
Abstract
Most sheep breeding programs designed for the tropics and sub-tropics have to take into account the impacts of environmental adaptive traits. However, the genetic mechanism regulating the multiple biological processes driving adaptive responses remains unclear. In this study, we applied a selective sweep analysis by combing 1% top values of Fst and ZHp on both altitude and geographic subpopulations (APS) in 636 indigenous Tibetan sheep breeds. Results show that 37 genes were identified within overlapped genomic regions regarding Fst significantly associated with APS. Out of the 37 genes, we found that 8, 3 and 6 genes at chromosomes (chr.) 13, 23 and 27, respectively, were identified in the genomic regions with 1% top values of ZHp. We further analyzed the INDEL variation of 6 genes at chr.27 (X chromosome) in APS together with corresponding orthologs of 6 genes in Capra, Pantholops, and Bos Taurus. We found that an INDEL was located within 5′UTR region of HAG1 gene. This INDEL of HAG1 was strongly associated with the variation of APS, which was further confirmed by qPCR. Sheep breeds carrying “C-INDEL” of HAG1 have significantly greater body weight, shear amount, corpuscular hemoglobin and globulin levels, but lower body height, than those carrying “CA-INDEL” of HAG1. We concluded that “C-INDEL” variation of HAG1 gene confers better hypoxia tolerance in the highlands of Tibetan and explains well geographic distributions in this population. These results contribute to our understanding of adaptive responses to altitude and geographic adaptation in Tibetan sheep populations and will help to guide future conservation programs for Tibetan sheep native to Qinghai-Tibetan Plateau.
Similar content being viewed by others

Introduction
Recent research data and model predictions indicate that increasing frequencies of abnormal weather events due to global climate change will have a fundamental impact on agricultural production1. For example, livestock experience numerous environmental stressors that have effects on both production traits, such as growth, reproductive performance, meat quality and adaptive traits such as cold tolerance2,3. In particular, the influence of climate change, i.e., cold stress on living organisms is exacerbated at the interface of extreme environments that typically occur in high altitude, plateaus and desert regions4. At such breeding programmes for livestock in these regions should include both adaptive and production traits5.
Tibetan sheep (Ovis aries) have the ability to adapt to a wide range of agro-ecological conditions, and represent an excellent model to gain new insights into genetic mechanisms underlying the adaptive response of livestock to extreme environments 6,7. This is also helpful to develop appropriate breeding programs under various scenarios of future climate change. Tibetan sheep have lived on the Tibetan plateau for thousands of years; however, the process and consequences of adaptation to this extreme environment conditions have not yet been elucidated 8.
Artificial selection during domestication and production-oriented breeding has greatly shaped the level of genomic variability in sheep. The genome of Tibetan sheep provides a unique opportunity for identifying signatures associated with selection. Array- or sequencing-based detection of the signatures of selection process has been described in cattle9, chicken10, dogs11, goat12, and in sheep8,13. Recently, the variation of many genes were identified to be strongly associated with as high-altitude adaptations in the Tibetan Mastiff (EPAS1)8, in yaks (ADAM17, ARG2 and MMP3), Tibetan antelopes (PKLR, ATP12A and NOS3) and Tibetan wild boars (ALB, GNG2 and PIK3C2G)14,15,16. However, each species may have different candidate genes responsible for high altitude adaptations, and investigations on selection signatures with respect to altitude and geographic adaptation were also less reported.
This study aimed to identify signatures evidence of recent selection among Tibetan sheep population for both altitude and geographic adaptation. We investigated selection signatures using whole-genomic variants in 15 indigenous Tibetan sheep populations. These populations consist of 636 sheep individuals living in the Qinghai-Tibetan Plateau areas in China. The overlapped genes underlying both altitude and environmental response were identified by screening genomic regions with significant values of both ZHp and Fst for autosome and hetersomes SNPs. We observed a small number of strong selection signatures near genes known to under strong artificial selection in other animals. Our findings can be used to better understand genomic signatures under selection controlling both adaptive traits in Tibetan sheep populations. The key genes identified in this study would be helpful to guide breeding practices for improvement of adaptive ability to extreme environments for sheep.
Methods
Ethics statement
All animals were handled according to the Guidelines for the Biological Studies Animal Care and Use Committee, People’ s Republic of China. Animal experiments were approved by the Animal Ethics Committee of the Institute of Animal Sciences of Chinese Academy of Agricultural Sciences.
Classification of population from different adaptive traits
15 Chinese indigenous Tibetan sheep populations, consisting of 636 individuals derived from different altitudes ranging from 3000-5000 m were used. These indigenous Tibetan sheep populations living in the Qinghai-Tibetan Plateau areas in China were classified into different subpopulations to estimate the candidate genes responsible for altitude and geographic adaptation (Table 1). According to originated altitude information, two subpopulations were divided, i.e., lowland Tibetan sheep (< 3500 m): Guide Black Fur sheep (GD), Qilian White Tibetan sheep (QL), Tianjun White Tibetan sheep (TJ), Qinghai Oula Tibetan sheep (QH), Minxian Black Fur sheep (MX), Ganjia Tibetan sheep (GJ), Qiaoke Tibetan sheep (QK), and Gannan Oula Tibetan sheep (GN); highland Tibetan sheep (> 4500 m): Langkazi Tibetan sheep (LKZ), Jiangzi Tibetan sheep (JZ), Gangba Tibetan sheep (GB), Huoba Tibetan sheep (HB), Duoma Tibetan sheep (DM), Awang Tibetan sheep (AW), and Linzhou Tibetan sheep (LZ). For geographic regions, three different provinces in China were classified, Qinghai (QH, GD, QL, and TJ); Gansu (MX, GJ, QK, and GN); Tibetan (LKZ, JZ, GB, HB, DM, AW and LZ) (Table 1).
Physiological measurements of samples from different altitudes
To further elucidate the physiological significances of INDEL variation of HAG1 gene based on the values of Fst and ZHp, we compared 4 out of 15 Tibetan subpopulations (GD, QL, AW and HB) due to the available phenotypic information. This selected subpopulations consist of 122 samples as listed in Table 2. Six parameters related to body size were determined: body weight/height/length, bust, chest depth, neck size, and two other traits, such as wool length, and shear amount (defined as the weight of the fleece post shearing)17; Thirteen physiological parameters: body temperature (Temperature), breathe rates (Breathe), pulse rates (Pulse), pulse interval (pulse interval), red-cell numbers (RBC), hematocrit, HCT, erythrocyte mean corpuscular volume (MCV)17, mean corpuscular hemoglobin (MCH), Mean corpuscular hemoglobin concentration (MCHC)18, Hemoglobin (HGB), Platelet (PLT), Red blood cell volume distribution width coefficient variation (RGW-CV), White blood cell count (WBC). Biochemical parameters: Glutamic pyruvic transaminase (ALT)19, Glutamic oxalacetic transaminase (AST) 19, Total protein (TP), Albumin (ALB), Globulin (GLO), Alkaline phosphatase (ALP), Lactate dehydrogenase (LDH), Cholinesterase (PCHE)20, Glucose (GLU), Total cholesterol (CHOL)21, Total calcium (CA); Nine blood-gas parameters: blood pH, pressure CO2 (pCO2), O2 saturation (O2S), concentration of HCO3-, standard bicarbonate, total CO2 concentration, Base excess (BE), Standard base excess (SBE); structural parameters related to Lung tissue: bronchial, thin bronchial, end thin bronchial, alveolar numbers per area (AN/m2), thick alveolar interval (TAI).
Sample collection
We used 15 Chinese indigenous Tibetan sheep populations, consisting of 636 individuals, and four samples were included in each pool for each population. The detailed sampling information for the 15 indigenous Tibetan sheep populations, i.e., population code, sample number, altitude, longitude and latitude, sampling location, and geographic location, is shown in Table 2. The procedure of sampling liver and lung tissues was followed as mentioned previously22.
DNA extraction and Sequencing
Briefly, for each Tibetan sheep, genomic DNA was extracted from liver and lung tissues using the QIAamp DNA blood mini kit (Qiagen, Germany) as described previously23. A260/280 ratio and agarose gel electrophoresis were used to evaluate the quality and integrity of the DNA. Genomic DNA was digested to a 300-400 bp fragment for library preparation, followed by end repair and ligated with an Illumina sequencing linker. The ligated products with sizes of 400-500 bp were loaded on 2% agarose gels and subsequently amplified via PCR. Libraries were sequenced by HiSeq 2500 sequencer (Illumina) in a 2 × 100 bp paired end mode. The sequencing coverage for each sample is averaged 10.3 × , within a range of 9–14 fold.
Reads alignment and variant calling
Reads were aligned to the sheep reference genome ‘Ovis aries’ v4.0 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000298735.2) using BWA (0.6.2-r126 version) followed by duplicate removal using Picard-Tools-1.55 (https://broadinstitute.github.io/picard/). The Genome Analysis Toolkit (GATK-2.6) was used to perform local realignment around existing INDELs and base quality score recalibration. Variant detection was performed using the GATK Unified Genotype Caller. To filter SNPs for downstream analysis, at least three reads with different mapping locations supporting the non-reference allele were present.
Population genetics analysis
The pairwise genetic distance was determined relying on the number of allelic differences as described previously23. Briefly, the neighbor join tree was calculated based on the distance matrix using PHYLIP (version 3.69) 24. SNP pair with high correlation were removed via PLINK (PLINK, RRID: SCR 001,757) 25. The PCA and population structure were performed using EIGEN-SOFT (version 6.0.1) and FRAPPE software (version 1.1), respectively26,27. TreeMix was applied to estimate migration events, with migration numbers m = 0–5 as previously reported28. The selective sweep analysis was performed using VCFtools, v0.1.12b29, including θ (number of isolated sites), π (paired nucleotide difference), and Tajima's D. There are 500,000 SNPs randomly selected from the genome, and these SNPs were used to analyze the linkage disequilibrium r2 with Haploview30.
Genome-wide Selective Sweep Test
To identify the functional genes underlying adaptive traits including altitude and geographic regions, we compared the overlapped genomic-regions by analyzing the signatures of selective effects on significant peaks. Two parameters relating to genome-wide selective sweep were used, i.e., Fst and ZHp. We calculated the genome-wide distribution of Fst values as previous reports31, for two subpopulations pairs as mentioned above, as to originated altitude information, and geographic regions (Table 2), using a sliding-window approach (100-kb windows with 50-kb increments). To identify regions that were likely to be or have been under selection, the “Z transformed heterozygosity” (ZHp) approach was used, as previously described 32. Individual Hp values were Z transformed as follows: ZHp = (Hp-μHp)/σHp, where μHp is the overall average heterozygosity and σHp is the standard deviation for all windows within each population. We calculated the ZHp value in sliding 150-kb windows along the autosomes from sequence reads corresponding to the most and least frequently observed alleles at all SNP positions as previously described33.
Bioinformatics analysis of population specific SNPs/INDELs
Overlapped candidate genes across altitudes and geographic regions were chosen for further analysis. In addition to this, we compared gene sequence of Capra hircus, Pantholops hodgsonii, and Bos taurus from NCBI database (https://www.ncbi.nlm.nih.gov/genome). We specifically focused on SNPs within genes and 1,000-bp upstream and downstream flanking regions of the genes (defined as the coordinates for the 3′ and 5′ UTR from ‘Ovis aries’ v4.0). Protein–protein interaction network was used to compare potential interactive genes with key overlapped genes in Homo sapiens from STRING database34.
Validation of interactive genes
Expression of putative proteins interacted with key overlapped candidates related to genetic variation of originated altitudes and geographic regions were confirmed by qPCR. The liver and lung tissues from four euthanized individual sheep for each population were collected. Lower edge of either left liver or left lung were sampled, and immediately stored in liquid nitrogen. Total RNA was extracted from liver and lung samples with Trizol reagent (Invitrogen, Carlsbad, CA, USA). Complementary DNA (cDNA) from 1 μg of total RNA was synthesized using the ExScript RT reagent Kit (Takara Biomedicals, Tokyo, Japan). Detailed procedures for real-time PCR amplification were performed following standard protocol of SYBR Green real-time PCR kit (Applied Biosystems SYBR Green, MN, USA). The amplification reaction conditions were as follows: 95 °C for 3 min followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The primer is listed in Table S1. GAPDH gene was used as internal control, using \(2^{{ - \Delta {\text{Ct}}}}\) method35. Four biological replicates and three technical replicates for each biological replicate were conducted.
Results and discussion
Genomic sequencing and PCA in 15 indigenous Tibetan sheep populations
In this study, we aimed to identify the candidate genes regulating multiple adaptive traits, including altitude and geographic regions. To accomplish this, we sequenced 15 indigenous Tibetan sheep populations consisting of 636 individuals, originated from different geographic locations (Table 2), via high-resolution whole genomic sequences techniques (WGS). To dissect the genomic heterozygosity levels and recombination events, we classified the populations into different altitude and geographic subpopulations (APS), including high altitude hypoxia (> 4500 m) vs. low altitude hypoxia (< 3500 m), and three different geographic locations, i.e., Qinghai, Gansu, and Tibetan (Table 1). WGS was performed on an Illumina HiSeq 2500 platform by using the pooled DNA from each population. Genome sequencing yielded an average of 40.84 Gb raw data, and produced 204 to 362 million sequence reads per population (Table S2). Over 90.75% of clean sequence reads were mapped to the newly annotated sheep reference genome (‘Ovis aries’ v4.0), indicating that high quality sequences were obtained (Table S3). Our efforts yielded an average sequence coverage of 10.3 × per sample, within a range of 9–14 fold. Single-nucleotide polymorphisms (SNPs) varied from 13–16 million for each population (Table S4).
More than 9 million SNPs for each Tibetan sheep population that confidently remained after filtering were used in the subsequent analyses. Results from SNPs statistics showed that 63% SNPs were identified at intergenic regions, whereas only 0.7% SNPs cases were found within exon regions (Table S5).
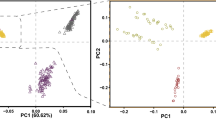
Principal components analysis (PCA) was performed to examine the genetic separation of 15 indigenous Tibetan sheep populations originated from the Qinghai-Tibetan Plateau areas in China (Fig. 1A). PCA results from SNPs after quality control clearly showed that most Tibetan sheep populations have high genetic homogeneity, and cumulative explained variance for genetic differentiation in 15 Tibetan sheep populations from PC1 and PC2 was 92.2% (Fig. 1B). After calling SNP, we obtained ~ 9.3 million SNPs across the Tibetan sheep populations. The distribution of minor allelic frequency (MAF) with 10 continued classes from 0–0.05 to 0.45–0.50 for each population was observed (Fig. 1C). MAF of SNPs across 15 indigenous Tibetan sheep populations showed MAF within the range of 0–5% had most relative abundance with around 20% of total SNPs, whereas 5–15% MAF showed relatively low abundance with less than 3% across 15 indigenous Tibetan sheep populations. The distribution pattern of MAF among Tibetan sheep populations was similar except for LKZ (Fig. 1C). Similarity index (IS) value was used to estimate the homogeneity of SNPs across 15 indigenous Tibetan sheep populations. LKZ showed lowest similarity with other Tibetan sheep populations, which is consistent with MAF distribution results (Fig. 1D).

SNP analysis and population structure for 15 indigenous Tibetan sheep populations. A: distributive map of 15 indigenous Tibetan sheep populations living in the Qinghai-Tibetan Plateau areas in China used in this study; The map inserted in panel A was referred to the Fig. 2 from Deng et al.53 with slight modifications. B: Principal component analysis on SNP dataset after quality control in different Tibetan sheep populations; C: Distribution of minor allelic frequency (MAF) with 10 continued classes from 0–0.05 to 0.45–0.50; D: Relatedness of similarity index (IS) values in 15 Tibetan sheep populations.
Overlapped selective signals regarding genomic heterozygosity among altitude and geographic regions distributions
Fst values were used to analyze the overlapped genes in the signatures of selection analysis on significant peaks for each of the different APS. It also named fixation index, typically used to evaluate differentiated genomic regions and identify selective signals among whole-genomic sequences31,36. Fst values over the whole genome based on genetic differentiation in APS were classified by altitude, and geographic locations (Fig. 2A-B). We found in total 248 and 244 SNPs with top 1% Fst values for APS, respectively (Fig. 2C). The numbers of overlapped candidate genes were presented based on top 1% Fst values for APS from 150-kb windows (Fig. 2D). There are three chromosomes (chr.) including chr.13, chr.23, and chr.27 (X chromosome), harboring significant SNPs, corresponding to 14, 3, and 20 genes, respectively (Fig. 2D). In addition, we compared these overlapped genomic regions with top 1% values for both ZHp and Fst to better understand genomic heterozygosity in specific populations. We found that ZEB1 gene at chr.13 is identified with overlapping in lowland subpopulation with top 1% values for both ZHp and Fst (Table S6). It was reported that ZEB1 is a target gene of hypoxia-inducible factors (HIFs), which is critical in the regulation of the macrovascular angiogenic response but not that of microvascular angiogenesis37. This indicates that HIFs can potentially be important transcription factors that involved in regulatory pathways of the available oxygen in the cellular environment under high-altitude conditions37. Moreover, it was reported that HIF-1α can stimulate the expression of TWIST1 and ZEB1 genes under hypoxic conditions38,39, suggesting potential functions of ZEB1 gene in adaptive response of Tibetan sheep to hypoxic conditions.

Manhattan analysis and candidate genes exploration. A-B: Manhattan plot representing Fst values for each SNP across Chromosomes for two subpopulations of altitude and geography. The genomic region with significant Fst values were highlighted in arrows. C: Venn diagram representing overlapped SNPs for two subpopulations of altitude and geography. D: Numbers of corresponding genes harboring overlapped SNPs for two subpopulations of altitude and geography. E: overlapped genes with 1% top values between Fst and ZHp.
We also identified six genes (LOC101110166, PANK2, RNF24, VPS16, PCED1A and PTPRA) with top 1% values for both ZHp and Fst in Tibetan geographic region of China (Fig. 2E; Table S6). Notably, one gene (RALY) is identified with overlapping with Gansu geographic region of China (Table S6). This gene (RALY) has been reported as a candidate gene involved in spliceosomal complex pathway in high-altitude adaption Tibetan sheep populations, indicating its important roles in hypoxia response in addition to ZEB113,37. Moreover, we found three genes (PSMA8, TAF4B, LOC105605498) and six genes (LOC101119700, LOC101119112, APOOL, ZNF711, LOC101123097 and POF1B) were identified in chr.23, and chr.27, respectively, with top 1% values for both ZHp and Fst in lowland subpopulation (Fig. 2E; Table S6).
Haplotypes of HAG1 are related to adaptive response to environments and body size
Heteromorphic X chromosome, where one sex has two different types of sex chromosomes, face very different evolutionary consequences than do the autosomes. To further analyze the natural variations of these 6 overlapped genes on X chromosome (chr.27), we analyzed INDELs of these genes including 2 k-bp promoter and CDS regions together with other species, such as Capra, Pantholops, and Bos Taurus (Fig. 3). Notably, these species, such as Bos Taurus are considered as ideal models to uncover the mechanism of animal adaption to highland altitude hypoxia16. Results from INDEL information showed that 62% INDEL events were identified at intergenic regions, whereas only 0.3% INDEL cases were found within exon regions (Table S7). The INDEL happens in different types, such as deletion and insertion compared to reference genomes across the 6 overlapped genes (Fig. 3). Interestingly, our findings suggested that among 6 genes, the variation of INDEL at 5′-flank region of HAG1 gene (LOC101123097) at 72,792,241 is in well line with the variations of APS (Fig. 3C); while the INDEL in other genes were either found in gene intron region or mixed INDELs types (Fig. 3A-F). INDEL of “C” on 5′UTR of HAG1 gene was observed in Tibetan sheep from highland, and Tibetan geographic region, we named here as “C-INDEL”, while INDEL of “CA” is occurred in Qinghai, we hence named as “CA-INDEL”. Interestingly, we found there are two INDEL types mixed in lowland altitude (Fig. 3C).

Gene INDEL analysis for 6 candidate overlapped genes at hetersomes within overlapped genomic regions by combing 1% top values of Fst and ZHp. A-F: INDEL analysis on 6 genes within clustered subpopulations in addition to three other species including Capra, Pantholops, and Bos taurus. The 6 genes were listed in Table S6.
We further analyzed the protein structure and gene expression variation of HAG1 among different indigenous Tibetan sheep (Fig. 4). According to bioinformatics analysis, we predicted the protein sequences of HAG1 using protein translation tool from ExPASy database (https://web.expasy.org/translate/), and found the similarities score reached up to 99% in either mRNA or protein sequences between CSDE1 and HAG1 protein, indicating both HAG1 with CSDE1 are likely to be involved in similar regulatory pathway. Notably, the CSDE1 (Cold Shock Domain Containing E1), alternative name: UNR, is a conserved RBP containing five cold-shock domains (CSDs) that bind single-stranded RNA40,41. Function of proteins possessing CSDs are involved in two processes: transcriptional and translational control. Two major cold shock proteins CspA and CspB in Escherichia coli and Bacillus subtilis, respectively, were intensively reported40. CspA and CspB are massively and transiently induced after a temperature downshift and are involved in the adaptation to cold shock42.

Comparison on sequence alignments and expression pattern between HAG1 and CSDE1. A: Secondary structure of HAG1 and differentiated amino acids between HAG1 and CSDE1. The location of 7 differentiated amino acids were depicted in symbol “*”, which are majorly on beta-strand and turn. Secondary structure of protein is predicted via Uniprot database from different PDB entries. B: Differentiated location of amino acids on representative 3D protein structure; C-D: Comparison on expression levels of CSDE1 and HAG1 in given 6 indigenous Tibetan sheep populations in this study. The 6 indigenous Tibetan sheep populations include Guide Black Fur sheep (GD), Qilian White Tibetan sheep (QL), Minxian Black Fur sheep (MX), Ganjia Tibetan sheep (GJ), Huoba Tibetan sheep (HB) and Awang Tibetan sheep (AW). Expression levels were calculated against GAPDH using mRNA from live and lung tissues, and four biological replicates were conducted.
HAG1 protein consists of 798 amino acids. We then compared the secondary structure of HAG1 with known CSDE1, and found most identified residues are conserved, the similarity between HAG1 and CSDE1 proteins were 87% from NCBI protein blast website (blast.ncbi.nlm.nih.gov/). Most differential regions were observed at N-terminal of protein containing 7 different amino acids, and these amino acids were located in either Beta strand or Turn domains, but not Helix (Fig. 4A). These domains were reported to represent the most evolutionarily conserved nucleic acid-binding protein domain, as found in bacteria and eukaryotes43,44. Interestingly, this kind of Beta strand domain can also mediate the binding affinity to single-stranded DNA and RNA45,46. Three different amino acids between HAG1 and CSDE1, i.e., S30, R89 and V202, were highlighted and mapped at 3D protein structure from two PDB entries (1WFQ and 2YTX) according to Uniprot database (CSDE1_Human Protein ID: 075,534) (Fig. 4B).
The 5′ untranslated region (5′-UTRs) are known to regulate gene expression. INDEL between different subpopulations at the region of 5′-UTRs is expected to influence different expression of HAG1 gene. To compare the expression levels between HAG1 and CSDE1, we used 6 indigenous Tibetan sheep populations, including Guide Black Fur sheep (GD), Qilian White Tibetan sheep (QL), Minxian Black Fur sheep (MX), GanjiaTibetan sheep (GJ), Huoba Tibetan sheep (HB) and Awang Tibetan sheep (AW). We sampled from liver and lung tissues for qPCR as same position for DNA sequencing. Notably, it was reported that CSDE1 gene is high expressed in muscle and fetal ovary according to sheep gene expression atlas dataset (https://biogps.org/sheepatlas/) and as documented elsewhere47. We also found that HAG1 gene from liver and lung tissues is highly expressed in Tibetan sheep from highland regions, rather than that from lowland regions (Fig. 4C). HAG1 gene also showed distinct expression pattern among different geographic locations (Fig. 4C). Interestingly, CSDE1 gene exhibited clear differences, measured in liver and lung tissues for the Tibetan sheep between highland and lowland, but not for the sheep from different geographic regions (Fig. 4D).
Function of HAG1 in adaptive characteristics
Therefore, an interesting question arising here is whether interactive proteins with CSDE1 could show similar expression pattern between the subpopulations of altitude and geographic region? Therefore, we reconstructed the CSDE1-centerized module according to STRING protein-interaction database (Figure S3A-B; Table S8). Differential gene expression analysis via qPCR suggested that 9 genes in the module have strong correlation with CSDE1 and HAG1 (Figure S3B; Table S8). These are: MAX, MYC, YBX1, HNRNPU, DHX9, PABPC1, PAIP1, STRAP, and SYNCRIP. Previous reports showed that the mRNA of CSDE1 is up-regulated in a high percentage of skin and ovary cancers, and UNR/CSDE1 regulates critical Melanoma genes, including PABPC1 gene48. Besides, CSDE1 regulates internal ribosome entry site (IRES)-dependent translation of the transcripts encoding the oncogene MYC49. Notably, PABPC1 and MYC show significantly distinct difference between highland and lowland subpopulations as CSDE1 (p < 0.05), but do not show clear difference in different geographic distribution as HAG1 (Figure S3C-D), although expression levels of HAG1 exerted strongly positive correlation with that of CSDE1 as well as its 9 interactive genes (Figure S3B; Table S8). These evidences reveal that the function of HAG1 gene might be also involved in other biological process.
Morphological and physiological parameters of different haplotypes
Finally, to interpret the biological functions of HAG1, we selected 4 indigenous Tibetan sheep populations containing 15 individuals in total with contrasting expression pattern of HAG1. AW and HB belonging to “C-INDEL” show high expression levels of HAG1, as presented in Fig. 5A, while GD and QL belonging to “CA-INDEL” show low expression levels from liver and lung tissues (Fig. 5B). We then compared the ratio of “C-INDEL” over “CA-INDEL” in 47 physiological and biochemical parameters (Fig. 5B). Results show two Tibetan sheep breeds possessing “C-INDEL” of HAG1 have significantly greater body weight, shear amount, chest width and body length, but have lower body height, relative to sheep in subpopulations with “CA-INDEL” of HAG1 (P < 0.005) (Fig. 5B).

Comparison on morphological and physiological traits in 15 Tibetan sheep populations from 4 representative Tibetan sheep subpopulations (GD, QL, AW and HB), consisting of 122 samples with different INDEL of HAG1 gene. A: Representative picture of indigenous Tibetan sheep (Huoba Tibetan sheep, HB for “C-INDEL”, and Guide Black Fur sheep, GD for “CA-INDEL” originated between two haplotypes promoter of HAG1; B: Comparison on different physiological parameters of 15 Tibetan sheep between “CA-INDEL” and “C-INDEL” of HAG1. The first types include GD and QL, while the latter types include AW and HB. The averaged values for each parameter were derived from at least 10 biological replicates.
Our study showed that certain haplotype of HAG1 could be an indicator for both adaptive response and production traits, such as altitude hypoxia (adaptive response), body weight (production traits), shear amount (production traits), and body length (production traits) (Fig. 5B). This is consistent with the conclusion that large physical size in highland as observed in human50. In particular, body weight of Tibetan sheep in “C-INDEL” population is 1.7 times higher than that in “CA-INDEL” group. For body haematological parameters, body temperature, pulse interval, MCH, MCHC, HGB levels are at least 1.5 times higher in Tibetan sheep population with “C-INDEL” of HAG1 than that with “CA-INDEL”; this trend is similar to that observed in Tibetan sheep and dogs from different altitudes13,51. Interestingly, breathe rates, pulse rates, RBC, HCT, MCV, PLT, and RDW-CV are relatively lower in “C-INDEL” of HAG1 than that in “CA-INDEL” Tibetan sheep population. Values of blood-gas parameters were similar or relatively lower in “C-INDEL” population than that in “CA-INDEL” population, except for pCO2. For lung tissue structure parameters, values are relatively higher in “C-INDEL” population than that in “CA-INDEL” population. In particular, elevated lactate dehydrogenase (LDH) in “C-INDEL” population relative to “CA-INDEL” population support the findings in other sheep studies on hypoxemia response52.
Conclusion
This study presents a strategy to uncover the potential genes that underlie both altitude and geographic adaption through selective sweep analysis on genomic sequence of 15 indigenous Tibetan sheep populations. INDEL “C” within 5′-UTR of HAG1 gene was strongly associated with higher expression levels of HAG1 gene and better adaptive ability to high altitude, than that in those INDEL “CA” Tibetan sheep breeds. We concluded that the elite INDEL of HAG1 gene could potentially enhance Tibetan sheep breeding programmes.
Abbreviations
- HAG1:
-
Heterosome altitude and geographic gene
- Temperature:
-
Body temperature
- Breathe:
-
Breathe rates
- Pulse:
-
Pulse rates
- pulse interval:
-
Pulse interval
- RBC:
-
Red-cell numbers
- HCT:
-
Hematocrit
- MCV:
-
Erythrocyte mean corpuscular volume
- MCH:
-
Mean corpuscular hemoglobin
- MCHC:
-
Mean corpuscular hemoglobin concentration
- HGB:
-
Hemoglobin
- PLT:
-
Platelet
- RGW-CV:
-
Red blood cell volume distribution width coefficient variation
- WBC:
-
White blood cell count
- ALT:
-
Glutamic pyruvic transaminase
- AST:
-
Glutamic oxalacetic transaminase
- TP:
-
Total protein
- ALB:
-
Albumin
- GLO:
-
Globulin
- ALP:
-
alkaline phosphatase
- LDH:
-
Lactate dehydrogenase
- PCHE:
-
Cholinesterase
- GLU:
-
Glucose
- CHOL:
-
Total cholesterol
- CA:
-
Total calcium
- pCO2 :
-
Pressure CO2
- O2S:
-
O2 saturation
- BE:
-
Base Excess
- SBE:
-
Standard Base Excess
- AN/m2 :
-
End thin bronchial, alveolar numbers per area
- TAI:
-
Thick alveolar interval
- GD:
-
Guide Black Fur sheep
- QL:
-
Qilian White Tibetan sheep
- TJ:
-
Tianjun White Tibetan sheep
- QH:
-
Qinghai Oula Tibetan sheep
- MX:
-
Minxian Black Fur sheep
- GJ:
-
Ganjia Tibetan sheep
- QK:
-
Qiaoke Tibetan sheep
- GN:
-
Gannan Oula Tibetan sheep
- LKZ:
-
Langkazi Tibetan sheep
- JZ:
-
Jiangzi Tibetan sheep
- GB:
-
Gangba Tibetan sheep
- HB:
-
Huoba Tibetan sheep
- DM:
-
Duoma Tibetan sheep
- AW:
-
Awang Tibetan sheep
- LZ:
-
Linzhou Tibetan sheep
- MAF:
-
Minor allelic frequency
- HIFs:
-
Hypoxia-inducible factors
- CSDE1 :
-
Cold Shock Domain Containing E1
- CSDs:
-
Cold-shock domains
References
Bernstein, L., Bosch, P. & Canziani, O. IPCC, Climate Change 2007: Synthesis Report. Contribution of Working Groups I, II and III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change (Intergovernmental Panel on Climate Change, Geneva, 2007).
Brown, M. E. & Funk, C. C. Food security under climate change. Science 319, 580–581 (2008).
Hoffmann, I. Climate Change in Context: Implications for Livestock Production and Diversity. In Sustainable Improvement of Animal Production and Health (eds Odongo, N. E. et al.) 33–44 (IAEA-FAO, Vienna, 2010).
Easterling, D. R. et al. Climate extremes: observations, modeling, and impacts. Science 289, 2068–2074 (2000).
König, E. Z. et al. Participatory definition of breeding objectives for sheep breeds under pastoral systems—the case of Red Maasai and Dorper sheep in Kenya. Trop. Anim. Health Prod. 48, 9–20 (2016).
Lustick, S., Adams, M. & Hinko, A. Interaction between posture, color, and the radiative heat load in birds. Science 208, 1052–1053 (1980).
Walsberg, G. E., Campbell, G. S. & King, J. R. Animal coat color and radiative heat gain: a re-evaluation. J. Comp. Physiol. 126, 211–222 (1978).
Hamilton, W. J. III. Life’s Color Code 214–221 (McGraw-Hill, New York, 1973).
Lustick, S. Plumage color and energetics. The Condor 73, 121–222 (1971).
Lustick, S., Battersby, B. & Kelty, M. Behavioral thermoregulation: orientation toward the sun in Herring Gulls. Science 200, 81–83 (1978).
Howden, S. M., Soussana, J. F., Tubiello, F. N. & Chhetri, N. Adapting agriculture to climate change. Proc. Natl. Acad. Sci. USA 104, 19691–19696 (2007).
Lobell, D. B. et al. Prioritizing climate change adaptation needs for food security in 2030. Science 319, 607–610 (2008).
Wei, C. et al. Genome-wide analysis reveals adaptation to high altitudes in Tibetan sheep. Sci. Rep. 6, 2677–2681 (2016).
Ge, R. L. et al. Draft genome sequence of the Tibetan antelope. Nat. Commun. 4, 1858 (2013).
Li, M. et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 45, 1431–1438 (2013).
Qiu, Q. et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 44, 946–949 (2012).
Egbe-Nwiyi, T. N., Nwaosy, S. C. & Salami, H. A. Hematological values of apparently health sheep and goat as influenced by age and sex in Arid zone of Nigeria. Afr. J. Biomed. Res. 3, 109–115 (2000).
Žaja, ŽI. et al. A new method of assessing sheep red blood cell types from their morphology. Animals 9, 1130 (2019).
Yeh, M. Y. et al. Chitosan promotes immune responses, ameliorates glutamic oxaloacetic transaminase and glutamic pyruvic transaminase, but enhances lactate dehydrogenase levels in normal mice in vivo. Exp. Ther. Med. 11, 1300–1306 (2016).
Agrawal, A., Gandhe, M. B., Gupta, D. & Reddy, M. V. Preliminary study on serum lactate dehydrogenase (LDH)-prognostic biomarker in carcinoma breast. J. Clin. Diagn. Res. 10, BC06-BC8 (2016).
Kozat, S. & Denizhan, V. Glucose, lipid, and lipoprotein levels in sheep naturally infected with Fasciola hepatica. J. Parasitol. 96, 657–659 (2010).
Liu, J. et al. Genetic diversity and phylogenetic evolution of Tibetan Sheep based on mtDNA D-Loop sequences. PLoS ONE 11(7), e0159308 (2016).
Pan, Z. Y. et al. Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization. Gigascience 24, 42–47 (2018).
Felsenstein, J. PHYLIP—phylogeny inference package (version 3.2). Cladistics 5, 164–6 (1989).
Sabeti, P. C. et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–8 (2007).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigen analysis. PLoS Genet. 2, 2074–93 (2006).
Tang, H., Peng, J., Wang, P. & Risch, N. J. Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol. 28, 289–301 (2005).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8(11), e1002967 (2012).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–8 (2001).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–5 (2005).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984).
Rubin, C. J. et al. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA 109, 19529–19536 (2012).
Rubin, C. J. et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591 (2010).
Mering, C. et al. String: known and predicted protein–protein associations, integrated and transferred across organisms. Nucl. Acids Res. 33, 433–437 (2005).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25, 402–8 (2001).
Akey, J. M. et al. Tracking footprints of artificial selection in the dog genome. Proc. Natl. Acad. Sci. USA 107, 1160–1165 (2010).
Luo, Z. et al. MicroRNA-and play an important role in endothelial cell differentiation and vasculo genesis by targeting transcription repressor ZEB1. Stem Cells. 31, 1749–1762 (2013).
Martin, A. & Cano, A. Tumorigenesis: twist links EMT to self-renewal. Nat Cell Biol. 12, 924–925 (2010).
Semenza, G. L., Nejfelt, M. K., Chi, S. M. & Antonarakis, S. E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 88, 5680–5684 (1991).
Goroncy, A. K. et al. The NMR solution structures of the five constituent cold-shock domains (CSD) of the human UNR (upstream of N-ras) protein. J. Struct. Funct. Genomics 11, 181–188 (2010).
Triqueneaux, G., Velten, M., Franzon, P., Dautry, F. & Jacquemin-Sablon, H. RNA binding specificity of Unr, a protein with five cold shock domains. Nucl. Acids Res. 27, 1926–1934 (1999).
Thieringer, H. A., Jones, P. G. & Inouye, M. Cold shock and adaptation. Bioessay 20, 49–57 (1998).
Graumann, P., Wendrich, T. M., Weber, M. H., Schröder, K. & Marahiel, M. A. A family of cold shock proteins in Bacillus subtilis is essential for cellular growth and for efficient protein synthesis at optimal and low temperature. Mol. Microbiol 25, 741–756 (1997).
Graumann, P. & Mahariel, M. A. A case of convergent evolution of nucleic acid binding modules. BioEssays 18, 309–315 (1996).
Wolffe, A. P. Structural and functional properties of the evolutionary ancient Y-box family of nucleic acid binding proteins. BioEssay 16, 245–251 (1994).
Sommerville, J. & Ladomery, M. Transcription and masking of mRNA in germ cells: involvement of Y-box proteins. FASEB J. 10, 435–443 (1996).
Clark, E. L. et al. A high resolution atlas of gene expression in the domestic sheep (Ovis aries). PLoS Genet. 13(9), e1006997 (2017).
Wurth, L. et al. UNR/CSDE1 drives a post-transcriptional program to promote melanoma invasion and metastasis. Cancer Cell 30, 694–707 (2016).
Evans, J. R. et al. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene 22, 8012–8020 (2003).
Lenfant, C. High altitude adaptation in mammals. Am. Zool. 13, 447–456 (1973).
Gou, X. et al. Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Res. 24, 1308–1315 (2014).
Ohtsuka, T. & Gilbert, R. D. Cardiac enzyme activities in fetal and adult pregnant and nonpregnant sheep exposed to high-altitude hypoxemia. J. Appl. Physiol. 79, 1286–1289 (1995).
Deng, N. et al. Closing yield gaps for rice self-sufficiency in China. Nat. Commun. 10, 1725 (2019).
Funding
This work was supported by the National Natural Science Foundation for General Program of China (Grant No. 31872981), the National Key Research and Development Program of China (Grant No. 2018YFD0502103), the Agricultural Science and Technology Innovation Program of China (Grant no. CAAS-ASTIP-2015-LIHPS), the Modern China Wool Cashmere Technology Research System (Grant no. CARS-39-02), Gansu Provincial Natural Science Foundation (Grant no. 18JR3RA016), the Modern Agricultural Technology Research System of Gansu (Grant no. GARS08), and the Central Level of Scientific Research Institutes for Basic R & D Special Fund Business (Grant no. 1610322020004). We thank Shanghai Orizymes Biotech. Co., Ltd, for whole-genome sequencing services and bioinformatics support.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: LJ, DX, ZH, YC, YY, DK. Performed the experiments: LJ, WF, SX, NC, LZ. Analyzed the data: ZY, YC, GT, ZH, XX. Wrote the paper: LJ, DZ, YB.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, J., Yuan, C., Guo, T. et al. Genetic signatures of high-altitude adaptation and geographic distribution in Tibetan sheep. Sci Rep 10, 18332 (2020). https://doi.org/10.1038/s41598-020-75428-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75428-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
