
Abstract
The combustion of hydrocarbon fuels within the automotive industry results in harmful and reactive incomplete combustion byproducts. Specifically, nitric oxide emissions (NO) lead to increased smog, acid rain, climate change, and respiratory inflammation within the population [Nitrogen Dioxide | American Lung Association]. Current methods for treating combustion exhaust include the catalytic converter in conjunction with nitrogen oxide traps. However, there is no active, continuous reduction method that does not require restrictions on the combustion environment (Hirata in Catal Surv Asia 18:128–133, 2014). Here, a small voltage potential oscillation across a newly designed electro-chemical catalytic membrane significantly reduces NO emissions. A ceramic membrane consisting of two dissimilar metal electrodes, sandwiching a dielectric layer, is able to achieve an NO reduction in excess of 2X that of a platinum group metal (PGM) three way catalytic converter. An analysis of the exhaust effluent from the membranes indicates N2O as a precursor to N2 and O2 formation, without the introduction of ammonia (NH3), during the reaction of NO indicating a divergence from current literature. Our results demonstrate how an oscillatory electric potential on a catalytic surface may alter anticipated reaction chemistry and interaction between the catalytic surface and fluid flow.
Similar content being viewed by others

Introduction
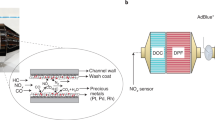
Increased concern over climate change, harmful combustion emissions and poor air quality has created a greater need for efficient catalytic emission processing systems1,2,3. Traditionally, three-way catalysts, such as those found in automobiles, and nitrogen oxide traps have been employed to reduce pollutants such as hydrocarbons, carbon monoxide and nitric oxide (NO). However, the performance of these devices is highly dependent upon the equivalence ratio of the exhaust. Three-way catalysts require that the exhaust remain at stoichiometric conditions4,5,6. If the exhaust becomes fuel lean, a nitrogen oxide trap is required, which will eventually become clogged with NO emission, calling for the direct injection of a fuel into the trap in order to react the stored gas6,7,8,9,10,11,12,13,14,15,16. A new technology must be developed that can more efficiently reduce harmful emissions while continuously operating in any exhaust condition. Therefore the potential of a layered perovskite membrane is investigated as an alternative catalyst to platinum group metals (PGM). The electrochemical catalytic membrane stack will be placed just downstream of the exhaust manifold of the engine. If needed, additional air may be injected into the housing canister in the case of a rich combustion environment during startup. The combustion engine’s exhaust gases will then flow through the first tubular support layer of the membrane stack, as illustrated by Fig. 1.

Schematic of electrochemical catalytic membrane integration into automotive exhaust. Patent Pending: US 2020–0,052,316 A1.
The tubular support layer is tuned to primarily convert unburned hydrocarbons, H2, and CO into water and carbon dioxide. The exhaust gas, upon exiting the tubular first layer, will be recycled to the outer perovskite layer of the membrane. This layer will then be primarily focused on the reaction of NO into N2 and O2. The support and outer layer will be separated by a dielectric ceramic layer. The exhaust gas will then leave the electrochemical catalytic stack, continuing within the automotive exhaust system.
The initial work was designed to directly compare the potential of the electrochemical-catalytic membrane for emission control against a traditional PGM catalytic converter. The PGM based catalytic converter is a commercially available catalytic converter purchased from the Volkswagen-Audi Group (VAG). It is a modern three-way catalyst, with an internal honeycomb structure consisting of a platinum combined with palladium and rhodium. Each specimen was held at 600 °C and subjected to 10 ml/min of two extremely lean (excessive O2 and limited hydrocarbon presence) model combustion exhausts, creating the most challenging environment for the electrochemical-catalytic membrane, and one stoichiometric exhaust, utilizing CH4 as a model hydrocarbon component of the exhaust. Although modern internal combustion engine control systems would not allow for such extreme conditions, the test is of academic interest to investigate how catalysts perform beyond normal operating conditions. The model exhausts were created by combining O2, CH4, CO, CO2, N2, and NO via benchtop flow meters.
The results shown below in Table 1 represent the time averaged results of each specimen. The specimen was introduced into the furnace 60 min prior to the collection of any effluent, for analysis to allow each sample to obtain steady state operation. The effluent of each sample specimen was then collected, analyzed, and recorded by the E-Instruments emission analyzer and gas chromatography-mass spectroscopy (GC–MS). The results were averaged over a constant 3 h test for each specimen.
The electrochemical-catalytic membrane is able to significantly outperform the PGM catalytic converter in the reduction of NO and CO, and closely matches the PGM catalytic converter in the reduction of hydrocarbons. Additionally, the electrochemical-catalytic membrane is able to maintain function in the presence of high concentrations of oxygen. The ability of the electrochemical-catalytic converter to maintain reactivity of NO at varying levels of NO has been previously reported17.
The novel electrochemical-catalytic membrane is able to consistently outperform traditional catalytic membranes for continuous NO reduction across multiple operation conditions. However, during testing, a minute oscillatory electrical voltage was seen to develop across the membrane.
Therefore, this work is designed to investigate the fundamental relation between electrical activity and reaction mechanism by which the novel electrochemical-catalytic membrane is able to react NO.
Catalytic perovskite materials are arranged such that two metal-based catalytic layers are separated by and in contact with a dielectric membrane for the study of NO reduction. The resulting electrochemical catalytic membrane is subjected to model exhaust flows within a controlled laboratory to serve as an initial investigation into the emission control potential.
Materials and methods
The support layer is comprised of nickel oxide-gadolinium doped ceria (48.9 vol% NiO 51.1 vol% Gd0.10Ce0.90O2-x (NiO-GDC)), the dielectric material of gadolinium doped ceria (Gd0.10Ce0.90O1.95 (GDC)) and the outer layer of lanthanum strontium cobalt ferrite-gadolinium doped ceria (52.4 vol% (La0.60Sr0.40)0.95Co0.20Fe0.80O3-X 47.6 vol% Gd0.10Ce0.90O1.95 (LSCF-GDC))18,19,20,21,22,23. Although these materials are commonly utilized in the production of high temperature solid oxide fuel cells, their purpose here is to act only as a catalytic membrane. As such, the nickel oxide was not reduced to nickel prior to testing. Pure silver wire electrodes were then added to both the support and outer layers via silver paste at the edge of the layer.
A test was constructed in order to investigate the fundamental mechanism by which the cathode layer of the electrochemical-catalytic membrane was able to significantly breakdown NO. The new electrochemical-catalytic membrane, with 0.81 cm2 of reactive surface area, is tested for NO reduction against: 1. VAG catalytic converter with 0.81 cm2 of reactive surface area; 2. The same electrochemical-catalytic membrane with the anode and cathode wired together to resemble a short circuit condition (hereafter referred to as an external short circuit or short circuited); and 3. The same electrochemical-catalytic membrane with an external 3 V, 8000 μs (4% duty cycle) pulse width modulation (PWM) signal with 0.81 cm2 of reactive surface area.
Each specimen was held at 600 °C and subject to 5 ml/min flow of a certified premixed gas cylinder of 10 vol% NO and 90 vol% N2. All testing equipment was cleaned, dried, and flushed with Ar for 1 h within the furnace at 600 °C prior to testing, to ensure no contamination of results. Only N2 and NO were supplied to the testing apparatus. During each test, the internal furnace environment surrounding the testing equipment was flooded with Ar, in order to ensure an external inert environment and to detect any leakage. For the electro-chemical catalyst, the NO flow was sent to the cathode side of the membrane. The voltage signal for both the electro-chemical catalytic membrane and the short-circuited membrane were recorded with a 100 MHz oscilloscope.
For both of the experimental setups, the effluent of each specimen, recorded with a Hiden Analytic QGA MS with a 100 ppb detection limit, verified via GC by a SRI 8610C and an E-instruments E-8500 emission analyzer, is compared to the baseline effluent. The experimental setup for the second, one sided test is depicted below in Fig. 2. The experimental setup for the initial study is the same, except before the effluent leaves the testing chamber it is recycled over the opposite side of the test specimen and then is exhausted.

Schematic of membrane testing apparatus.
Results and discussion
Experimental results of NO reduction phenomenon
The electro-chemical catalytic membrane, consisting of two metal electrode surfaces separated by a dielectric electrolyte, was able to achieve a reduction in NO concentration greater than 2X that of a traditional PGM catalyst as seen in Table 2 above. Additionally, wiring an external oscillating voltage signal between the two metal electrodes of the membrane increased reduction potential by an additional 10%. This behavior is in stark contrast to that of the electro-chemical catalytic membrane with the addition of an external short circuit, which was only able to convert an amount of NO similar to a traditional PGM catalyst.
Figure 3 shows the comparison of the voltage signal generated by the electro-chemical catalytic membrane and the electro-chemical catalytic membrane with an external short circuit during the NO reduction. The externally short-circuited membrane is centered on a voltage potential of 0, whereas the membrane without a short circuit is centered around -6.0 mV, under the standard convention of the cathode being the electrically positive electrode.

Electrical Signal Recording Overlays of the Novel Electrochemical Catalytic Membrane with (shown in orange) and without (shown in blue) an External Electrical Short Circuit at 600 °C and Subjected to 5 ml/min Flow of a Certified Premixed Gas Cylinder of 10 vol% NO and 90 vol% N2. Signal Noise Amplitude of ~ 35 mV with Electrical Short (orange) and ~ 50 mV without the Short Circuit (blue).
Additionally, the oscillations of the membrane without a short circuit are 2X the magnitude of those seen in the short-circuited membrane. Amplification of voltage oscillations within the membrane, as with the addition of a PWM external source, increases the total amount of NO reduction. Therefore, the ability of the electro-chemical catalytic membrane to charge and discharge while interacting with an external flow is the key difference allowing for significantly increased reaction rates.
The charge, discharge behavior of the electro-chemical catalytic membrane significantly differs from the reaction pathway previously accepted in literature.
The electro-chemical catalytic membrane shows significant deviations from all other specimens in the mass spectrum at 17, 18, 30, 32 and 44 amu, as shown in Fig. 4. The difference at 30 and 32 amu represent a reduction in NO and production of O2, respectively. The large peak at 18 amu, followed by sub-peaks at 17 and 16 amu indicates lone and charged oxygen. The peak at 44 amu indicates a concentration of N2O. The decrease in magnitude at 18 amu and the substantial peak at 44 amu represents a significant deviation from the majority of literature for NO breakdown. NO reduction has traditionally been separated into two categories: 1. Chemical gradient and equilibrium with or without a catalyst present24,28; 2. Combustion reaction29,30,31,32.

Mass Spectrum Analysis Comparison of Exhaust Effluent after Interacting with Each Specimen at 600 °C and Subject to 5 ml/min Flow of a Certified Premixed Gas Cylinder of 10 vol% NO and 90 vol% N2. Baseline Represents the Effluent of the Same Gas through the Testing Apparatus at Temperature without Any Reactive Membrane in Place. **The electro-chemical catalytic membrane with external short circuit is not shown, as it nearly coincides with the traditional PGM catalytic converter, reducing the clarity of the plot.
A high concentration of NO will drive an equilibrium reaction given sufficient energy and/or the presence of a catalyst31,32,33.
NO may also undergo a combustion reaction with ammonia (NH3), air and/or hydrocarbons (CxHy) in which NO will typically pass through an intermediary of NO229,34,35,36,37,38.
In this study, there is clear formation of N2O without additional reactants being sourced to aid the reduction of NO. Previously, an intermediary of N2O during the breakdown of NO had only been recorded while supplying ammonia to a platinum catalyst at 850 °C29,31,32,33,34,35,36,37. It is therefore believed that the electrical oscillation activity of the electro-chemical membrane is the reason for an altered reaction pathway and significant reduction of NO. This hypothesis is primarily investigated through a Gibbs free energy analysis. The following table lists potential reactions and respective changes in Gibbs free energy for a temperature range near the testing point of 600 °C in the breakdown of NO and eventual formation of N2 and O2.
For simplicity of analysis, it is assumed that the first step in the breakdown of NO is the formation of N2O2, as has been reported previously in literature17,18,19,20,21,22,23,24,25,26,27,31,32,33,34,35,36,37,38,39,40,41,42. Therefore, the key equations to investigate in Table 3, given sufficient concentration of N2O2, are reaction equations numbers 8 and 10. It is clear that at 600 °C the formation of NO2 is energetically favorable to the formation of NO2, reaction 10, if the Gibbs free energy minimization is the only driving factor. However, it also indicates that the formation of N2O, reaction 8, could also be spontaneous but is less probable to occur than reaction 10. Therefore, it is reasonable to assume that an electrochemical manipulation/influence could drive the likelihood of occurrence toward reaction 8 and the formation of N2O.
Additionally, the transfer efficiency from NO to N2 is considered in conjunction with the Gibbs free energy analysis. Table 3 indicates in reactions 12 and 14 that N2O as an intermediary species should result in a higher yield of N2 when compared to NO2.
Table 4 above calculates the transfer efficiency factor from NO reduction to N2 production. The ideal increase in N2 is calculated from the reduction of NO reported in Table 2, and represents the amount of N2 that could be produced if the transfer efficiency were equal to 1. That is, if all NO reduction resulted in the formation of N2. It is clear to see that the electrochemical catalytic membrane with enhanced electrical activity has a significantly better transfer efficiency at 0.94. There is a direct correlation between N2O production and increased N2 transfer efficiency. The negative factor for the PGM catalytic converter is not considered to be abnormal for catalytic converter operation. It has been reported that commercial PGM catalytic converters do have limited N2, NO storage capability, which would account for the discrepancy in N2 concentration in the exhaust4,5,6,7,8,9,10,11,12.
Formation of N2O is seen in the mass spectrum analysis of the electrochemical catalytic membrane and more predominately in the electrochemical catalytic membrane with forced electrical oscillations. It is not as evident in the effluent of the traditional PGM catalyst, nor the electrochemical catalytic membrane with an electrical connection between the anode and cathode. Therefore, when the electrical activity on the membrane is not present, the formation of N2O is significantly diminished.
The deposition, removal, and rearrangement of electrons from/on the catalytic surface is believed to be the key in altering the probability of reaction pathways. Traditional catalytic reaction chemistry with a solid, non-consumable catalyst without the presence or possibility of water formation or the presence of a proton acceptor/donor, assumes that the catalytic surface, after absorbing the reactant, acts primarily as an electron transport mechanism around the reactant molecule43,44. For the above experimental results, the apparatus was free of any water vapor, as described in the methods section, and the test flow consisted of only nitrogen and oxygen, eliminating the possibility of acid–base catalysis. Within any catalyst in which there is electron movement around the reactant, there must be an ohmic like resistance. However, if the charge on the catalytic surface is forced to vary, i.e. the surface is forced to charge and discharge, electron movement to and from the surface is established prior to the absorption of the reactant molecule. The addition and removal behavior of electrons that is established on the surface is believed to manipulate the natural reaction pathways increasing the overall reactivity of the system. In addition, this alternating voltage potential creates a fluctuating external electric field capable of doing work and exciting any incoming polar molecule. The external field may also manipulate the collisions of the incoming reacting species by forcing varying molecular alignment as the flow approaches the catalytic surface.
Conclusions
The unique configuration of a metal-based catalytic surface connected through a dielectric membrane to another metal-based electrode allows for the potential of surface charging and charge communication without significant internal current flow or internal charge balancing. This charging and discharging of the catalytic surface allows for the altered chemical reaction pathways demonstrated in this work. When the two metal electrodes of the electro-chemical catalytic membrane are electrically connected, the reactivity decreased by a factor of ~ 2. Any charge that began to develop on the surface was able to relax back into equilibrium via current flow. Therefore, the surface charge was no longer capable of reacting with the incoming flow, decreasing efficacy of NO conversion.
Further investigation into this phenomenon has led to the consideration of CO and CO2 conversion to particulate carbon and gaseous oxygen. Although this exploration is in its infancy, it does indicate potential for the electrochemical manipulation of carbon based emissions through electric potential oscillation. However, unlike the work described in the background of this work17, initial testing has shown significant carbon deposition and carbon coking of the electrode layer during prolonged operation in a purely CO and CO2 environment, decreasing reactivity over time. Additional work is required to develop a technology capable of continually reacting CO and CO2 into particulate carbon and gaseous oxygen without the addition of other reactants to prevent coking.
References
Nitrogen Dioxide | American Lung Association. Available at: https://www.lung.org/our-initiatives/healthy-air/outdoor/air-pollution/nitrogen-dioxide.html.
Tajuelo, M. et al. Secondary organic aerosol formation from α-methylstyrene atmospheric degradation: Role of NO level, relative humidity and inorganic seed aerosol. Atmos. Res. 230, 104631 (2019).
Wood, N. & Roelich, K. Tensions, capabilities, and justice in climate change mitigation of fossil fuels. Energy Res. Soc. Sci. 52, 114–122 (2019).
Podlovchenko, B. I., Manzhos, R. A. & Maksimov, Y. M. Kinetics and mechanism of interaction between methanol and adsorbed oxygen on a smooth polycrystalline platinum electrode: Transients of the open-circuit potential. Russ. J. Electrochem. 42, 1061–1066 (2006).
Previdello, B. A. F., Fernández, P. S., Tremiliosi-Filho, G. & Varela, H. Oscillatory electro-oxidation of methanol on platinum single crystal electrodes. Electrocatalysis. 7, 276–279 (2016).
Hirata, H. Recent research progress in automotive exhaust gas purification catalyst. Catal. Surv. Asia 18, 128–133 (2014).
Matsumoto, S. Recent advances in automobile exhaust catalysts. Catal. Today 90, 183–190 (2004).
Mizuno, N., Yamato, M. & Misono, M. Reactions of the monoxides of carbon and nitrogen over the superconducting lanthanoid mixed oxide YBa2Cu3Oy. J. Chem. Soc. Chem. Commun. 887–888 (1988).
Dong, X., Tsuneyama, K. & Hibino, T. Ultra-low loading Pt-Rh/Sn0.9In0.1P2O7 three-way catalyst for propane+NO+O2 reaction. Appl. Catal. B Environ. 106, 503–509 (2011).
Hori, T. et al. Electrolysis of humidified methane to hydrogen and carbon dioxide at low temperatures and voltages. Int. J. Hydrogen Energy 44, 2454–2460 (2019).
Iwamoto, M. & Hamada, H. Removal of nitrogen monoxide from exhaust gases through novel catalytic processes. Catal. Today 10, 57–71 (1991).
Liu, G. & Gao, P.-X. A review of NOx storage/reduction catalysts: mechanism, materials and degradation studies. Catal. Sci. Technol. 1, 552 (2011).
He, H., Zhang, C. & Yu, Y. A comparative study of Ag/Al2O3 and Cu/Al2O3 catalysts for the selective catalytic reduction of NO by C3H6. Catal. Today 90, 191–197 (2004).
Hamada, H., Kintaichi, Y., Sasaki, M., Ito, T. & Tabata, M. Highly selective reduction of nitrogen oxides with hydrocarbons over H-form zeolite catalysts in oxygen-rich atmosphere. Appl. Catal. 64, L1 (1990).
Suda, A. et al. Store and release of oxygen of ceria-zirconia solid solution synthesized by solid phase reaction at near room temperature. J. Ceram. Soc. Jpn. 109, 177–180 (2001).
Suda, A., Ukyo, Y., Sobukawa, H. & Sugiura, M. Improvement of oxygen storage capacity of CeO2-ZrO2 solid solution by heat treatment in reducing atmosphere. J. Ceram. Soc. Jpn. 110, 126–130 (2002).
Welles, T. S. & Ahn, J. A novel sold oxide fuel cell based catalytic converter replacement for enhanced emission control and power generation in automotive exhaust. Proceedings of the SAE 20PFL-0379 (2020).
Peters, R., Dahl, R., Klüttgen, U., Palm, C. & Stolten, D. Internal reforming of methane in solid oxide fuel cell systems. J. Power Sour. 106, 238–244 (2002).
Hydrogen Production: Natural Gas Reforming | Department of Energy. Available at: https://www.energy.gov/eere/fuelcells/hydrogen-production-natural-gas-reforming.
Beretta, D. et al. Thermoelectrics: from history, a window to the future. Mater. Sci. Eng. R Reports. 138, 100501 (2019).
Skinner, S. J. & Kilner, J. A. Oxygen ion conductors. Mater. Today 6, 30–37 (2003).
Lu, H., Rihko-Struckmann, L. & Sundmacher, K. Spontaneous oscillations of cell voltage, power density, and anode exit CO concentration in a PEM fuel cell. Phys. Chem. Chem. Phys. 13, 18179–18185 (2011).
Saleemi, A. S., Abdullah, A. & Anis-Ur-Rehman, M. Thermophysical properties of Gd doped ceria nanoparticles. Dig. J. Nanomater. Biostruct. 12, 19–28 (2017).
Nagai, Y. et al. X-ray absorption fine structure analysis of local structure of CeO2-ZrO2 mixed oxides with the same composition ratio (Ce/Zr = 1). Catal. Today 74, 225–234 (2002).
Ozawa, M., Kimura, M. & Isogai, A. The application of CeZr oxide solid solution to oxygen storage promoters in automotive catalysts. J. Alloys Compd. 193, 73–75 (1993).
Shelef, M. Selective catalytic reduction of NOx with N-free reductants. Chem. Rev. 95, 209–225 (1995).
Nagao, M., Yoshii, T., Hibino, T., Sano, M. & Tomita, A. Electrochemical reduction of NOx at intermediate temperatures using a proton-conducting In 3+–doped SnP2O7 electrolyte. Electrochem. Solid-State Lett. 9, J1–J4 (2006).
Bauerle, G. L., Wu, S. C. & Nobe, K. Catalytic reduction of nitric oxide with ammonia on vanadium oxide and iron-chromium oxide. Ind. Eng. Chem. Prod. Res. Dev. 14, 268–273 (1975).
Auxilia, F. et al. Low-temperature remediation of NO catalyzed by interleaved CuO nanoplates. Adv. Mater. 26, 4481–4485 (2014).
Grossale, A., Nova, I., Tronconi, E., Chatterjee, D. & Weibel, M. The chemistry of the NO/NO2–NH3 “fast” SCR reaction over Fe-ZSM5 investigated by transient reaction analysis. J. Catal. 256, 312–322 (2008).
Huang, T.-J. & Hsiao, I.-C. Nitric oxide removal from simulated lean-burn engine exhaust using a solid oxide fuel cell with V-added (LaSr)MnO3 cathode. Chem. Eng. J. 165, 234–239 (2010).
Walsh, F. C. & Ponce de León, C. Progress in electrochemical flow reactors for laboratory and pilot scale processing. Electrochim. Acta 280, 121–148 (2018).
Burch, R., Millington, P. J. & Walker, A. P. Mechanism of the selective reduction of nitrogen monoxide on platinum-based catalysts in the presence of excess oxygen. Appl. Catal. B Environ. 4, 65–94 (1994).
Kammer Hansen, K., Skou, E. M. & Christensen, H. Perovskites as cathodes for nitric oxide reduction. J. Electrochem. Soc. 147, 2007–2012 (2000).
Delmonde, M. et al. Production of volatile species during the oscillatory electro-oxidation of small organic molecules. J. Phys. Chem. C 118, 17699–17709 (2014).
Iwamoto, H., Kameoka, S., Xu, Y., Nishimura, C. & Tsai, A. P. Effects of Cu oxidation states on the catalysis of NO+CO and N2O+CO reactions. J. Phys. Chem. Solids 125, 64–73 (2019).
Ozensoy, E., Hess, C. & Goodman, D. W. Understanding the catalytic conversion of automobile exhaust emissions using model catalysts: CO + NO reaction on Pd(111). Top. Catal. 28, 13–23 (2004).
Bauerle, G. L., Wu, S. C. & Nobe, K. Parametric and durability studies of NOx reduction with NH3 on V2O5 catalysts. Ind. Eng. Chem. Prod. Res. Dev. 17, 117–122 (1978).
Otto, K. & Shelef, M. Studies of surface sections of NO by isotope labeling: VI The reduction of nitric oxide by ammonia and hydrogen over supported liuthenium. Zeitschrift fur Phys. Chemie 85, 308–322 (1973).
Inomata, M., Miyamoto, A. & Murakami, Y. Mechanism of the reaction of NO and NH3 on vanadium oxide catalyst in the presence of oxygen under the dilute gas condition. J. Catal. 62, 140–148 (1980).
Otto, K., Shelef, M. & Kummer, J. T. Studies of surface reactions of NO by isotope labelling. II. Deuterium kinetic isotope effect in the ammonia-nitric oxide reaction on a supported platinum catalyst. J. Phys. Chem. 75, 875–879 (1971).
Otto, K. & Shelef, M. Studies of surface reactions of nitric oxide by isotope labelling. IV. The reaction between nitric oxide and ammonia over copper surfaces at 150–200°. J. Phys. Chem. 76, 37–43 (1972).
Nagao, R., Cantane, D. A., Lima, F. H. B. & Varela, H. The dual pathway in action: decoupling parallel routes for CO2 production during the oscillatory electro-oxidation of methanol. Phys. Chem. Chem. Phys. 14, 8294 (2012).
Nagao, R., Cantane, D. A., Lima, F. H. B. & Varela, H. Influence of anion adsorption on the parallel reaction pathways in the oscillatory electro-oxidation of methanol. J. Phys. Chem. C 117, 15098–15105 (2013).
Acknowledgements
This material is based upon work supported by the National Science Foundation (NSF) Graduate Research Fellowship Program (GRFP) under Grant No. 2019265542. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the NSF.
Author information
Authors and Affiliations
Contributions
All authors wrote and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Welles, T.S., Ahn, J. Novel investigation of perovskite membrane based electrochemical nitric oxide control phenomenon. Sci Rep 10, 18750 (2020). https://doi.org/10.1038/s41598-020-75360-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75360-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
