
Abstract
A nutritional intervention, exclusive enteral nutrition (EEN) can induce remission in patients with pediatric Crohn’s disease (CD). We characterized changes in the fecal microbiota and metabolome to identify the mechanism of EEN. Feces of 43 children were collected prior, during and after EEN. Microbiota and metabolites were analyzed by 16S rRNA gene amplicon sequencing and NMR. Selected metabolites were evaluated in relevant model systems. Microbiota and metabolome of patients with CD and controls were different at all time points. Amino acids, primary bile salts, trimethylamine and cadaverine were elevated in patients with CD. Microbiota and metabolome differed between responders and non-responders prior to EEN. EEN decreased microbiota diversity and reduced amino acids, trimethylamine and cadaverine towards control levels. Patients with CD had reduced microbial metabolism of bile acids that partially normalized during EEN. Trimethylamine and cadaverine inhibited intestinal cell growth. TMA and cadaverine inhibited LPS-stimulated TNF-alpha and IL-6 secretion by primary human monocytes. A diet rich in free amino acids worsened inflammation in the DSS model of intestinal inflammation. Trimethylamine, cadaverine, bile salts and amino acids could play a role in the mechanism by which EEN induces remission. Prior to EEN, microbiota and metabolome are different between responders and non-responders.
Similar content being viewed by others


Introduction
Exclusive enteral nutrition (EEN) is the treatment of choice for patients with pediatric Crohn’s disease (CD) in Europe1. It is equally efficacious as corticosteroids in inducing clinical remission in children and adolescents with active CD2,3 and superior in inducing mucosal healing4. Despite solid evidence for efficacy of nutritional therapy in children, the mechanisms by which EEN induces remission remain elusive.
The gut microbiota and metabolomes of patients with CD are different from healthy individuals5,6. The implication is that perturbed gut microbial co-metabolism could play an important role in the pathogenesis of CD. Indeed many studies showed that interactions between the gut microbiota, metabolites and the intestinal immune system are critical for maintenance of a healthy intestine7. The intestinal microbiota of new-onset patients with pediatric CD typically exhibits a decreased microbial diversity8, with lower relative abundance of Firmicutes9. Moreover, patients with CD exhibit profound differences in the intestinal metabolome, including lower concentration of short chain fatty acids10,11, higher concentration of amino acids11, and a dysregulation of bile acid composition, including higher conjugated and lower secondary bile acid concentrations12.
EEN therapy likely modulates both the microbial and metabolic environments in the gut of patients with CD13,14,15. Several studies demonstrated that EEN reduces the alpha diversity of the intestinal microbiota of patients with CD14,15. This appears paradoxical, as higher alpha diversity is often associated with a more “healthy” microbiota. More specifically, EEN was found to induce a decline in numbers of presumably protective gut bacterial species (e.g. Faecalibacterium prausnitzii and Bifidobacterium spp.). In addition, decreased concentrations of fecal short chain fatty acids (SCFAs) such as butyrate, which is generally thought to be a beneficial metabolite for host health, have been found to be associated with disease improvement during EEN15. However, from a nutritional and microbiological point of view, these findings are to some extent expected. Because EEN contains relatively few components, when compared to a regular diet, a reduced alpha diversity of the gut microbiota is likely to result in people with this diet. In addition, removing complex carbohydrates from the diet reduces the amount of substrate available for fermentation into SCFAs by fiber-degrading bacterial taxa.
Whether changes in the intestinal microbiota and metabolome are a cause or consequence of CD remains uncertain, primarily due to the lack of longitudinal observations8,16. Two recent papers show that diets with either partial EEN17 or mimicking EEN composition with more solid ingredients18, are equally effective as EEN in inducing remission in patients with pediatric CD. These papers17,18 are important because the novel nutritional therapies are better tolerated and would thus be beneficial for a larger number of patients. Moreover, these papers17,18 also show that changes in the fecal microbiota and metabolome induced by these novel nutritional therapies are comparable to the changes that are induced by EEN. The findings reported thus also provide evidence that changes in the microbiota and metabolome could be causative in inducing remission in patients with pediatric CD disease.
In this study we prospectively followed a cohort of pediatric treatment-naïve patients with CD undergoing EEN therapy and investigated changes in their fecal microbiota and metabolome. The aims of this study were; (1) to describe gut microbial and metabolic changes during the course of EEN and the differences between responders and non-responders to EEN. (2) To evaluate these microbial and metabolic changes as potential mechanisms of EEN action.
Results
Patients
In total, 43 children with newly diagnosed CD were included (47% male, median age, 14 years [IQR 12–15], Fig. 1A,B). Patient and disease characteristics, and clinical and biochemical response are shown in Fig. 1B and Table 1 and were partially described previously19. In addition to EEN, all patients were started on concomitant thiopurines (i.e. azathioprine) following the international guidelines1. Eighteen healthy controls participated (50% male, median age years 13 [IQR 11–16]). For the microbiota analysis 96 samples were included, at baseline (T0) 27 samples, during EEN (T1) 22 samples, at end of EEN (T2) 19 samples, at follow up on habitual diet (T3) 13 samples, and 15 healthy control samples. For the metabolite analysis 122 samples were included, at baseline (T0) 43 samples, during EEN (T1) 37 samples, at end of EEN (T2) 22 samples, at habitual diet (T3) 20 samples, and 31 healthy control samples were included.

Study outline and patient selection. Panel (A) shows the therapy and time points of fecal sample collection. In panel (B) the selection of patients that were included in this study is outlined.
Some patients halted EEN due to various reasons. The samples of these patients were analyzed up to the latest time point at which they did receive EEN and were excluded from further study.
Regarding control samples used in this study; two time points of the 18 control subjects were analyzed in the metabolic analysis. Five samples had to be excluded because of preparation errors. For the microbiome analysis only 15 controls at a single time point were used because of limitations in sequence capacity. The overall study design is shown in Fig. 1A,B.
Responders to EEN were identified by a reduction of fecal calprotectin of more than 50% at T2, compared to T0. See Fig. 1B and Table 1 for patient inclusions. Pilot analysis did not reveal statistically significant correlations when using the clinical definitions for remission (data not shown).
Microbiota
Healthy controls versus patients with CD
To investigate the fecal microbiota of patients with CD and controls and the effect of EEN treatment, 16S rRNA gene amplicon sequence analysis was carried out.
At baseline, patients had reduced observed OTU richness, as compared to healthy controls (HC, mean OTU richness of 235.6 vs. CD T0, mean OTU richness of 192.9, p = 0.036, Mann–Whitney, Fig. 2A). Shannon and inverse Simpson diversity measures, which incorporate species evenness as well as richness, were not significantly different between controls and patients at baseline (HC 3.67 vs. T0 3.4, p = 0.098, and HC 20.37 vs. CD T0 16.75, p = 0.198, respectively).

Microbiota of patients versus controls, the effect of EEN and differences between responders and non-responders. Panel (A) shows the difference in richness between controls and patients at T0. Panel (B) shows that EEN reduced diversity. Panel (C) and (D) show the effect of EEN on the proportional abundance of the Blautia and Subdoligranulum genera. Principal coordinate (PCoA) plots in panels (E) and (F) show separation of controls (red) and patients (blue), and responders (red) and non-responders (blue) at T0. In panels (G,H) and (I) some of the species that comprise the signature of responders versus non-responders at T0 are depicted. HC healthy controls, R responders, NR non-responders.
As expected, the overall microbial composition of controls was highly different (p < 0.001) from that of patients at all time points (Bray Curtis with AMOVA, Table 2, Fig. 2E). Numerous taxa exhibited significantly different proportional abundances; Escherichia coli, Ruminococcus gnavus, Dorea longicatena, and Blautia spp. were present in greater relative abundance in CD, whereas Eubacterium rectale, Bifidobacterium longum and Ruminococcus bromii were proportionally higher in controls (all p values < 0.05 with both LEfSe and Benjamini–Hochberg-corrected p values generated by Metastats) Supplementary Table 4 details the differences of the most proportionally abundant species between patients with CD at T0 and controls.
The effect of EEN on the microbiota
EEN therapy caused a decrease in microbial diversity, with patients at T1 and T2 having a significantly lower diversity than at baseline (mean inverse Simpson Index score for T0 = 19.40 vs. T1 = 11.57, p = 0.003) (T0 = 19.40 vs. T2 = 12.85, p = 0.045) (Fig. 2B). At follow up (T3) there was no difference in diversity from baseline (mean T0 inverse Simpson index score = 19.40 vs. T3 mean = 16.87, p = 0.505).
The introduction of EEN was also significantly associated with a shift in the overall microbial composition (T0 vs. T1, p = < 0.001) and at the end of EEN (T0 vs. T2, p = < 0.001), with differences disappearing at follow up (T0 vs. T3, p = 0.99) (Bray Curtis-based AMOVA tests, Table 2). However, the response of individual taxa varied greatly between individuals. (Supplementary Figure 1). Only genera that were identified as being significantly different using the stringent, Benjamini–Hochberg-corrected, Metastats-based approach, and with a proportional abundance of more than 0.01% of the total observed microbiota, are detailed below. The genera significantly altered between baseline (T0) and during the EEN treatment (T1) were Blautia (10.8% of the total observed microbiota across all samples) and Subdoligranulum (2.5% of the total observed microbiota), both of which were reduced in relative abundance during EEN therapy, (Metastats-based p = 0.033 and p = 0.044, respectively) (Fig. 2C,D). However, neither of these genera was significantly reduced at timepoint T2 compared to T0. No bacterial taxa were shown to be significantly increased in proportional abundance with both the Metastats-based and LEfSe approaches as a result of the EEN therapy.
Responders versus non-responders
Prior to the onset of EEN therapy, the overall composition of the microbiota differed between subsequent responders and non-responders (Bray Curtis dissimilarity analysis and AMOVA tests, p = 0.008). A PCoA plot is shown in Fig. 2F.
Due to the high inter-individual variation in microbiota composition, there were no significant differences in alpha diversity measures, and few species or genera that differed between responders and non-responders prior to therapy (T0). However, Dorea longicatena (2.5% of the total observed microbiota) (Metastats-based p = 0.012; LEfSe p = 0.005), (Fig. 2G) Blautia obeum (1.3% of the total observed microbiota) (Metastats-based p = 0.019; LEfSe p = 0.006) (Fig. 2H), and Bifidobacterium longum (0.07% of the total observed microbiota) (Metastats-based p = 0.040; LEfSe p = 0.012) (Fig. 2I) were all associated with lack of response. Using three-way LEfSe analysis, comparing healthy controls versus responders (T0) versus non-responders (T0), multiple taxa appeared to be associated with both responders and non-responders at T0. Supplementary Table 5a details the most important differences between healthy controls, responders and non-responders at T0. The following taxa are examples of proportionally abundant components of the bacterial “signature” that differentiated non-responders from responders prior to therapy (T0): Dorea longicatena (Non-responders, p = < 0.001), Blautia obeum (Non-responders, p = 0.002), and Escherichia coli (Non-responders, p = < 0.001).
At the follow up period (T3), there were also differences between responders and non-responders. As with T0, the overall composition of the microbiota at T3 also differed between responders and non-responders (Bray Curtis dissimilarity analysis and AMOVA tests, p = 0.004). (PCoA plot shown in Supplementary Figure 3A). Of note, both Metastats-based and LEfSe analyses associated higher proportional abundances of the butyrate-producing genus Roseburia with responders (2.40% mean proportional abundance at T3 in responders vs. mean of 0.22% in non-responders, Benjamini–Hochberg-corrected Metastats-based p = 0.043, LEfSe p = 0.013), while another key butyrate-producing genus, Faecalibacterium, was also associated with responders by LEfSe only (10.48% mean proportional abundance in responders vs. mean of 1.42% in non-responders, p = 0.041). Three-way LEfSe analysis, comparing healthy controls versus responders (T3) versus non-responders (T3), associated, amongst others, Faecalibacterium prausnitzii (p = 0.034), Bifidobacterium adolescentis (p = 0.032), and Ruminococcus bromii (p = 0.009) with responders, and Escherichia coli (p = 0.004) and four Blautia OTUs (p = 0.003, 0.008, 0.026, and 0.009) with non-responders.
Supplementary Table 5b details the most important differences between healthy controls, responders and non-responders at T0 and at T3.
Thus, EEN significantly changes both alpha and beta diversity indices in patients with CD, and we provide evidence for differences between responders and non-responders both prior to and after therapy.
1H NMR spectroscopy-based metabolic profiling analysis
Healthy controls versus patients with CD
Metabolic differences between patients with CD and controls and the effects of EEN were analyzed using 1H NMR spectroscopy.
Significant differences were observed between patients with CD and HC at all time points based on Orthogonal Partial Least Squares-Discriminant Analysis (OPLS-DA) models. An example of the cross-validated scores plot of OPLS-DA between the HC and patients with CD at T0 is shown in Fig. 3A (R2X = 59.7%, Q2Y = 0.55, permutation p = 0.01). The corresponding ROC curve (Supplementary Figure 2A, AUC = 0.94) confirms that this model is highly predictive to distinguish patients from controls. Amino acids (alanine, tryptophan, tyrosine, valine, isoleucine, leucine, phenylalanine) and microbial metabolites (cadaverine, lactate, propionate, putrescine, trimethylamine (TMA)) were found in higher concentrations in patients with CD at T0 compared to healthy controls (Supplementary Table 6).

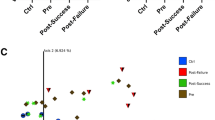
Differences in fecal metabolites between patients and controls, responders and non-responders and the effect of EEN on the fecal metabolome. Panels (A–C) show OPLS-DA plots. This analysis indicates that 1HNMR analysis can distinguish controls (blue) from patients (red) at T0, panel (A). A marginal EEN induced change in metabolome from T0 (red) to T2 (blue) is shown in panel (B). The difference in metabolome between responders (blue) and non-responders (red) at T0 is shown in panel (C). Panel (D) shows increased total fecal amino acids in patients that decrease during EEN therapy. Panel (E) shows that bile salts are not elevated in patients and unchanged during therapy. Panels (F) and (G) show decreased bile salt metabolism and hydrophobicity in patients at T0 that is partially normalized during therapy. Panels (H) and (I) show increased levels of trimethylamine and cadaverine respectively and show partial normalization of these compounds during EEN therapy. HC healthy controls, R responders, NR non-responders.
The effect of EEN on the metabolome
EEN affected the global metabolome with the fecal metabolic profile prior to EEN (T0) differing significantly from the end of EEN (T2) (R2X = 59.4%, Q2Y = 0.1, p = 0.04, Fig. 3B). However, the corresponding ROC curve (Supplementary Figure 2B, AUC 0.71) does not suggest a highly predictive value of the model at this time point. No significant differences were found between other time points.
Responders versus non-responders
When patients were stratified based on their responses to EEN therapy, there was a statistically significant difference between the fecal metabolic profiles of responders and non-responders at baseline (T0) (R2 = 60.4%, Q2 = 0.28, p = 0.030, Fig. 3C). The ROC curve (Supplementary Figure 2C, AUC = 0.8) suggests that the fecal metabolic profiles could predict the responses of patients to EEN prior to the treatment.
Although the global metabolic profiles of both responders (R2X = 58.0%, Q2Y = 0.33, p = 0.01) and non-responders (R2X = 67.5%, Q2Y = 0.69, p = 0.01) were different from healthy controls at follow-up (T3), several metabolites (i.e. leucine, propionate, valine, lactate, alanine, cadaverine, trimethylamine, tyrosine, phenylalanine, isovalerate, urocanate, succinate) were normalized in responders but not in non-responders (Supplementary Table 6).
Amino acids
Healthy controls versus patients with CD
Because the NMR analysis showed large differences in amino acids between patients with CD and controls, we investigated fecal amino acids in more detail using HPLC. At baseline (T0), there was a clear separation of the fecal amino acid profile in CD from HC, with a higher concentration of the total and most individual amino acids (Fig. 3D, Supplementary Table 7, Supplementary Table 8). Only glutamic acid, arginine and taurine were not elevated in patients.
The effect of EEN on amino acids
Total amino acid concentrations tended to decrease at T1 when compared to T0. However, the fecal amino acid profiles at baseline (T0) were not significantly different from time-points (T1, T2 & T3), for either the total or individual amino acid concentrations (Fig. 3D, Supplementary Table 7, Supplementary Table 8).
Responders versus non-responders
When patients were stratified based on response to EEN therapy, responders had a lower fecal concentration of histidine, citrulline, and isoleucine both at baseline (T0) and at the end of EEN therapy (T2), and a lower concentration of serine, glycine, and alanine at end of EEN therapy (T2) (Supplementary Table 9).
We also compared amino acids between healthy controls, responders and non-responders. Although some individual amino acids of both responders and non-responders had a higher concentration in healthy controls at follow-up (T3) (i.e. asparagine, tryptophan), many amino acid concentrations were normalized in responders and not in non-responders at T3 (i.e. serine, histidine, tyrosine, phenylalanine, leucine), (Supplementary Table 8).
CD does not affect systemic amino acids
To establish whether the elevated fecal amino acids in patients with CD are reflected in the systemic circulation we measured plasma amino acids in an independent cohort of pediatric IBD patients (N = 41, 63% male, median age 15 years [IQR 12–16], median disease duration 22 months [IQR 10–44], 76% CD, 24% ulcerative colitis). No correlation was found between plasma total, or individual amino acids concentrations and disease severity as measured by fecal calprotectin (r: 0.166, p = 0.306, Supplementary Figure 3B).
Bile acids
Healthy controls versus patients with CD
Bile salts are emerging as important mediators in immune function20. Because little is known about bile salt metabolism in CD and during EEN, we analyzed the fecal bile acid composition by HPLC. Although the total concentrations of bile acids were not different between patients with CD and controls (Fig. 3E, Supplementary Table 10), the metabolism of fecal bile acids was significantly changed in patients with CD (Fig. 3F, Supplementary Figure 3D–H). At baseline (T0), the relative concentration of primary bile acids was higher in patients with CD resulting in a reduced hydrophobicity of the bile acid pool (Fig. 3G, Supplementary Table 10).
The effect of EEN on bile acids
Comparing the fecal bile acids at baseline (T0) to during EEN (T1), or time-points after EEN cessation (T2 & T3), revealed no difference in bile acids hydrophobicity or the percentage of secondary bile acids (Fig. 3G, Supplementary Table 11).
Responders versus non-responders
When patients were stratified based on response to EEN therapy, there was no difference in the total bile acid concentration, bile acid hydrophobicity and the fraction of secondary bile acids between responders and non-responders at baseline (T0), or other time points (T1-3) (Supplementary Tables 10 and 11).
At follow-up (T3), the fraction of secondary bile acids and the total bile acid concentration in feces of responders and non-responders did not differ from HC (Supplementary Table 10). However, bile acid hydrophobicity was normalized in responders and not in non-responders at follow-up (T3) (Supplementary Table 10).
Effects of selected metabolites on physiological parameters
After establishing the effect of EEN on fecal metabolic composition we investigated if metabolites that were shown to be different between controls and patients with CD could have a causal role in disease development.
TMA and cadaverine inhibit Caco-2 proliferation
Since wound healing is a critical process in recovery from inflammation we used a real time assay of cell growth and attachment to study the effect of selected metabolites on this process.
TMA and cadaverine were metabolites of interest as they differed between patients with CD and healthy controls and are metabolic products of intestinal microbes (Fig. 3H,I)21,22,23. These compounds were especially interesting because they were normalized in responders only at follow up (Supplementary Table 6). As a control metabolite for TMA we selected TMA N-oxide (TMAO), which is produced from TMA by hepatic flavin monooxygenases 3 (FMO3)24.
Both TMA and cadaverine inhibited caco-2 cell proliferation, relative to standard DMEM medium (Fig. 4A,B, Table 3). The control metabolite TMAO, did not inhibit caco-2 cell proliferation (Fig. 4A, Table 3).

The effect of TMA and cadaverine on cell growth, differentiation and cytokine secretion. Panel (A) shows that TMA, but not its metabolite TMAO reduces CaCo2 cell growth in a concentration dependent manner. Panel (B) shows that cadaverine reduced CaCo2 cell growth in a concentration dependent manner. Panels (C) and (D) show that TMA, but not its metabolite TMAO, and cadaverine are both able to reduce LPS induced TNFα and IL-β secretion in primary human PBMC.
TMA and cadaverine reduce LPS induced TNFα and IL-6 secretion in primary human lymphocytes
A possible mechanism by which microbial metabolites induce remission is by modulating the host immune response. We therefore studied the effect of selected metabolites on LPS induced cytokine secretion in primary human lymphocytes.
Human peripheral blood monocytes were incubated with TMA, TMAO and cadaverine for 24 h and subsequently stimulated with LPS. IL-6 and TNFα release from primary human monocytes upon LPS stimulation was inhibited by TMA and cadaverine but not by TMAO (Fig. 4C,D), p < 0.0001. The differences in IL-6 and TNFα release by monocytes were not caused by increased cell death as shown by FACS viability staining (Supplementary Figure 3C).
Amino acid feeding worsens outcome of a mouse model of intestinal inflammation
Since amino acids were elevated in patients, and many amino acids were normalized in responders at T3 only (Supplementary Table 9) we wanted to investigate a causal role of these compounds in inflammation. As cell culture models are not suitable for studying the effect of amino acids, we determined if these compounds could affect the DSS model of intestinal inflammation.
Mice were fed amino acids or a diet containing a caloric equivalent amount of whole milk protein. Non-DSS treated mice fed with standard chow served as a control group. Weight loss, stool score, pathology, and endoscopy score were all worse in DSS treated mice (both amino acid or milk protein fed) compared to the control group (Fig. 5A–D). Amino acid-fed DSS mice had a lower body weight, and significantly (p < 0.0001) worse pathology and endoscopy score compared to milk-protein fed DSS mice (Fig. 5A–D).

A diet of amino acids worsens the murine DSS model of colitis. Mice received 1.5% DSS in their drinking water and were fed chow supplemented with milk protein (DSS milk protein) or an equivalent amount of amino acids (DSS Amino Acids). Control mice received milk protein (No DSS milk protein). Mice fed amino acids lost weight more rapidly (panel A), had increased stool, pathology and endoscopy scores, (panels B–D). Levels of TNFα, as measured by quantitative PCR, were elevated in both groups (panel E) whereas levels of IL-6, as measured by quantitative PCR, were only elevated in the group receiving amino acids (panels F).
TNF-α expression was higher in the colons of DSS treated mice (both amino acid or milk protein fed) compared to control mice (Fig. 5E), IL-6 was only higher in the colons of DSS-treated mice fed with amino acids (Fig. 5F). Amino acid-fed mice showed a significantly (p = 0.028) increased expression of IL-6 over those fed with milk protein (Fig. 5F). TNF-α, did not show differences between amino acid and milk protein fed mice (Fig. 5E).
Discussion
In this study, we report a comprehensive analysis of the fecal microbiota and metabolome during a course of EEN therapy in patients with CD.
Microbiota analysis
We found marked differences in microbiota between patients with CD and controls. The differences, as seen in this cohort, largely correspond to previous studies25,26,27,28.
The microbiota of children with CD was highly variable, as has been reported before13. Nonetheless, patients with CD had a reduced richness as compared to controls prior to therapy. In common with many studies29,30, we also detected increased proportional representation of Escherichia coli and Ruminococcus gnavus31 in patients with CD, and a reduction in the relative abundance of putatively beneficial organisms such as Bifidobacterium spp., and butyrate producers such as E. rectale. During EEN, diversity (as measured using the inverse Simpson Index) was further reduced, confirming results from a previous study14.
Although limited by our relatively small number of samples, our results suggest that increased proportional abundance of taxa such as Bifidobacterium longum, Dorea longicatena, and Blautia obeum may indicate a lower chance of disease remission following EEN. Our observations suggest that the Blautia genus is of particular interest since proportional abundances of this group were generally higher in patients with CD versus healthy controls at baseline, were reduced in patients with CD during the EEN period, and were associated with non-responders at both T0 and T3. In contrast, a randomized clinical trial comparing nutritional interventions in pediatric patients with CD reported an increase in the Blautia genus during EEN17.
A meta-analysis of microbiota profiling studies in adult IBD patients reported an association of increased proportional abundance of Blautia spp. in patients with CD32. A more recent study showed that Blautia spp were associated with a dysbiotic cluster that separated part of patients with pediatric CD from controls33 Another study in pediatric CD reported increased Blautia, comparable to controls in patients with sustained response to Infliximab treatment34. Observations on Blautia spp from individual studies of pediatric CD cohorts thus appear to be variable, possibly because the resolution of genus level comparisons is not sufficient and masking contributions of Blautia species, or because the inherent inter-individual variation in microbiota composition makes it more difficult to find reproducible findings across different cohorts. More mechanistic research is evidently required in future in order to determine whether or not these species are playing a role in the disease process.
Metabolites
In contrast to the microbiota, the metabolome trended towards normalization for many compounds as a result of EEN therapy. These observations are not completely surprising as the diversity measures used in this study reflects microbiota composition and not its metabolic activity.
Several microbial metabolites that could play a role in the pathogenesis of CD were identified in this study. Cadaverine and trimethylamine (TMA) were found in a higher concentration in patients with CD, tended to decrease during the course of EEN (Fig. 3H,I) and were normalized at T3 in responders only.
Increased amino acids have been previously observed in CD patients35,36,37. The most straightforward explanation for this phenomenon is a reduced uptake due to a damaged epithelium. However, because total bile salt concentrations were not different between patients with CD and controls, and glutamic acid and taurine levels were not elevated in patients with CD, reduced epithelial uptake capacity does not seem a likely explanation for increased amino acid levels.
Mechanism of EEN
A possible causal role of elevated amino acids in CD is suggested by our observations that a diet containing amino acids worsens DSS induced colitis in a mouse model. These results are surprising in the light of previous studies showing that supplementation of amino acids reduces DSS-induced colitis38,39.
As examples, supplementation of mice in de DSS model of colitis with dietary tryptophan40, glutamine41, serine42 or arginine43 reduced inflammation.
However, study designs are not completely comparable, since we fed mice a complete mixture of amino acids compared to supplementation with selected amino acids. A recent paper shows that elevated fecal amino acids in patients with CD depended on increased urease activity of the microbiota37. Inoculation of a murine colitis model with E. coli engineered to express urease led to a worsening of the colitis in these animals. Increased amounts of amino acids seen in patients with CD might thus have a role in pathogenesis.
Cadaverine and TMA inhibit epithelial growth and adherence. This indicates that high levels of TMA and cadaverine could be detrimental for gut homeostasis and that these metabolites may be important in CD pathogenesis. Decreased serum levels of TMAO have been observed in patients with inflammatory bowel disease, providing additional evidence that reduced TMA metabolism might play a role in CD pathogenesis44.
Perhaps surprisingly, TNFα and IL-6 production of LPS stimulated human monocytes was reduced by TMA and cadaverine. However, besides its role as a pro-inflammatory cytokine, multiple anti-inflammatory effects of TNFα have also been described45,46,47.
Our data from the DSS model of colitis also suggest a complex role of TNFα and IL-6 in intestinal inflammation. Whereas amino acid feeding increased severity of intestinal inflammation, as assessed by multiple parameters, it only increased IL-6 levels, TNFα levels were unchanged.
Short chain fatty acids (SCFA) have been extensively studied in patients with IBD48. In this cohort, propionate concentrations were higher in children with CD, while butyrate levels were unaltered (not shown). Data regarding the effect of EEN on SCFA levels are conflicting, with decreases and increases both observed15,49.
The fecal bile acid pool of patients with CD was characterized by a higher concentration of primary and conjugated bile acids and lower secondary bile acids. This likely results in a fecal bile salt pool that is less hydrophobic in patients with CD. During EEN therapy, this alteration in bile acid composition was partially restored (Supplementary Figures 3D–H). Our results expand on an earlier study12, that showed similar alterations of the CD gut bile acids.
The anti-inflammatory effects of the TGR5 bile acid receptor activation are well described48. Since hydrophobic bile acids such as lithocholic acid are more potent activators of TGR5 than the hydrophilic bile acids50, the hydrophilic bile salt pool of patients with CD could be an important factor in the inflammatory process.
Differentiating responders and non-responders prior to therapy
Fecal microbiota and metabolome of responders and non-responders differ before the start of EEN therapy. Microbial differences between responders and non-responders to immunomodulators or nutrition have been reported before51,52. This could indicate that there might be as yet unidentified subtypes of CD disease and open the possibility to identify non-responders before treatment. This approach seems feasible as a recent paper53 identifies microbial signatures that could predict long term responders to EEN.
Conclusions
Patients responding to EEN therapy have different microbiota and metabolomes prior to therapy than patients that do not respond. This may allow for future prediction of EEN response.
The mechanism by which EEN induces remission is complex, several metabolites (TMA, cadaverine, amino acids and bile salts) possibly have a causal role in the development of CD.
Patients, materials and methods
Ethics
The Medical Ethical Committee of the Amsterdam UMC (Medisch Ethische Toetsingscommissie) approved the analysis of human samples described in this study under NL39254.029.12. In accordance with this approval, the children and their parents both gave informed consent for the use of pediatric samples in this study. All analysis and further experiments in this study were performed according to this approved protocol and relevant regulations and guidelines.
Animal experiments described in this study were performed according to a protocol that was approved by the Animal Ethical Committee of the Amsterdam UMC, (Dierexperimentele commissie) under nr DMO65. All analysis and further experiments in this study were performed according to this approved protocol and relevant regulations and guidelines.
Patients
This was a prospective multi-center cohort study in two academic hospitals (Amsterdam UMC locations AMC and VUMC) performed between January 2010 and July 2014. All children (< 18 years) with newly diagnosed, therapy naïve CD according to the revised Porto criteria54 undergoing EEN induction treatment were included. The control group consisted of age and sex matched, healthy school children from the same geographic area, with no family history of inflammatory bowel disease (IBD), from a well-documented cohort55. An outline of analysis performed in this study can be found in Fig. 1A,B). Participants who received antibiotics or probiotics within 3 months prior to inclusion or during the study period were excluded. Moreover, participants with a proven bacterial gastroenteritis or who had taken immunomodulatory drugs 3 months prior to inclusion were also excluded. A range of polymeric EEN formulas with similar composition based on cow milk protein (Supplementary Table 1) was provided during a 6-week course, during which no other food or fluid (except water) was allowed, followed by a 2-week course of EEN tapering and gradual introduction of habitual diet. During follow-up, dose adjustments or switch of maintenance therapy (and reasons for therapy adjustment) were collected. Localization and disease behavior were classified using the Paris classification56.
Sample collection
Patient and healthy controls were instructed to collect fecal samples in provided sterile containers, to store the sample at − 20 °C within at least 2 h of collection, and to deliver these frozen samples in a cooled condition to the hospital. A maximum of 4 samples were collected per patient at the following time points: At the time of diagnosis, but prior to bowel preparation and endoscopy (T0), during EEN (± 21 days after EEN initiation) (T1), at the end of treatment (± 42 days after EEN initiation) (T2), and after patients returned to their habitual diet (± 4 months after EEN initiation) (T3) (Fig. 1A). To avoid heterogeneity, samples from patients who had discontinued EEN therapy prematurely were excluded: T1 samples were excluded if EEN was previously discontinued, T2 and T3 samples were excluded if the full course of 6 weeks EEN had not been completed. Healthy controls were instructed to collect 2 fecal samples with an interval of 6 weeks. Aliquots of fecal samples were stored at − 20 °C until analysis.
Biochemical and clinical disease activity
Biochemical disease activity was assessed using fecal calprotectin (FC), the most accurate fecal biomarker of intestinal inflammation currently available57. Fecal calprotectin levels were determined at baseline (T0) and end of EEN (T2) by the Amsterdam University Medical Centre hospital clinical chemistry laboratory. Biochemical response was defined as a reduction of ≥ 50% at T2 compared to T0, as this has the highest predictive value for endoscopic treatment response58. Because we did not obtain calprotectin from all patients and not all patients completed EEN therapy, for the analysis of responders versus non-responders not all samples could be used.
Clinical disease activity was assessed by the treating physician at T0 and T2 using the Physician Global Assessment (PGA) 4 point scale: inactive, mild, moderate, and severe59. Clinical response on EEN was defined as a PGA scored as inactive to mild after a full 6 week-course of EEN, without the need for additional remission induction treatment.
Microbiota analysis using 16S rRNA gene amplicon sequencing
Sequencing of bacterial 16S rRNA gene amplicons was performed to characterize microbial community composition, largely as described in60, of which a modified version is described below.
DNA extraction
DNA extraction was carried out on fecal samples, ranging from 25 to 265 mg in weight, using the FastDNA SPIN Kit for Soil (116560200, MP Biomedicals) per kit instructions. Samples were eluted in 50 µl of DES then quality checked by running on an agarose gel.
PCR amplification for sequencing
PCR amplification of the V1-V2 hypervariable regions of the 16S rRNA gene was carried out using primers adapted for Illumina MiSeq, 27F (AATGATACGGCGACCACCGAGATCTAC-ACTATGGTAATTCCAGGTTYGATYMTGGCTCAG) and 338R individually barcoded (CAAGCAGAAGACGGCATACGAGAT-barcode-AGTCAGTCAGAAGCTGCCTCCCGTAGGAGT) primers. 1 µl of extracted DNA was run in a 25 µl PCR reaction containing 1.25 µl of each primer (10 µM), 5 µl of 5X Q5 Reaction Buffer, 0.25 µl of Q5 High-Fidelity DNA Polymerase (M0491, New England Biolabs), 0.5 µl of 10 mM dNTPs (N0447, New England Biolabs) and 15.75 µl of Nuclease-Free water as per Q5 standard protocol. Four PCR reactions were run for each sample using thermal cycler conditions of 98 °C for 2 min, then 20 cycles of 98 °C for 30 secs, 50 °C for 30 secs, 72 °C for 90 secs before a final extension of 72 °C for 5 min. Sample reactions were pooled and checked by running on an agarose gel before being cleaned by ethanol precipitation then quantified using the Qubit dsDNA HS Assay Kit (Q32854, Invitrogen/Life Technologies). An equimolar mix of all samples was prepared before carrying out a final clean up step using the Wizard SV Gel and PCR Clean-Up System (A9281, Promega). The equimolar mix was then eluted in 50 µl of Nuclease-Free Water, of which 25 µl was submitted for Illumina MiSeq sequencing (paired end 250 bp read length) at the Wellcome Sanger Institute in Cambridgeshire, UK.
Analysis of microbiota sequence data
Illumina MiSeq data was analyzed using mothur software (v 1.39.5)61, similar to as described by Dalby et al.60. Contigs were assembled using the forward and reverse reads, and only those which were between 280 and 470 bases were taken forward in the analysis pipeline. Sequences were aligned against the SILVA reference database and operational taxonomic units (OTUs) generated (97% similarity) using the default OptiClust option in mothur62. Representative sequences were obtained for each OTU and these were then able to be run through the BLAST database for species identification. Unlike Dalby et al. 201760, no chimera removal software was used and instead a cut-off to remove all sequences with 10 reads or less applied. All samples were sub-sampled to 4237 reads to allow comparison across all samples at the same depth of coverage. The median Good’s coverage estimate at this sequence depth was 98.07% (range from 96.55 to 99.74%). Sequence data have been deposited in the European Nucleotide Archive under study accession number PRJEB14084. The final OTU table of the 500 most abundant OTU is shown as Supplementary Table 2, alongside results for each sample at the genus, family and phylum levels. Principal Coordinate Analysis (PCoA) plots, and dendrograms were created from the OTU-level data using the Bray Curtis dissimilarity calculator in mothur62.
Quantitative reverse transcriptase-PCR
RNA was isolated using an Isolate II RNA micro kit (Bioline). cDNA was synthesized by means Superscript II reverse transcriptase (Invitrogen) for colon with oligo (dT) and random primers. A SYBR green-based real-time PCR technique was used to detect the expression of transcripts (SensiFAST master mix, GC-Biotech). Real-time PCR was performed using the Light cycler 480 (Roche) detection system. Data were analyzed using LinregPCR software63 and results were expressed as fold difference relative to the geometric mean expression of the reference genes ubiquitin and cyclophilin for murine colon. The following human primers sets were used:
β-actin F: AATGTGGCCGAGGACTTTGA, R: TGGCTTTTAGGATGGCAAGG, GAPDH F: GAGTCAACGGATTTGGTCGT, R: TTGATTTTGGAGGGATCTCG, mTNF F: TGGAACTGGCAGAAGAGGCACT, R: CCATAGAACTGATGAGAGGGAGGC, mIL-6 F: AGTTGCCTTCTTGGGACTGA, R: TCCACGATTTCCCAGAGAAC, mIL-1b F: GCCCATCCTCTGTGACTCAT, R: AGGCCACAGGTATTTTGTCG.
Metabolome analysis
1H Nuclear magnetic resonance (NMR) spectroscopy
Fecal samples were weighted and mixed with water (UPLC grade) in a ratio of 1 mg : 4 μl (fecal weight : water volume). The samples were mixed by pipetting, vortexing and centrifuged at 18,000g for 10 min at 4 °C. The fecal water supernatant was then separated from the pellet and stored at − 40 °C. Fecal water samples were thawed and centrifuged at 18,000g for 10 min at 4 °C. A total of 100 μl fecal water was mixed with 400 μl of deuterium oxide (D2O) and 100 μl of 0.2 M sodium phosphate buffer (pH = 7.4) containing 0.01% TSP (3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt) and 3 mM NaN3. After vortexing, 580 μl was transferred to an NMR tube with an outer diameter of 5 mm pending 1H NMR spectral acquisition.
1H NMR spectra of fecal water samples were acquired using a Bruker 600 MHz spectrometer (Bruker, Rheinstetten, Germany) at the operating 1H frequency of 600.13 MHz at a temperature of 300 K. A standard NMR pulse sequence (recycle delay-90°-t1-90°-tm-90°-acquisition) was applied to acquire one-dimensional 1H NMR spectral data, where t1 was set to 3 μs and tm (mixing time) was set to 10 ms. The water peak suppression was achieved using selective irradiation during a recycle delay of 2 s and tm. A 90° pulse was adjusted to ~ 10 μs. A total of 128 scans were collected into 64 k data points with a spectral width of 20 ppm. The standard parameters used for these spectral acquisitions have previously been reported64,65.
Metabolic data have been submitted to the MetaboLights database66 under accession number MTBLS2051.
High-performance liquid chromatography (HPLC)
Amino acids
Amino acids were quantified in fecal water prepared by weighing the fecal sample and mixing it with four parts of sodium phosphate buffer [pH = 7.4], vortexing and centrifugation at 20,000g at 4 °C for 30 min. Fecal water (50 µl) was mixed with 24% sulfosalicylic acid and centrifuged at 20,000g at 4 °C to remove proteins. Amino acids were measured using a gradient reversed-phase HPLC system with precolumn derivatization with o-phtalaldehyde (Pierce) and 3-mercaptopropionic acid (Sigma), and fluorescence detection. Separation was done using 2 serial coupled BDS Hypersil C18 columns (150 × 4.8 mm, 3 µm particles, Thermo Scientific, flowrate 0.7 ml/min) and linear gradient of solvent A and B (from 10% B at start to 100% B at the end linearly). Solvent A was 12.5 mM sodium phosphate (pH 7.0) + 0.005% tetrahydrofuran and solvent B was 6 mM sodium phosphate (pH 7.0) + 0.07% tetrahydrofuran + 40% acetonitrile67. For normalization purposes we used norvaline as an internal standard.
Bile acids
Feces was freeze dried overnight. Feces were diluted 1:10 on a weight:volume basis with 50% Tert-Butanol solution, mixed, sonicated, and centrifuged at 3500 g. Supernatant was freeze-dried overnight and re-suspended in 300 µl 25% Methanol. Bile acids were separated and quantified by reverse-phase HPLC, which was an adaptation to the method used by Kunne et al.68. 100 µl sample was applied to a Hypersil C18 HPLC column (internal diameter: 3 µm, column length: 15 cm; Thermo Scientific, Breda, The Netherlands) operated at 20 °C. The starting eluent consisted of 6.8 mM ammonium formiate (pH 3.9), followed by a linear gradient or isocratic elution with acetonitrile at the indicated concentration: 28% (1 min), 38% (13 min), 42% (19 min), 61% (20 min), 63% (25 min), 80% (28 min), 80% (31 min) and 0% (33 min). The flow rate was 0.8 mL per minute. Detection was performed using a Charged aerosol detector (ThermoFisher). Quantification of the different bile salt species was performed by using a calibration curve for all different bile salt species.
Real time cell growth and adherence assay
The effect of metabolites on epithelial cell proliferation and adhesion was performed using a label-free real-time cell analysis platform (xCELLigence; Roche Applied Science, Indianapolis, IN) as previously described69. Each well of a 16 well E-plate received, 100 μl complete Dulbecco's Modified Eagle Medium (DMEM) culture medium supplemented with metabolites of interest, and background impedance was measured over 24 s. Caco-2 cells were cultured until confluent, trypsinized and suspended in DMEM supplemented at 20,000 cells/ml. Resuspended cells (100 μl) were added to the E-plate wells. After 30 min of incubation at room temperature, the plate was placed into the cell culture incubator (37 °C, 5% CO2). Control wells received DMEM medium only. Cell proliferation and adhesion was measured every 30 min for 72 h by determining the cell index using the xCELLigence system software version 1.2.170.
Monocyte immunological response assay
Primary monocytes were isolated from whole blood buffy coats in 2 steps: (1) Ficoll was added under the buffy coat layer and spun down (2000 RCF, acc. 3, decl. 0, 20 min) and the separated layer of peripheral blood mononuclear cells (PBMC) were aspirated an re-suspended, (2) PBMC were incubated (90 min, 37 °C, 5% CO2) after which culture plates were washed and remaining monocytes loosened with EDTA and re-suspended and plated at 500.000 cells/well in 6 well plates (1.5 ml/plate). Monocytes were co-cultured with metabolites of interest for 24 h, after which LPS (100 ng/ml) was added. The medium was subsequently collected after 4 h and Tumor Necrosis Factor-alpha (TNFα) and Interleukin-6 (IL-6) were measured using sandwich ELISA (R&D Systems, Minneapolis, Minn., USA).
Dextran sulfate sodium (DSS) colitis mouse model
C57BL/6 N mice (Charles River Laboratories) were housed and maintained under specific pathogen free conditions in our animal facility at the AMC in Amsterdam. Mice were females between 8 and 12 weeks of age at the time of study. Eleven days prior to inducing intestinal inflammation with DSS, mice were given chow supplemented with milk protein (n = 10) or chow supplemented with amino acids (n = 10) (Mead Johnson Nutrition). Intestinal inflammation was induced using 1.5% (w/v) DSS (TdB Consultancy, Uppsala, Sweden) added to the drinking water for 7 days. Fresh DSS solutions were prepared daily. Body weights were recorded daily. At the end of the study endoscopy was done according to the scoring system described by Becker et al.71. Five features of endoscopic severity were scored: ‘mucosal thickening’, ‘vasculature’, ‘granularity of the mucosal surface’, ‘fibrin deposits’ and ‘stool appearance’. The total endoscopic disease severity score was calculated from the disease components, excluding the stool component score (as it was only clearly determinable in 158 of 201 of the videos), with a total score between 0 and 12. Subsequent to endoscopy mice were killed and organs collected. Wet weights of colons were recorded together with total colon length. Colon weight per cm was used as a disease parameter. Stools were scored as follows: (0) normal feces, (1) soft pellets, (2) thin feces, (3) watery diarrhea, (4) bloody diarrhea. Colons were divided in two parts longitudinally; one part was used for histology the other part for qPCR analysis.
Histology
Histology was performed as described previously72. Longitudinally divided colons were rolled, fixed in 4% formalin and embedded in paraffin for routine histology. An experienced pathologist evaluated formalin-fixed hematoxylin tissue sections microscopically, in a blinded fashion. Colons were evaluated, and graded from 0 to 4 as an indication of incidence and severity of inflammatory lesions based on the extent of the area involved, the number of follicle aggregates, edema, fibrosis, hyperplasia, erosion/ulceration, crypt loss and infiltration of granulocytes and mononuclear cells. The pathology score was calculated as the total score of the above.
Statistical analysis
Microbiota and metabolomic data were compared between (1) CD patients at T0 and healthy controls, (2) between different time points during EEN in CD patients (T0 vs. T1 vs. T2 vs. T3), (3) between biochemical responders and non-responders, and (4) between responder or non-responders at follow-up (T3) and healthy controls.
The statistical analysis for the microbiota data was carried out as described by Dalby et al. 201760, using LEfSe73 and Metastats74 (p values corrected with Benjamini–Hochberg to account for multiple comparisons)75 to assess changes in proportional abundance across OTUs, Genus, Family and Phylum Levels. Differences in overall community compositions were analyzed at the OTU level using the Analysis of Molecular Variance (AMOVA) function within the mothur software package76, based on Bray Curtis dissimilarity. The Shannon and inverse Simpson diversity indices, which are commonly used to characterizes species diversity in a community based on proportional abundance and evenness of the species present, were used to calculate the bacterial diversity within each sample using the mothur software package76. These were compared between cohorts using Mann–Whitney tests for CD versus healthy control comparisons, and using the Wilcoxon test for the longitudinal comparisons across the term of the EEN intervention. The subsets of samples that were included in each of the comparative analyses included in the Results section are shown in Supplementary Table 3.
Multivariate statistical analysis of 1H NMR spectral data were pre-processed (phasing, baseline correction and calibration) and imported into MATLAB (R2014a) using an in-house MATLAB script from Imperial College London. The shift ranges from − 0.02 to 0.02 ppm, 3.70 to 3.72 ppm and 4.78 to 4.84 ppm were removed to exclude TSP, polyethylene glycol (PEG) and water peaks, respectively. Due to the high intensity of PEG compared to other peaks in the spectrum, PEG was deemed to be a remnant of the bowel cleansing procedure present in some patients and therefore excluded from further analysis. NMR spectra were then aligned using recursive segment-wise peak alignment77, normalized using the probabilistic quotient normalization78 and log-transformed prior to Principal Component Analysis (PCA) and Orthogonal Partial Least Squares-Discriminant Analysis (OPLS-DA).
Unit variance scaling method and sevenfold cross validation were used in OPLS-DA models. The model parameters were presented as R2 (percentage of variation explained by the model) and Q2 (predictivity of the models) and p value generated from the permutation tests. Metabolites that are associated with CD, EEN treatment or EEN response were extracted from OPLS-DA models using statistical total correlation spectroscopy (STOCSY), the Human Metabolite Database and other literature documenting faecal water metabolites obtained from NMR79,80,81. Correlation coefficient values (r) were provided based on a selected signal from each metabolite. p- and q-values represent the significance of the metabolite changes and Benjamini–Hochberg correction-adjusted p-values, respectively.
Bile acids were categorized into primary and secondary bile acids. The hydrophobicity of the bile acid pool per fecal sample was calculated using the bile acid hydrophobicity index82. Differences in amino acids (individual and total concentration) and bile acids (primary and secondary concentration, and bile acid hydrophobicity index) were analyzed using t test (2 groups) and one-way ANOVA with Turkey post-hoc test (> 2 groups) for normally distributed or Mann–Whitney U (2 groups) and Kruskal–Wallis test with Dunn’s post-hoc test (> 2 groups) for non-normally distributed variables.
Cell indexes deriving from XCELLigence and cytokine concentrations deriving from the monocyte immunological response assay were analyzed identical to amino and bile acid data.
References
Ruemmele, F. M. et al. Consensus guidelines of ECCO/ESPGHAN on the medical management of pediatric Crohn’s disease. J. Crohn’s Colitis 8, 1179–1207 (2014).
Heuschkel, R. B., Menache, C. C., Megerian, J. T. & Baird, A. E. Enteral nutrition and corticosteroids in the treatment of acute Crohn’s disease in children. J. Pediatr. Gastroenterol. Nutr. 31, 8–15 (2000).
Dziechciarz, P., Horvath, A., Shamir, R. & Szajewska, H. Meta-analysis: enteral nutrition in active Crohn’s disease in children. Aliment. Pharmacol. Ther. 26, 795–806 (2007).
Borrelli, O. et al. Polymeric diet alone versus corticosteroids in the treatment of active pediatric Crohn’s disease: a randomized controlled open-label trial. Clin. Gastroenterol. Hepatol. 4, 744–753 (2006).
Sartor, R. B. Microbial influences in inflammatory bowel diseases. Gastroenterology 134, 577–594 (2008).
Kolho, K.-L., Pessia, A., Jaakkola, T., de Vos, W. M. & Velagapudi, V. Fecal and serum metabolomics in pediatric inflammatory bowel disease. J. Crohns. Colitis https://doi.org/10.1093/ecco-jcc/jjw158 (2016).
Haberman, Y. et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Investig. 124, 3617–3633 (2014).
Hansen, R. et al. Microbiota of De-Novo Pediatric IBD: Increased Faecalibacterium Prausnitzii and Reduced Bacterial Diversity in Crohn’s But Not in Ulcerative Colitis. Am. J. Gastroenterol. 107, 1913–1922 (2012).
Hedin, C. R. et al. Altered intestinal microbiota and blood T cell phenotype are shared by patients with Crohn’s disease and their unaffected siblings. Gut 63, 1578–1586 (2014).
Treem, W. R., Ahsan, N., Shoup, M. & Hyams, J. S. Fecal short-chain fatty acids in children with inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 18, 159–164 (1994).
Bjerrum, J. T. et al. Metabonomics of human fecal extracts characterize ulcerative colitis, Crohn’s disease and healthy individuals. Metabolomics 11, 122–133 (2015).
Duboc, H. et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62, 531–539 (2013).
Schwerd, T. et al. Exclusive enteral nutrition in active pediatric Crohn disease: Effects on intestinal microbiota and immune regulation. J. Allergy Clin. Immunol. 138, 592–596 (2016).
Quince, C. et al. Extensive modulation of the fecal metagenome in children With Crohn’s disease during exclusive enteral nutrition. Am. J. Gastroenterol. 110, 1718–1729 (2015) (quiz 1730).
Gerasimidis, K. et al. Decline in presumptively protective gut bacterial species and metabolites are paradoxically associated with disease improvement in pediatric Crohn’s disease during enteral nutrition. Inflamm. Bowel Dis. 20, 861–871 (2014).
Hall, L. J., Walshaw, J. & Watson, A. J. M. Gut microbiome in new-onset crohn’s disease. Gastroenterology 147, 932–934 (2014).
Levine, A. et al. Crohn’s disease exclusion diet plus partial enteral nutrition induces sustained remission in a randomized controlled trial. Gastroenterology 157, 440-450.e8 (2019).
Svolos, V. et al. Treatment of active Crohn’s disease with an ordinary food-based diet that replicates exclusive enteral nutrition. Gastroenterology 156, 1354-1367.e6 (2019).
de Meij, T. G. J. et al. Variability of core microbiota in newly diagnosed treatment-naïve paediatric inflammatory bowel disease patients. PLoS ONE 13, e0197649 (2018).
Perino, A. & Schoonjans, K. TGR5 and immunometabolism: insights from physiology and pharmacology. Trends Pharmacol. Sci. 36, 847–857 (2015).
Rauscher-Gabernig, E. et al. Dietary exposure assessment of putrescine and cadaverine and derivation of tolerable levels in selected foods consumed in Austria. Eur. Food Res. Technol. 235, 209–220 (2012).
Koeth, R. A. et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585 (2013).
Smith, E. A. & Macfarlane, G. T. Studies on amine production in the human colon: enumeration of amine forming bacteria and physiological effects of carbohydrate and pH. Anaerobe 2, 285–297 (1996).
Koeth, R. A. et al. γ-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 20, 799–812 (2014).
Park, S. H., Park-Min, K.-H., Chen, J., Hu, X. & Ivashkiv, L. B. Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat. Immunol. 12, 607–615 (2011).
Frank, D. N. et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm. Bowel Dis. 17, 179–184 (2011).
Joossens, M. et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60, 631–637 (2011).
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 (2014).
Pascal, V. et al. A microbial signature for Crohn’s disease. Gut 66, 813–822 (2017).
Mishina, D. et al. On the etiology of Crohn disease. Proc. Natl. Acad. Sci. USA 93, 9816–9820 (1996).
Hall, A. B. et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 9, 103 (2017).
Walters, W. A., Xu, Z. & Knight, R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 588, 4223–4233 (2014).
Lewis, J. D. et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s disease. Cell Host Microbe 18, 489–500 (2015).
Wang, Y. et al. Characteristics of faecal microbiota in paediatric Crohn’s disease and their dynamic changes during infliximab therapy. J. Crohn’s Colitis 12, 337–346 (2018).
Marchesi, J. R. et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J. Proteome Res. 6, 546–551 (2007).
Jansson, J. et al. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS ONE 4, e6386 (2009).
Ni, J. et al. A role for bacterial urease in gut dysbiosis and Crohn’s disease. Sci. Transl. Med. 9, eaah6888 (2017).
Vidal-Lletjós, S. et al. Dietary protein and amino acid supplementation in inflammatory bowel disease course: what impact on the colonic mucosa?. Nutrients 9, 310 (2017).
Zhang, H., Hu, C.-A.A., Kovacs-Nolan, J. & Mine, Y. Bioactive dietary peptides and amino acids in inflammatory bowel disease. Amino Acids 47, 2127–2141 (2015).
Islam, J. et al. Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J. Nutr. Biochem. 42, 43–50 (2017).
Hsiung, Y. C., Liu, J. J., Hou, Y. C., Yeh, C. L. & Yeh, S. L. Effects of dietary glutamine on the homeostasis of CD4+ T cells in mice with dextran sulfate sodium-induced acute colitis. PLoS ONE 9, e84410 (2014).
Zhang, H. et al. Serine alleviates dextran sulfate sodium-induced colitis and regulates the gut microbiota in mice. Front. Microbiol. 9, 3062 (2018).
Ren, W. et al. Serum amino acids profile and the beneficial effects of L-arginine or L-glutamine supplementation in dextran sulfate sodium colitis. PLoS ONE 9, e88335 (2014).
Wilson, A. et al. Trimethylamine-N-oxide: a novel biomarker for the identification of inflammatory bowel disease. Dig. Dis. Sci. 60, 3620–3630 (2015).
Naito, Y. et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J. Gastroenterol. Hepatol. 18, 560–569 (2003).
Masli, S. & Turpie, B. Anti-inflammatory effects of tumour necrosis factor (TNF)-α are mediated via TNF-R2 (p75) in tolerogenic transforming growth factor-β-treated antigen-presenting cells. Immunology 127, 62–72 (2009).
Scheller, J., Chalaris, A., Schmidt-Arras, D. & Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta Mol. Cell Res. 1813, 878–888 (2011).
Keitel, V., Donner, M., Winandy, S., Kubitz, R. & Häussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 372, 78–84 (2008).
Tjellström, B. et al. Effect of exclusive enteral nutrition on gut microflora function in children with Crohn’s disease. Scand. J. Gastroenterol. 47, 1454–1459 (2012).
Duboc, H., Taché, Y. & Hofmann, A. F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 46, 302–312 (2014).
Shaw, K. A. et al. Dysbiosis, inflammation, and response to treatment: a longitudinal study of pediatric subjects with newly diagnosed inflammatory bowel disease. Genome Med. 8, 75 (2016).
Douglas, G. M. et al. Multi-omics differentially classify disease state and treatment outcome in pediatric Crohn’s disease. Microbiome 6, 13 (2018).
Jones, C. M. A. et al. Bacterial taxa and functions are predictive of sustained remission following exclusive enteral nutrition in pediatric Crohn’s disease. Inflamm. Bowel Dis. 26, 1026–1037 (2020).
Levine, A. et al. ESPGHAN revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J. Pediatr. Gastroenterol. Nutr. 58, 795–806 (2014).
de Meij, T. G. J. et al. Composition and stability of intestinal microbiota of healthy children within a Dutch population. FASEB J. 30, 1512–1522 (2016).
Sherlock, M. E. et al. Phenotypic evolution of pediatric inflammatory bowel disease using the new Paris classification. Gastroenterology 140, S90–S91 (2011).
D’Haens, G. R. A. M. et al. Fecal calprotectin is a surrogate marker for endoscopic lesions in inflammatory bowel disease. Inflamm. Bowel Dis. 18, 2218–2224 (2012).
Zubin, G. & Peter, L. Predicting endoscopic Crohn’s disease activity before and after induction therapy in children: a comprehensive assessment of PCDAI, CRP, and fecal calprotectin. Inflamm. Bowel Dis. 21, 1386–1391 (2015).
Samson, C. M. et al. Improved outcomes with quality improvement interventions in pediatric inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 55, 679–688 (2012).
Dalby, M. J., Ross, A. W., Walker, A. W. & Morgan, P. J. dietary uncoupling of gut microbiota and energy harvesting from obesity and glucose tolerance in mice. Cell Rep. 21, 1521–1533 (2017).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120 (2013).
Westcott, S. L. & Schloss, P. D. OptiClust, an Improved Method for Assigning Amplicon-Based Sequence Data to Operational Taxonomic Units. mSphere 2, (2017).
Ruijter, J. M. et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37, e45–e45 (2009).
Nicholson, J. K., Foxall, P. J., Spraul, M., Farrant, R. D. & Lindon, J. C. 750 MHz 1H and 1H–13C NMR spectroscopy of human blood plasma. Anal. Chem. 67, 793–811 (1995).
Beckonert, O. et al. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2, 2692–2703 (2007).
Haug, K. et al. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 48, D440–D444 (2020).
van Wenum, M. et al. Selecting cells for bioartificial liver devices and the importance of a 3D culture environment: a functional comparison between the HepaRG and C3A cell lines. Int. J. Biol. Sci. 12, 964–978 (2016).
Kunne, C. et al. Defective bile salt biosynthesis and hydroxylation in mice with reduced cytochrome P450 activity. Hepatology 57, 1509–1517 (2013).
Limame, R. et al. Comparative analysis of dynamic cell viability, migration and invasion assessments by novel real-time technology and classic endpoint assays. PLoS ONE 7, e46536 (2012).
Atienza, J. M. et al. Dynamic and label-free cell-based assays using the real-time cell electronic sensing system. Assay Drug Dev. Technol. 4, 597–607 (2006).
Becker, C. et al. In vivo imaging of colitis and colon cancer development in mice using high resolution chromoendoscopy. Gut 54, 950–954 (2005).
Heinsbroek, S. E. M. et al. Orally delivered β-glucans aggravate dextran sulfate sodium (DSS)-induced intestinal inflammation. Nutr. Res. 35, 1106–1112 (2015).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
White, J. R., Nagarajan, N. & Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5, e1000352 (2009).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Veselkov, K. Computational approaches to improve information recovery from biological NMR spectra. (2009).
Dieterle, F., Ross, A., Schlotterbeck, G. & Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in1H NMR metabonomics. Anal. Chem. 78, 4281–4290 (2006).
Lamichhane, S. et al. Strategy for nuclear-magnetic-resonance-based metabolomics of human feces. Anal. Chem. 87, 5930–5937 (2015).
Wishart, D. S. Characterization of biopharmaceuticals by NMR spectroscopy. TrAC Trends Anal. Chem. 48, 96–111 (2013).
Cloarec, O. et al. Statistical total correlation spectroscopy: an exploratory approach for latent biomarker identification from metabolic 1H NMR data sets. Anal. Chem. https://doi.org/10.1021/AC048630X (2005).
Heuman, D. M. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J. Lipid Res. 30, 719–730 (1989).
Acknowledgements
GD and AWW receive core funding support from the Scottish Government’s Rural and Environmental Science and Analytical Services (RESAS) Division. JW was funded by the Wellcome Trust [Grant No. 098051]. JVL is funded by MRC New Investigator Grant (MR/P002536/1) and ERC Starting Grant (715662). JK is funded by NIHR: II-OL-1116-10027, NIH: R01-CA204403-01A1, Horizon H2020: ITN GROWTH. Imperial Biomedical Research Centre, SAGES research grant. Infrastructure support for this research was provided by the NIHR Imperial biomedical Research Centre (BRC). Microbiota analyses were carried out using the Maxwell computer cluster at the University of Aberdeen. We thank the Illumina MiSeq team at the Wellcome Sanger Institute for their assistance. This work was partially described in the Ph.D. thesis of KD (Retrieved 2020, Pediatric inflammatory bowel disease Monitoring, nutrition and surgery, https://pure.uva.nl/ws/files/23176012/Thesis_complete_.pdf).
Author information
Authors and Affiliations
Contributions
K.D. and J.S. designed the study and wrote the paper. K.D. performed and analyzed the studies with CaCo cells and PBMC. J.S. and W.J.d.J. supervised the study. J.S. coordinated co-author contributions and wrote the final draft. J.V.L., V.A. and J.K. performed and analyzed the NMR metabolome studies, and drafted the relevant sections and edited the manuscript. G.D., J.W. and A.W.W. performed and analyzed the 16S rRNA gene amplicon-based microbial studies, and drafted the relevant sections and edited the manuscript. Tg.d.M., A.K., A.At.V. and M.A.B. were responsible for sample collection and clinical management of the patients. D.R.dW. and K.D. performed and analyzed the bile salt studies. T.B.M.H. and K.D. performed and analyzed the amino acid studies. S.E.M.H. performed and analyzed the murine DSS study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Diederen, K., Li, J.V., Donachie, G.E. et al. Exclusive enteral nutrition mediates gut microbial and metabolic changes that are associated with remission in children with Crohn’s disease. Sci Rep 10, 18879 (2020). https://doi.org/10.1038/s41598-020-75306-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75306-z
This article is cited by
-
Dietary interventions in Crohn’s disease: A simple solution to refractory disease
Indian Journal of Gastroenterology (2024)
-
Dietary management of adults with IBD — the emerging role of dietary therapy
Nature Reviews Gastroenterology & Hepatology (2022)
-
Mechanisms of mucosal healing: treating inflammatory bowel disease without immunosuppression?
Nature Reviews Gastroenterology & Hepatology (2022)
-
Metabolic changes during exclusive enteral nutrition in pediatric Crohn’s disease patients
Metabolomics (2022)
-
Changes of intestinal microbiota and microbiota-based treatments in IBD
Archives of Microbiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
