxidase I sequences. J. Eukaryot. Microbiol. 50, 20–25 (2006).
Doerder, F. P. Barcodes Reveal 48 New Species of Tetrahymena, Dexiostoma, and Glaucoma: phylogeny, ecology, and biogeography of new and established species. J. Eukaryot. Microbiol. 66, 182–208 (2019).
Nygren, A. et al. A mega-cryptic species complex hidden among one of the most common annelids in the North East Atlantic. PLoS ONE 13, 0198356 (2018).
Ya’cob, Z., Takaoka, H., Low, V. L. & Sofian-Azirun, M. First description of a new cryptic species, Simulium vanluni from Peninsular Malaysia: an integrated morpho-taxonomical and genetic approach for naming cryptic species in the family Simuliidae. Acta. Trop. 167, 31–39 (2017).
Ilyasov, R. A., Poskryakov, A. V. & Nikolenko, A. G. Seven genes of mitochondrial genome enabling differentiation of honeybee subspecies Apis mellifera. Russ. J. Genet. 52, 1062–1070 (2016).
Lavrov, D. V., Brown, W. M. & Boore, J. L. Phylogenetic position of the Pentastomida and (pan) crustacean relationships. Proc. R. Soc. Lond. B Biol. Sci. 271, 537–544 (2004).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Diller, W. F. & Kounaris, D. Description of a zoochlorella-bearing form of Euplotes, E. daidaleos n. sp. (Ciliophora Hypotrichida). Biol. Bull. 131, 437–445 (1966).
Fokin, S. I., Di Giuseppe, G., Erra, F. & Dini, F. Euplotespora binucleata n. gen., n. sp. (Protozoa: Microsporidia), a parasite infecting the hypotrichous ciliate Euplotes woodruffi, with observations on microsporidian infections in Ciliophora. J. Eukaryot. Microbiol. 55, 214–228 (2008).
Heckmann, K., Hagen, R. T. & Görtz, H. D. Freshwater Euplotes species with a 9 type 1 cirrus pattern depend upon endosymbionts. J. Protozool. 30, 284–289 (1983).
Vannini, C., Rosati, G., Verni, F. & Petroni, G. Identification of the bacterial endosymbionts of the marine ciliate Euplotes magnicirratus (Ciliophora, Hypotrichia) and proposal of ‘Candidatus Devosia euplotis’. Int. J. Syst. Evol. Microbiol. 54, 1151–1156 (2004).
Vannini, C., Ferrantini, F., Verni, F. & Petroni, G. A new obligate bacterial symbiont colonizing the ciliate Euplotes in brackish and freshwater: ‘Candidatus Protistobacter heckmanni’. Aquat. Microb. Ecol. 70, 233–243 (2013).
Boscaro, V. et al. Symbiont replacement between bacteria of different classes reveals additional layers of complexity in the evolution of symbiosis in the ciliate Euplotes. Protist 169, 43–52 (2018).
Boscaro, V., Husnik, F., Vannini, C. & Keeling, P. J. Symbionts of the ciliate Euplotes: diversity, patterns and potential as models for bacteria–eukaryote endosymbioses. Proc. Roy. Soc. B Biol. Sci. 286, 20190693 (2019).
Heckmann, K. & Schmidt, H. J. Polynucleobacter necessarius gen. nov., sp. nov., an obligately endosymbiotic bacterium living in the cytoplasm of Euplotes aediculatus. Int. J. Syst. Evol. Microbiol. 37, 456–457 (1987).
Boscaro, V., Vannini, C., Fokin, S. I., Verni, F. & Petroni, G. Characterization of “Candidatus Nebulobacter yamunensis” from the cytoplasm of Euplotes aediculatus (Ciliophora, Spirotrichea) and emended description of the family Francisellaceae. Syst. Appl. Microbiol. 35, 432–440 (2012).
Boscaro, V., Petroni, G., Ristori, A., Verni, F. & Vannini, C. “Candidatus Defluviella procrastinata” and “Candidatus Cyrtobacter zanobii”, two novel ciliate endosymbionts belonging to the “Midichloria clade. Microb. Ecol. 65, 302–310 (2013).
Schrallhammer, M. et al. ‘Candidatus Megaira polyxenophila’ gen. nov., sp. nov.: considerations on evolutionary history, host range and shift of early divergent rickettsiae. PLoS ONE 8, e72581 (2013).
Vannini, C. et al. “Candidatus Anadelfobacter veles” and “Candidatus Cyrtobacter comes”, two new Rickettsiales species hosted by the protist ciliate Euplotes harpa (Ciliophora, Spirotrichea). Appl. Environ. Microbiol. 76, 4047–4054 (2010).
Vannini, C. et al. Flagellar movement in two bacteria of the family Rickettsiaceae: a re-evaluation of motility in an evolutionary perspective. PLoS ONE 9, e87718 (2014).
Chiellini, C. et al. Harmful effect of Rheinheimera sp. EpRS3 (Gammaproteobacteria) against the protist Euplotes aediculatus (Ciliophora, Spirotrichea): insights into the ecological role of antimicrobial compounds from environmental bacterial strains. Front. Microbiol. 10, 510. https://doi.org/10.3389/fmicb.2019.00510 (2019).
Lanzoni, O. et al. Diversity and environmental distribution of the cosmopolitan endosymbiont “Candidatus Megaira”. Sci. Rep. https://doi.org/10.1038/s41598-018-37629-w (2019).
Senra, M. V. et al. A house for two—double bacterial infection in Euplotes woodruffi Sq1 (Ciliophora, Euplotia) sampled in Southeastern Brazil. Microb. Ecol. 71, 505–517 (2016).
Schrallhammer, M., Schweikert, M., Vallesi, A., Verni, F. & Petroni, G. Detection of a novel subspecies of Francisella noatunensis as endosymbiont of the ciliate Euplotes raikovi. Microb. Ecol. 61, 455–464 (2011).
Vallesi, A. et al. A new species of the γ-Proteobacterium Francisella, F. adeliensis sp. nov., endocytobiont in an antarctic marine ciliate and potential evolutionary forerunner of pathogenic species. Microb. Ecol. 77, 1–10 (2019).
Zhang, L. & Xu, Z. Assessing bacterial diversity in soil. J. Soils Sediments 8, 379–388 (2008).
Freitas, S. et al. Global distribution and diversity of marine Verrucomicrobia. ISME J. 6, 1499 (2012).
Pearce, D. A., van der Gast, C. J., Lawley, B. & Ellis-Evans, J. C. Bacterioplankton community diversity in a maritime Antarctic lake, determined by culture-dependent and culture-independent techniques. FEMS Microbiol. Ecol. 45, 59–70 (2003).
Hou, S. et al. Complete genome sequence of the extremely acidophilic methanotroph isolate V4, Methylacidiphilum infernorum, a representative of the bacterial phylum Verrucomicrobia. Biol. Direct. 3, 26 (2008).
Vandekerckhove, T. T., Willems, A., Gillis, M. & Coomans, A. Occurrence of novel verrucomicrobial species, endosymbiotic and associated with parthenogenesis in Xiphinema americanum-group species (Nematoda, Longidoridae). Int. J. Syst. Evol. Microbiol. 50, 2197–2205 (2000).
Sakai, T., Ishizuka, K. & Kato, I. Isolation and characterization of a fucoidan-degrading marine bacterium. Mar. Biotechnol. 5, 409–416 (2003).
Collins, A. J. et al. Diversity and partitioning of bacterial populations within the accessory nidamental gland of the squid Euprymna scolopes. Appl. Environ. Microbiol. 78, 4200–4208 (2012).
Yildirim, S. et al. Characterization of the fecal microbiome from non-human wild primates reveals species specific microbial communities. PLoS ONE 5, 13963 (2010).
Romero-Pérez, G. A., Ominski, K. H., McAllister, T. A. & Krause, D. O. Effect of environmental factors and influence of rumen and hindgut biogeography on bacterial communities in steers. Appl. Environ. Microbiol. 77, 258–268 (2011).
Wang, M., Ahrne, S., Jeppsson, B. & Molin, G. Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol. Ecol. 54, 219–231 (2005).
Rosati, G. et al. Epixenosomes, peculiar epibionts of the ciliated protozoon Euplotidium itoi: What kind of organisms are they?. Protoplasma 201, 38–44 (1998).
Petroni, G., Spring, S., Schleifer, K. H., Verni, F. & Rosati, G. Defensive extrusive ectosymbionts of Euplotidium (Ciliophora) that contain microtubule-like structures are bacteria related to Verrucomicrobia. Proc. Natl. Acad. Sci. 97, 1813–1817 (2000).
Fuerst, J. A. & Sagulenko, E. Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat. Rev. Microbiol. 6, 403 (2011).
Devos, D. P. PVC bacteria: variation of, but not exception to, the Gram-negative cell plan. Trends Microbiol. 22, 14–20 (2014).
Lee, K. C. et al. Phylum Verrucomicrobia representatives share a compartmentalized cell plan with members of bacterial phylum Planctomycetes. BMC Microbiol. 9, 5 (2009).
Pinos, S., Pontarotti, P., Raoult, D., Baudoin, J. P. & Pagnier, I. Compartmentalization in PVC super-phylum: evolution and impact. Biol. Direct 11, 38 (2016).
Medeiros, J. M., Böck, D. & Pilhofer, M. Imaging bacteria inside their host by cryo-focused ion beam milling and electron cryotomography. Curr. Opin. Microbiol. 43, 62–68 (2018).
Bennett, G. M. & Moran, N. A. Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol. Evol. 5, 1675–1688 (2013).
Kotak, M. et al. Complete genome sequence of the Opitutaceae bacterium strain TAV5, a potential facultative methylotroph of the wood-feeding termite Reticulitermes flavipes. Genome Announc. 3(2), e00060-e115 (2015).
Brown, A. M. et al. Comparative genomics of a plant-parasitic nematode endosymbiont suggest a role in nutritional symbiosis. Genome Biol. Evol. 7, 2727–2746 (2015).
Kobiałka, M., Michalik, A., Walczak, M., Junkiert, Ł & Szklarzewicz, T. Sulcia symbiont of the leafhopper Macrosteles laevis (Ribaut, 1927) (Insecta, Hemiptera, Cicadellidae: Deltocephalinae) harbors Arsenophonus bacteria. Protoplasma 253, 903–912 (2016).
Wilson, A. C. C. et al. Genomic insight into the amino acid relations of the pea aphid, Acyrthosiphon pisum, with its symbiotic bacterium Buchnera aphidicola. Insect. Mol. Biol. 19, 249–258 (2010).
Boscaro, V. et al. Polynucleobacter necessarius, a model for genome reduction in both free-living and symbiotic bacteria. Proc. Natl. Acad. Sci. U.S.A. 110, 18590–18595 (2013).
Boscaro, V. et al. Parallel genome reduction in symbionts descended from closely related free-living bacteria. Nat. Ecol. Evol. 1, 1160 (2018).
McCutcheon, J. P., McDonald, B. R. & Moran, N. A. Origin of an alternative genetic code in the extremely small and GC–rich genome of a bacterial symbiont. PLoS Genet. 5, 1000565 (2009).
Osawa, S. & Jukes, T. H. Evolution of the genetic code as affected by anticodon content. Trends Genet. 4, 191–198 (1988).
Yoon, J., Matsuo, Y., Matsuda, S., Kasai, H. & Yokota, A. Cerasicoccus maritimus sp. nov. and Cerasicoccus frondis sp. nov., two peptidoglycan-less marine verrucomicrobial species, and description of Verrucomicrobia phyl. nov., nom. rev. J. Gen. Appl. Microbiol. 56, 213–222 (2010).
Yoon, J. Phylogenetic studies on the bacterial phylum ‘Verrucomicrobia’. Microbiol. Cult. Coll. 27, 61–65 (2011).
Kim, M. et al. Luteolibacter arcticus sp. nov., isolated from high Arctic tundra soil, and emended description of the genus Luteolibacter. Int. J. Syst. Evol. Microbiol. 65, 1922–1928 (2015).
Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004 (2018).
Bergmann, G. T. et al. The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol. Biochem. 43, 1450–1455 (2011).
Takahashi, S., Tomita, J., Nishioka, K., Hisada, T. & Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next generation sequencing. PLoS ONE 9, e105592 (2014).
Rosenberg, E., Koren, O., Reshef, L., Efrony, R. & Zilber-Rosenberg, I. The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 5, 355–362 (2007).
Zilber-Rosenberg, I. & Rosenberg, E. Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbial. Rev. 32, 723–735 (2008).
Rosenberg, E., Sharon, G. & Zilber-Rosenberg, I. The hologenome theory of evolution contains Lamarckian aspects within a Darwinian framework. Environ. Microbiol. 11, 2959–2962 (2009).
Rosenberg, E. & Zilber-Rosenberg, I. Symbiosis and development: the hologenome concept. Birth Defects Res. C 93, 56–66 (2011).
Bordenstein, S. R. & Theis, K. R. Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol. 13, e1002226 (2015).
Bosch, T. C. & Miller, D. J. The Holobiont Imperative: Perspectives from Early Emerging Animals (Springer, Vienna, 2016).
Roughgarden, J., Gilbert, S. F., Rosenberg, E., Zilber-Rosenberg, I. & Lloyd, E. A. Holobionts as units of selection and a model of their population dynamics and evolution. Biol. Theory 13, 44–65 (2018).
Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Le Van, A. & Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206 (2015).
Sanchez-Canizares, C., Jorrin, B., Poole, P. S. & Tkacz, A. Understanding the holobiont: the interdependence of plants and their microbiome. Curr. Opin. Microbiol. 38, 188–196 (2017).
Egan, S. et al. The seaweed holobiont: understanding seaweed–bacteria interactions. FEMS Microbiol. Rev. 37, 462–476 (2012).
Guerrero, R., Margulis, L. & Berlanga, M. Symbiogenesis: the holobiont as a unit of evolution. Int. Microbiol. 16, 133–143 (2013).
Minard, G., Mavingui, P. & Moro, C. V. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasite Vector 6, 146 (2013).
Berlanga, M. & Guerrero, R. The holobiont concept: the case of xylophagous termites and cockroaches. Symbiosis 68, 49–60 (2016).
Vezzulli, L., Pezzati, E., Huete-Stauffer, C., Pruzzo, C. & Cerrano, C. 16SrDNA pyrosequencing of the Mediterranean gorgonian Paramuricea clavata reveals a link among alterations in bacterial holobiont members, anthropogenic influence and disease outbreaks. PLoS ONE 8, e67745 (2013).
Thompson, J. R., Rivera, H. E., Closek, C. J. & Medina, M. Microbes in the coral holobiont: partners through evolution, development, and ecological interactions. Front. Cell. Infect. Microbiol. 4, 176 (2015).
van de Water, J. A., Allemand, D. & Ferrier-Pagès, C. Host-microbe interactions in octocoral holobionts-recent advances and perspectives. Microbiome 6, 64 (2018).
Postler, T. S. & Ghosh, S. Understanding the holobiont: how microbial metabolites affect human health and shape the immune system. Cell Metab. 26, 110–130 (2017).
Šlapeta, J., Moreira, D. & López-García, P. The extent of protist diversity: insights from molecular ecology of freshwater eukaryotes. Proc. R. Soc. Lond. B Biol. Sci. 272, 2073–2081 (2005).
Foissner, W. Biogeography and dispersal of micro-organisms: a review emphasizing protists. Acta Protozool. 45, 111–136 (2006).
Weisse, T. Distribution and diversity of aquatic protists: an evolutionary and ecological perspective. Biodivers. Conserv. 17, 243–259 (2008).
Molmeret, M., Horn, M., Wagner, M., Santic, M. & Kwaik, Y. A. Amoebae as training grounds for intracellular bacterial pathogens. Appl. Environ. Microbiol. 71, 20–28 (2005).
Horn, M. Chlamydiae as symbionts in eukaryotes. Annu. Rev. Microbiol. 62, 113–131 (2008).
Ohkuma, M. Symbioses of flagellates and prokaryotes in the gut of lower termites. Trends Microbiol. 16, 345–352 (2008).
Schmitz-Esser, S. et al. The genome of the amoeba symbiont “Candidatus Amoebophilus asiaticus” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J. Bacteriol. 192, 1045–1057 (2010).
Gast, R. J., Sanders, R. W. & Caron, D. A. Ecological strategies of protists and their symbiotic relationships with prokaryotic microbes. Trends Microbiol. 17, 563–569 (2009).
Görtz, H.-D. Microbial infections in free-living protozoa. Crit. Rev. Immunol. 30(1), 95–106 (2010).
Nowack, E. C. & Melkonian, M. Endosymbiotic associations within protists. Philos. Trans. R. Soc. B 365, 699–712 (2010).
Schweikert, M., Fujishima, M. & Görtz, H.-D. Symbiotic associations between ciliates and prokaryotes. In The Prokaryotes—Prokaryotic Biology and Symbiotic Associations (eds Rosenberg, E. et al.) 427–458 (Springer, Berlin, 2013).
Scheid, P. Relevance of free-living amoebae as hosts for phylogenetically diverse microorganisms. Parasitol. Res. 113, 2407–2414 (2014).
Chen, Y. et al. Mitochondrial DNA genomes revealed different patterns of high-altitude adaptation in high-altitude Tajiks compared with Tibetans and Sherpas. Sci. Rep. 10, 1–9 (2020).
Jones, B. P. et al. Divergence across mitochondrial genomes of sympatric members of the Schistosoma indicum group and clues into the evolution of Schistosoma spindale. Sci. Rep. 10, 1–14 (2020).
Landeryou, T., Ropiquet, A., Kett, S. M., Wildeboer, D. & Lawton, S. P. Characterization of the complete mitochondrial genome of Diplostomum baeri. Parasitol. Int. 79, 102166 (2020).
Bian, D. et al. Complete mitochondrial genome of Spilosoma lubricipedum (Noctuoidea: Erebidae) and implications for phylogeny of noctuid insects. Genomics 112, 4577–4584 (2020).
Mullens, N. et al. Mitogenomic characterization and systematic placement of the Congo blind barb Caecobarbus geertsii (Cypriniformes: Cyprinidae). Int. J. Biol. Macromol. 161, 292–298 (2020).
Xu, H., Wu, Y., Wang, Y. & Liu, Z. Comparative analysis of five mitogenomes of Osmylinae (Neuroptera: Osmylidae) and their phylogenetic implications. Int. J. Biol. Macromol. 164, 447–455 (2020).
Zou, Y. et al. Taxonomy and phylogenetic relationship of zokors. J. Genet. 99, 38 (2020).
Arnaiz, O., Meyer, E. & Sperling, L. ParameciumDB 2019: integrating genomic data across the genus for functional and evolutionary biology. Nucleic Acids Res. 48, D599–D605 (2020).
Vannini, C., Ferrantini, F., Ristori, A., Verni, F. & Petroni, G. Betaproteobacterial symbionts of the ciliate Euplotes: origin and tangled evolutionary path of an obligate microbial association. Environ. Microbiol. 14, 2553–2563 (2012).
Nitla, V. et al. Critical revision of the family Plagiopylidae (Ciliophora: Plagiopylea), including the description of two novel species, Plagiopyla ramani and Plagiopyla.narasimhamurthii, and redescription of Plagiopyla nasuta Stein, 1860 from India. Zool. J. Linnean. Soc. 186, 1–45. https://doi.org/10.1093/zoolinnean/zly041 (2019).
Skovorodkin, I. N. A device for immobilization of small biological objects during light microscopical observation. Tsitologiia 32, 87–91 (1990).
Corliss, J. O. Silver impregnation of ciliated protozoa by the Chatton-Lwoff technic. Stain Technol. 28, 97–100 (1953).
Dragesco, J. & Dragesco-Kernéis, A. Ciliés libres de l’ Afrique Intertropicale. Introduction á la connaissance et á l’étude des ciliés. Faune tropicale. In L’Orstom ed. Paris 26, 1–559 (1986).
Ferreira T., Rasband W. 2012. ImageJ user guide. ImageJ/Fiji, 1. (https://imagej.nih.gov/ij/docs/guide/user-guide.pdf).
Montesanto, G. A fast GNU method to draw accurate scientific illustrations for taxonomy. ZooKeys 515, 191 (2015).
Curds, C. R. A guide to the species of the genus Euplotes (Hypotrichida, Ciliatea). Bull. Br. Mus. (Nat. Hist.) Zool. 28, 1–61 (1975).
Berger, H. Monograph of the Urostyloidea (Ciliophora, Hypotricha). Monogr. Biol. 85, 1–1303 (2006).
Lynn, D. H. The Ciliated Protozoa: Characterization, Classification, and Guide to the Literature (Springer, Berlin, 2008).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Bioinform. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Kumar, S., Jones, M., Koutsovoulos, G., Clarke, M. & Blaxter, M. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front. Genet. 4, 237. https://doi.org/10.3389/fgene.2013.00237 (2013).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Seemann T. barrnap 0.5: rapid ribosomal RNA prediction. https://www.vicbioinformatics.com/. (2013).
Galperin, M. Y., Kristensen, D. M., Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Microbial genome analysis: the COG approach. Brief Bioinf https://doi.org/10.1093/bib/bbx117 (2017).
Westram, R. et al. ARB: a software environment for sequence data. In Handbook of Molecular Microbial Ecology I: Metagenomics and Complementary Approaches (ed. de Bruijn, F. J.) 399–406 (Wiley, Hoboken, 2011). https://doi.org/10.1002/9781118010518.ch46.
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl. Acids Res. 41, D590–D596. https://doi.org/10.1093/nar/gks1219 (2013).
Wagner, M. & Horn, M. The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr. Opin. Biotechnol. 17, 241–249 (2006).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 9, 772. https://doi.org/10.1038/nmeth.2109 (2012).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. https://doi.org/10.1080/10635150390235520 (2003).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Boscaro, V., Fokin, S. I., Schrallhammer, M., Schweikert, M. & Petroni, G. Revised systematics of Holospora-like bacteria and characterization of “Candidatus Gortzia infectiva”, a novel macronuclear symbiont of Paramecium jenningsi. Microb. Ecol. 65, 255–267 (2013).
Daims, H., Brühl, A., Amann, R., Schleifer, K. H. & Wagner, M. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444 (1999).
Amann, R. I. et al. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56, 1919–1925 (1990).
Cole, J. R. et al. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145. https://doi.org/10.1093/nar/gkn879 (2009).
Greuter, D., Loy, A., Horn, M. & Rattei, T. probeBase—an online resource for rRNA-targeted oligonucleotide probes and primers: new features 2016. Nucleic Acids Res. 44, D586–D589. https://doi.org/10.1093/nar/gkv1232 (2016).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009).
Medlin, L., Elwood, H. J., Stickel, S. & Sogin, M. L. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71, 491–499 (1988).
Rosati, G., Modeo, L., Melai, M., Petroni, G. & Verni, F. A multidisciplinary approach to describe Protists: a morphological, ultrastructural, and molecular study on Peritromus kahli (Ciliophora, Heterotrichea). J. Eukaryot. Microbiol. 51, 49–59 (2004).
Acknowledgements
The authors are grateful to the Marine Biology Laboratory, Andhra University, India, for providing the research facilities. In particular, Professor Kalavati, Professor Raman Akkur, and Professor Prabhakara Rao Yallapragada are acknowledged for their valuable aid. Special acknowledgements to Simone Gabrielli for tree and photo-editing support.
Funding
This work was supported by the European Commission FP7-PEOPLE-2009-IRSES project CINAR PATHOBACTER (247658) (to GP); this project has received funding from the European Union’s Horizon 2020 research and innovation programme under Grant Agreement n° 872767 (to GP); the University of Pisa PRA_2018_63 project (to GP); the present study was supported by the Fondazione Cassa di Risparmio di Pistoia e Pescia—giovani@ricercascientifica2019 fellowship (No. 2019.0380) (to VS and GP); the Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022)—Dept. of Biology and Biotechnology "L. Spallanzani", University of Pavia (to DS).
Author information
Authors and Affiliations
Contributions
Conceptualization: G.P., V.S., L.G. Sampling and culturing: V.N., B.V.S. Morphological and ultrastructural analyses: V.N., L.M. Molecular analyses: V.S., L.G., V.N. Bioinformatics: L.G., M.C., D.S. Phylogeny and phylogenomic analyses: V.S., L.G., M.C., G.P. Environmental screening on I.M.N.G.S.: O.L. Visualization: V.N., V.S., L.G., L.M., O.L., F.V., G.P. Funding acquisition: G.P., F.V., D.S., C.B., V.S. Work supervision: G.P., L.M. Writing—original draft: V.S., L.G., L.M., O.L. Writing—review and editing: G.P., M.C., D.S., C.B., V.N., F.V., B.V.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Serra, V., Gammuto, L., Nitla, V. et al. Morphology, ultrastructure, genomics, and phylogeny of Euplotes vanleeuwenhoeki sp. nov. and its ultra-reduced endosymbiont “Candidatus Pinguicoccus supinus” sp. nov.. Sci Rep 10, 20311 (2020). https://doi.org/10.1038/s41598-020-76348-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-76348-z
This article is cited by
-
Exploring the biogeography, morphology, and phylogeny of the condylostomatid ciliates (Alveolata, Ciliophora, Heterotrichea), with establishment of four new Condylostoma species and a revision including redescriptions of five species found in China
Marine Life Science & Technology (2024)
-
Symbionts of Ciliates and Ciliates as Symbionts
Indian Journal of Microbiology (2024)
-
“Candidatus Intestinibacterium parameciiphilum”—member of the “Candidatus Paracaedibacteraceae” family (Alphaproteobacteria, Holosporales) inhabiting the ciliated protist Paramecium
International Microbiology (2023)
-
Single-cell Microbiomics Unveils Distribution and Patterns of Microbial Symbioses in the Natural Environment
Microbial Ecology (2023)
-
Taxonomy and SSU rRNA gene-based phylogeny of two new Euplotes species from China: E. chongmingensis n. sp. and E. paramieti n. sp. (Protista, Ciliophora)
BMC Microbiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
