
Abstract
The most downstream elements of the Hippo pathway, the TEAD transcription factors, are regulated by several cofactors, such as Vg/VGLL1-3. Earlier findings on human VGLL1 and here on human VGLL3 show that these proteins interact with TEAD via a conserved amino acid motif called the TONDU domain. Surprisingly, our studies reveal that the TEAD-binding domain of Drosophila Vg and of human VGLL2 is more complex and contains an additional structural element, an Ω-loop, that contributes to TEAD binding. To explain this unexpected structural difference between proteins from the same family, we propose that, after the genome-wide duplications at the origin of vertebrates, the Ω-loop present in an ancestral VGLL gene has been lost in some VGLL variants. These findings illustrate how structural and functional constraints can guide the evolution of transcriptional cofactors to preserve their ability to compete with other cofactors for binding to transcription factors.
Similar content being viewed by others


Introduction
The Hippo pathway, which controls organ size and tissue regeneration via the regulation of cell division and apoptosis1,2, is an intense field of research in regenerative medicine3,4,5,6. Furthermore, deregulation of Hippo signalling in several cancers suggests that molecules targeting this pathway may have antineoplastic properties7,8,9,10,11 (but see12,13). The Hippo pathway regulates, via a signalling cascade involving the MST1/2 and LATS1/2 protein kinases, the TEAD (TEA/ATTS domain) transcription factors that, upon binding to YAP (Yes associated protein) or VGLL1-3 (Vestigial-like), become transcriptionally active1,14,15,16. The elucidation of the structures of the YAP:TEAD17,18 and of the VGLL1:TEAD19 complexes has shed some light on how these proteins bind to TEAD. The TEAD-binding domain of YAP is formed of a β-strand:α-helix:Ω-loop motif while the TEAD-binding domain of VGLL1 contains only a β-strand:α-helix motif, known as TONDU domain, which binds to TEAD in a fashion similar to the β-strand:α-helix region of YAP (Fig. S1)18. The residues present in the TONDU domain of VGLL1 are well-conserved amongst the members of the Vg/VGLL1-3 family (Vg, vestigial, Drosophila homolog of VGLL1-3)20 and no other regions of these proteins outside this motif have so far been reported to play a direct role in the interaction with TEAD proteins. Therefore, the main structural difference between the TEAD-binding domain of these two cofactor families is the presence of an Ω-loop in the Yki/YAP proteins (Yki, Yorkie, Drosophila homolog of YAP), but not in the Vg/VGLL1-3 proteins. However, we made an observation which suggests that some members of the Vg/VGLL1-3 family may also possess an Ω-loop in their TEAD-binding domain. We identified a region in the amino acid sequence of the Vg protein from Drosophila melanogaster, residues 323–334 (Vg323–334), which has a strong homology with the Ω-loop of YAP and is separated from the TONDU domain (Vg288–311) by a similar number of residues compared with the Ω-loop from the β-strand:α-helix region in YAP (Fig. 1A). This unexpected finding, which suggests that Vg contains an Ω-loop, echoes earlier findings. Simmonds et al. have mapped the Sd-binding domain of Vg (Sd, Scalloped, Drosophila homolog of human TEAD) to Vg279–335, which contains not only the TONDU domain but also the putative Ω-loop, Vg323–33421. However, these authors did not conduct experiments with shorter fragments to determine whether the entire Vg279–335 is required for efficient binding to Sd. In their study of the ladybird beetle Vg protein, Ohde et al. observed that, in addition to the TONDU domain, this protein also contains a small sequence conserved in the Vg protein of other species, including D. melanogaster22. This sequence corresponds to the putative Ω-loop, Vg321–334, but these authors report that the function of this region is unknown. Altogether, these observations prompted us to investigate the interaction between Vg and Sd in more detail and, in the light of our findings, to revisit the interaction between TEAD and VGLL1-3.

Primary sequence alignments. (A) The structures of the YAP:TEAD (PDB 3KYS18) and of the VGLL1:TEAD (PDB 5Z2Q19) complexes were used to manually align the primary sequences of YAP (UniProt P46937), Vg (UniProt Q26366) and Yki (UniProt Q45VV3). The secondary structure adopted by YAP upon binding to TEAD is indicated. β: β-strand; α: α-helix; Ω: Ω-loop. Gaps are indicated by dashes (−). (B) The primary sequences of human VGLL1 (UniProt Q99990), VGLL2 (UniProt Q8N8G2) and VGLL3 (UniProt A8MV65) were manually aligned to the primary sequence of Vg to obtain an optimal alignment of the residues in the β-strand:α-helix and of the conserved serine and tryptophan residues located near the C-terminus. The asterisks (*) indicate strictly conserved residues. (C) The primary sequence of the peptides used in the TR-FRET assay are given at the bottom of the figure. The residues in bold and normal fonts in the sequence of Flyman are from VGLL2 and Vg, respectively.
Results
Biophysical characterization of the Vg:Sd interaction
The alignment of the primary sequence of TEAD and Sd and the analysis of the structure of the YAP/VGLL1:TEAD complexes17,18,19 allowed us to tentatively map the Vg/Yki-binding domain of Sd between residues 223 and 440 (Fig. S2). This protein fragment, hereafter referred to as wtSd, was purified from Escherichia coli as a mixture of acylated and non-acylated proteins (Fig. S3). The acylation of the human TEAD proteins has been described23,24,25 and the physiological role of Sd acylation on tissue overgrowth has been shown in Drosophila23. A circular dichroism (CD) analysis shows that recombinant wtSd is well-folded and its CD spectrum is similar to that of TEAD4217–434 (corresponding TEAD4 fragment) (Fig. S4A). The structure of the YAP:TEAD complex18 and the alignment of the amino acid sequence of YAP and Yki enabled us to delineate the Sd-binding domain of Yki to Yki31–79 (Fig. 1A). The affinities (Kd) of Vg288–311 (β-strand:α-helix, TONDU domain) and Yki31–79 (β-strand:α-helix:Ω-loop) for wtSd, as measured by Surface Plasmon Resonance (SPR), are 291 and 23 nM, respectively (Table 1). These Kd values are similar to those of their counterparts VGLL127–51 (160 nM) and YAP50–99 (11 nM) measured with TEAD426,27.
We next studied Vg288–337, which contains the TONDU domain and the putative Ω-loop (Vg323–334). Binding of Vg288–337 to wtSd was so tight (Kd < 2 nM) that we were unable to accurately determine its affinity using a standard SPR procedure. Therefore, we used the kinetic titration method, which allows Kd determination for high affinity ligands28. To validate this methodology for our proteins, we purified Yki30–146, which according to results obtained with the equivalent YAP50–171 fragment should have a high affinity for wtSd26. Since the Kd values of Yki30–146 measured by both SPR methods are similar (Table 1), kinetic titration was applied to Vg288–337 and a KdKT = 1.6 nM was determined (Table 1). Vg288–337 binds 190 times more tightly to wtSd than the TONDU domain alone (Vg288–311), showing that the region Vg312–337 (including the putative Ω-loop) dramatically enhances the affinity for wtSd. To determine whether additional regions outside Vg288–337 contribute to the binding to wtSd, the longer protein fragment Vg263–382 was purified. The affinity of Vg263–382 − KdKT = 0.38 nM (Table 1)—is similar (fourfold difference) to that of Vg288–337, suggesting that no other regions from Vg263–382 contribute significantly to the interaction with wtSd. The residues in the β-strand of the Sd/TEAD-binding domain of YAP/Yki are variable across species and their contribution to the interaction with Sd/TEAD is limited18,26. In contrast, the amino acids from the β-strand in the Sd/TEAD-binding domain of Vg/VGLL1-3 are well conserved20 suggesting that they might contribute more to the interaction with these transcription factors. The Kd values for wtSd of Yki41–79 and Vg298–337, which both lack the putative β-strand region (Fig. 1A), are 69 nM and 61 nM, respectively (Table 1). As observed for YAP, the deletion of the putative β-strand of the Sd-binding domain of Yki has little effect on binding to wtSd (only a threefold change in Kd). In contrast, the deletion of the putative β-strand from the Sd-binding domain of Vg leads to a 40-fold reduction in binding, indicating that this secondary structure element plays an important part in the interaction with wtSd.
Structural characterization of the Vg:Sd interaction
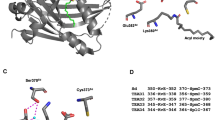
To study the interaction between Vg and Sd at the atomic level, we set up co-crystallization experiments between wtSd and Vg288–337 as well as Vg298–337 and we obtained crystals of the Vg298–337:wtSd complex diffracting at 1.85 Å (PDB 6Y20; Table S1). In agreement with their high amino acid sequence homology and in line with our CD data (Fig. S4A), Sd and TEAD have a very similar three-dimensional structure (Fig. S6). The superimposition of the structure of the Vg:Sd complex on that of the YAP:TEAD and VGLL1:TEAD complexes show that the α-helix of the Sd/TEAD-binding site of these three co-factors bind to a similar location at the surface of Sd/TEAD (Fig. 2A). The axes of the α-helix of Vg and YAP are not aligned, as also previously noticed between VGLL1 and YAP29, but the α-helices of Vg and VGLL1 bind in a similar fashion (Fig. 2B). The three conserved residues in the VxxHF motif from the TONDU domain of VGLL1 and Vg occupy the same position and His305Vg forms the same two hydrogen bonds with Sd (with Ser342Sd and Val395Sd) as His44VGLL1 does with TEAD in the VGLL1:TEAD complex19 (Fig. 2C). The structure of the Vg:Sd complex shows that the Sd-binding domain of Vg indeed contains an Ω-loop, which binds to Sd in a similar fashion than the Ω-loop of YAP binding to TEAD (Fig. 2A). The linker region connecting the α-helix and the Ω-loop of Vg is not completely resolved in our structure probably because of its flexibility (Fig. 2A). The three hydrophobic residues Met324Vg:Phe329Vg:Phe333Vg present in the Ω-loop of Vg occupy the same position at the binding interface as Met86YAP:Leu91YAP:Phe95YAP do (Fig. 2D). They also create a hydrophobic core within the bound Ω-loop, which is stabilized in the YAP:TEAD complex by Phe96YAP that makes a π-cation interaction with Arg87YAP (Fig. 2E). In the Vg:Sd complex, Trp334Vg is at the position of Phe96YAP but it does not engage in a π-cation interaction because Ser325Vg replaces Arg87YAP. This suggests that the presence of a larger aromatic residue at this position might be sufficient to stabilize and shield the hydrophobic core of the bound Ω-loop from solvent. The key salt bridge between Arg89YAP and Asp272TEAD4 found in the YAP:TEAD complex30 is also observed between Arg327Vg and Asp276Sd in the Vg:Sd complex (Fig. 2F). Finally, Ser332Vg is within hydrogen bond distance of Glu267Sd:Tyr435Sd, as is the case for Ser94YAP and Glu263TEAD4:Tyr409TEAD4 in the YAP:TEAD complex (Fig. 2F). Overall, the structure of the Vg298–337:Sd223–440 complex clearly demonstrates the presence of an Ω-loop in the Sd-binding domain of Vg and that it interacts with Sd in a manner similar to that of the Ω-loop of YAP with TEAD.

Structure of the Vg:Sd complex. The structure of the Vg298–337:wtSd complex (PDB 6Y20) is superimposed on that of the YAP:TEAD (PDB 3KYS18) and VGLL1:TEAD (PDB 5Z2Q19) complexes. (A) wtSd, Vg298–337, VGLL126–50 and YAP50–100 are represented in grey, green, magenta and orange, respectively. (B,C) α-helix binding pocket. The α-helices present in the Sd/TEAD-binding domain of Vg, VGLL1 and YAP are represented in green, magenta and orange, respectively. Sd is in grey. The red arrows indicate the approximate axes of the α-helices. The three conserved residues of the VxxHF motif are represented by sticks and the hydrogen bonds made between His305Vg and Sd are indicated by yellow dots. (D–F) Ω-loop binding pocket. The Ω-loop present in the Sd/TEAD-binding domain of Vg and YAP are represented in green and orange, respectively. Sd is in grey. The different interactions are described in the text. The residue labelled Met324Vg is a norleucine. This amino acid was introduced in Vg298–337 to enhance its stability against oxidation. The yellow dots indicate hydrogen bonds. The figures were drawn with PyMOL (Schrödinger Inc., Cambridge, MA).
Effect of mutations in the Ω-loop binding pocket of Sd
The β-strand:α-helix region of YAP (YAP44–74) has a low affinity for TEAD27 while its Ω-loop (YAP85–99), binds more tightly to it26,31. Consequently, mutations in the Ω-loop binding pocket of TEAD are more destabilizing than mutations in the β-strand:α-helix binding pocket18,30. Since the β-strand:α-helix region of Vg (Vg288–311) has a high affinity for wtSd (Table1), we hypothesized that the Vg:Sd complex might be more permissive to mutations in the Ω-loop binding pocket of Sd than the Yki:Sd complex, which should behave like the YAP:TEAD complex. To test this hypothesis, we engineered mutations disrupting key interactions at the Ω-loop binding interface. The Asp276AlaSd mutation prevents the formation of a salt bridge with Arg327Vg in the Vg:Sd complex (Fig. 2F) and probably with Arg69Yki in the Yki:Sd complex. The same mutation in TEAD4, Asp272AlaTEAD4, has a strong destabilizing effect on the YAP:TEAD complex32. The Tyr435HisSd deletes the hydrogen bond with Ser332Vg in the Vg:Sd complex (Fig. 2F) and probably with Ser74Yki in the Yki:Sd complex. This mutation in TEAD1, Tyr421HisTEAD1, which is at the root of Sveinsson’s chorioretinal atrophy33, has a major negative impact on the YAP:TEAD complex34. Asp246AlaSd and Tyr435HisSd were purified, and their CD spectrum shows that the mutations do not affect the structure of Sd (Fig. S4B) as previously observed with the corresponding TEAD4 mutations30,34. The affinity of Vg263–382 and of Yki30–146 for Asp276AlaSd is reduced by 1.58 and 3.6 kcal/mol, respectively (Table 2) and the Tyr435HisSd mutation lowers the binding of Vg263–382 and of Yki30–146 by 2.44 and 4.11 kcal/mol, respectively (Table 2). Therefore, the Vg:Sd complex is less destabilized by these two mutations than the Yki:Sd complex, suggesting that the higher affinity of the β-strand:α-helix region of Vg for wtSd mitigates the impact of disruptive mutations at the Ω-loop binding pocket.
Presence of an Ω-loop in other proteins of the Vg/VGLL1-3 subfamily
The presence of an Ω-loop in the Sd-binding domain of Vg from D. melanogaster could be specific for this species or genus. In agreement with the initial observation made by Ohde et al.22, our analysis shows that the Vg proteins of other species belonging to the same or different insect orders contain a sequence that has strong homology with the residues located in the Ω-loop of Vg from D. melanogaster (Fig. S7). Therefore, Vg may not be the only member of the Vg/VGLL1-3 family to possess an Ω-loop. This prompted us to turn our attention to the VGLL proteins of higher animal species and more particularly to human VGLL1-3. As previously reported20, the β-strand:α-helix region is very well conserved between Vg and VGLL1-3 (Fig. 1B). VGLL1 has no homology with Vg323–334 (Fig. 1B) indicating that it does not possess an Ω-loop in agreement with the published structural data19. VGLL3140–151 shows some level of homology with Vg323–334, but several residues (e.g. Met324Vg or Arg327Vg) required for the interaction between Vg and Sd are missing in VGLL3 (Fig. 1B) suggesting that VGLL3140–151 should only bind very weakly (if at all) to TEAD. In contrast, VGLL2135–146 shows significant sequence homology with Vg323–334 (Fig. 1B) indicating that this region of VGLL2 may form an Ω-loop upon binding to TEAD. To measure the affinity for TEAD4 of VGLL2135–149, VGLL3140–154, YAP85–99 (Ω-loop) and Vg323–337 (Fig. 1C), we used a TR-FRET assay26,27 because VGLL2135–149 and VGLL3140–154 at high concentration bind in a non-specific manner to SPR sensor chips. In the TR-FRET assay, we found that VGLL3140–154 has a very weak potency (IC50 > 500 μM; Table 3) while the IC50 of VGLL2135–149 (133 μM; Table 3) is similar to that of YAP85–99 (98 μM; Table 3) and Vg323–337 (157 μM; Table 3). This finding suggests that VGLL2, in contrast to VGLL1 and VGLL3, possesses an Ω-loop. Since VGLL2-derived peptides are rather unstable in solution (presence of three cysteines in their sequence), making them difficult to handle in crystallization experiments, we built up a molecular model of the VGLL2:TEAD complex by homology to existing crystal structures of the YAP:TEAD4 (Ω-loop region; PDB 6GE3) and VGLL1:TEAD4 (β-strand:α-helix region; PDB 5Z2Q) complexes. The fact that no structural inconsistency was observed by making the appropriate mutations and that the model was stable in a molecular dynamics simulation is consistent with the notion that VGLL2 forms an Ω-loop upon binding to TEAD and that it binds to TEAD4 in a manner similar to the way Vg binds to Sd as illustrated by a superposition (Fig. 3). According to the model, the main structural difference between bound Vg and VGLL2 resides in the linker region connecting the β-strand:α-helix and the Ω-loop. As the linker is much longer in VGLL2 (27 residues instead of 12), it adopts a more globular conformation in the VGLL2:TEAD4 complex. On the basis of these observations, we next measured the affinity of VGLL285–108 (β-strand:α-helix) and VGLL285–149 (β-strand:α-helix:Ω-loop) (Fig. 1C) to TEAD4. VGLL285–108 has an affinity (590 nM, Table 3) similar to that of its Vg counterpart, Vg288–311 (291 nM, Table 1). The Kd of VGLL285–149 is in the low nanomolar range (34 nM, Table 3), showing that the presence of the Ω-loop substantially increases complex formation between VGLL2 and TEAD4. However, VGLL285–149 binds less tightly to TEAD4 (Table 3) than Vg288–337 to Sd or TEAD4 (Tables 1, 3). Since the isolated β-strand:α-helix and Ω-loop regions of VGLL2 and Vg have a similar affinity for TEAD or Sd, we hypothesized that the longer linker region connecting these elements in VGLL2 (Fig. 1B, 3) has a negative impact on the interaction with TEAD4. To test this hypothesis, we designed a hybrid peptide (hereafter referred to as Flyman) formed of the β-strand:α-helix and Ω-loop of VGLL2 connected by the linker from Vg (Fig. 1C). Flyman is more potent than VGLL285–149 and its potency is similar to that of Vg288–337 (Table 3) suggesting that the linker found in VGLL2 has a negative effect on the binding of VGLL285–149 to TEAD4.

Molecular model of the VGLL2:TEAD4 complex. A molecular model of the VGLL285–149:TEAD4217–434 complex was superimposed on that of the crystal structure of the Vg298–337:wtSd complex (PDB 6Y20). VGLL285–149, Vg298–337 and TEAD4217–434 are represented in magenta, green and grey, respectively. The secondary structure elements of VGLL285–149 are indicated. The figure was drawn with PyMOL (Schrödinger Inc., Cambridge, MA).
To determine whether the Ω-loop region of VGLL2, VGLL2135–146 also contributes to the interaction with TEAD in cells in the context of the full-length proteins, N-terminally V5-tagged wtTEAD4 and (FLAG)3-wtVGLL2 were co-transfected into HEK293FT cells harboring a genomic deletion of YAP and TAZ (transcriptional co-activator with PDZ-binding motif, a paralog of YAP) (see below). Following V5-mediated immunoprecipitation, wtTEAD4 and bound wtVGLL2 were monitored using Western blot with V5 and FLAG-antibodies, respectively. wtVGLL2 was efficiently co-immunoprecipitated with wtTEAD4, while it was not detected in similar experiments carried out in the absence of V5-tagged TEAD4 (empty vector control) (Fig. 4A). To test whether VGLL2 interacts with TEAD4 via its Ω-loop region, we used the delΩVGLL2 mutant protein, in which the residues 135–146 have been deleted. The deletion significantly decreases the amount of VGLL2 protein co-immunoprecipitated with wtTEAD4 indicating that the region 135–146 is required for efficient binding to TEAD4 (Fig. 4A). To determine whether VGLL2 interacts with TEAD4 via its Ω-loop binding pocket, we used the Asp272AlaTEAD4 mutant. According to our molecular model, this mutation should prevent the formation of a salt bridge with Arg139VGLL2 and it should have the same disruptive effect on the VGLL2:TEAD4 complex as the Asp246AlaSd mutation on the Vg:Sd complex (Table 2). The amount of wtVGLL2 co-immunoprecipitated with Asp272AlaTEAD4 is lower than that detected in the presence of wtTEAD4 (Fig. 4A). Altogether, these findings show that the Ω-loop region, VGLL2135–146, is required for an efficient interaction between full-length VGLL2 and TEAD4 in cells. Upon quantification of the immunoprecipitated fraction (IP) over input ratio across three separate experiments which exhibited a similar pattern, the Asp272AlaTEAD4 mutation or the Ω-loop deletion of VGLL2 led to a 2 to 2.5-fold reduction in the VGLL2:TEAD4 interaction (Fig. 4B). The combination of these two alterations (Fig. 4A) did not further decrease the extent of co-immunoprecipitation (~ 2-fold reduction, Fig. 4B). This supports the notion that the two mutations affect the same binding interface; if they disrupted interactions at distinct interfaces, then an additive or a synergistic effect would be expected.

Interaction between VGLL2 and TEAD4 in cells. (A) V5-tagged wtTEAD4 or Asp272AlaTEAD4 constructs were co-transfected with (FLAG)3-tagged wtVGLL2 or delΩVGLL2 in the indicated combinations into HEK293FT double YAP/TAZ knockout cells. TEAD4 was immunoprecipitated with a V5-specific antibody and co-immunoprecipitated VGLL2 was detected by anti-FLAG Western blot (left panel). Empty: Empty vector control. IP: Immunoprecipitated fraction. Three different exposures (at 6, 10 and 45 s) of the anti-FLAG Western blot are presented. (B) Image J-based (https://imagej.nih.gov/ij/) quantification was completed on the Western blot signals of indicated lysates and co-IP combinations, and IP/lysate ratios are shown relative to the wtTEAD4/wtVgll2 interaction set to 100% (right panel). The data presented correspond to the average and SEM derived from three independent experiments (2 exposures each).
Functional role of the Ω-loop from Vg in Drosophila
To investigate the functional contribution of the Ω-loop in an in vivo context, we generated several transgenic drosophila fly lines that conditionally express different HA-tagged forms of Vg. We obtained UAS lines containing cDNAs encoding either full-length Vg, or mutant versions bearing a deletion of the Ω-loop (delΩ), a deletion of the β-strand:α-helix (delβ:α) or the deletion of the whole Sd-binding domain of Vg (delβ:α:Ω). In the larval wing primordium, the wing imaginal disc, Vg-Sd interaction plays a major role in the specification of the wing cell fate; the deletion of Vg leads to a complete loss of the wing pouch and subsequently the adult wing35. Strikingly, ectopic expression of Vg outside its wild-type expression pattern leads to ectopic wing outgrowths in the wing imaginal disc, whereas ectopic expression in other imaginal discs leads to their transformation into wing fate in a Sd dependent manner21,36. We used such in vivo ectopic expression assays to probe the function of the different forms of Vg mentioned above.
Upon 24 h ectopic expression using an en-gal4 driver line expressed in the posterior compartment of the wing imaginal disc, we detected comparable level of expression of the four HA::Vg proteins (Fig. 5A). Expression of HA::Vg or HA::VgdelΩ resulted in tissue outgrowth and repression of the hinge specific protein Homothorax (Hth) in the posterior compartment (Fig. 5B), suggesting hinge to wing transformation via these two functional Vg proteins. Expression of HA::Vgdelα:β and HA:Vgdelα:β:Ω did not lead to morphological defects and no change in the distribution of Hth were observed. Consistently, the presumptive wing region, delimited by a Wingless (Wg) ring pattern, was expanded upon expression of HA::Vg or HA::VgdelΩ, but not when HA::Vgdelα:β and HA:Vgdelα:β:Ω where expressed (Fig. 5C). These results show a strong dependency on the β-strand and α-helix for full Vg function, but fail to reveal differences between wild-type Vg and VgdelΩ.

Functional characterization of Vg mutant proteins. (A) Immunostaining of N-terminal tagged HA::Vg constructs upon 24 h expression with an en-Gal4 driver, active in posterior compartment cells. Dashed line delimits the wing disc. (B) Hth immunostaining of same discs as in (A) HA::Vg and HA::VgdelΩ expressing discs show a disruption of the Hth localization pattern accompanied with tissue outgrowth. Asterisks point the affected area. Scalebars 100 μm. (C) Wg localization pattern in disc of same genotype as in (A,B). Asterisks show expansion of presumptive wing tissue in HA::Vg and HA::VgdelΩ discs. (D) Phenotypical classes scored upon expression of the Vg constructs with the ey-Gal4 driver. Arrowhead points the small outgrowth and arrow points to the large outgrowth. (E) Quantification of the observed phenotypes. *p value < 0.05.
Therefore, we turned our attention to a different ectopic expression assay and expressed the four Vg constructs under the eye-specific driver line ey-Gal4. As expected based on previous studies37, expression of HA::Vg resulted in different eye phenotypes (Fig. 5D), including wing-like tissue outgrowths in the eye, with a high phenotypic penetrance (Fig. 5E). Expression of HA::VgdelΩ was also able to generate such outgrowths, but the size and penetrance of these was lower than upon HA::Vg expression. Expression of HA::Vgdelα:β and HA::Vgdelα:β:ω failed to change the fate of eye cells. Together, these results show again a strong dependence on the TONDU domain (β-strand:α-helix region), and also reveal a moderate but significant role of the Ω-loop in the function of Vg.
Discussion
VGLL1 and YAP bind via a similar β-strand:α-helix motif to an overlapping region at the surface of the TEAD transcription factors17,18,19. This motif, which is highly conserved amongst the Vg/VGLL1-3 proteins (TONDU domain)20, is the only structural element known to be involved in the interaction between these co-factors and the TEAD transcription factors. However, YAP (and its paralog TAZ38) requires in addition to the β-strand:α-helix motif an Ω-loop for efficient binding to TEAD18,26. Therefore, the main difference between the TEAD-binding domain of the Vg/VGLL1-3 and YAP-like proteins is the presence of an Ω-loop in the latter11,19,39. The data presented in this article challenge this view, revealing that the Sd/TEAD-binding domain of some Vg/VGLL1-3 proteins also contains an Ω-loop. We demonstrate that Vg from D. melanogaster has an Ω-loop that is required for tight binding to Sd and our in vivo assays show that in some ectopic assays (such as the transformation of the eye to wing-like structures), full function of the Vg protein requires the Ω-loop. Furthermore, we show that human VGLL2, in contrast to human VGLL1 and VGLL3, also possesses an Ω-loop that contributes to its interaction with TEAD. Altogether, these findings indicate that the Sd/TEAD-binding domain of the Vg/VGLL1-3 proteins exists in two different forms: a β-strand:α-helix motif or a β-strand:α-helix:Ω-loop motif. This suggests that to be fully functional, all the Vg/VGLL1-3 proteins must possess a TONDU domain but that the presence of an Ω-loop is not required for some of them (VGLL1, 3) while for others (Vg, VGLL2) it might be needed to reach complete activity. In the following, we shall discuss a hypothesis to explain the evolution of this protein family.
The whole genome duplications that occurred at the origin of the vertebrate lineage40,41 probably led to the presence of duplicates of an ancestor Vg gene in the genome of early vertebrates. As VGLL1-3 from vertebrates derive from Drosophila Vg42, it is likely that the duplicated VGLL proteins present in early vertebrates contained an Ω-loop. The presence of multiple forms of the same gene in genomes enables the accumulation of mutations that can lead to the modification of the ancestral gene43,44,45. The nanomolar affinity of the isolated TONDU domain of Vg/VGLL2 and the triple-digit micromolar affinity of their isolated Ω-loop indicate that the TONDU domain is the “hot spot” for the interaction of these proteins with Sd/TEAD. Since isolated TONDU domains and the TEAD-binding domain of Yki/YAP (or TAZ) have a similar affinity for TEAD (here and34), the VGLL proteins need to contain only a TONDU domain to bind to TEAD and to compete with YAP (or TAZ) for binding to TEAD, as is observed in cells with VGLL1 and VGLL319,46. Therefore, mutations in the Ω-loop region of the VGLL proteins from early vertebrates could take place and be tolerated during evolution because the mutated proteins would still be able to bind with sufficient affinity to TEAD via their TONDU domain. We propose that the accumulation of mutations during this evolutionary process led to the loss of the Ω-loop in VGLL1 and VGLL3. VGLL2 followed a different trajectory since it still contains a functional Ω-loop. Nonetheless, the linker connecting this Ω-loop to the TONDU domain has a negative contribution to the interaction with TEAD. It is difficult to determine whether the ancestral VGLL variant leading to VGLL2 had a similar linker or whether mutations have progressively transformed it from a shorter Vg-like linker to a longer one. In the second case, the transformation of the linker could be the first step in leading to the loss of the Ω-loop and VGLL2 could be an evolutionary intermediate between an ancestral VGLL protein and VGLL1/VGLL3.
The duplication of genes and their subsequent transformation by mutations is a well-accepted concept in the literature, and the Vg/VGLL1-3 and Yki/YAP families provide a beautiful example of how structural and functional requirements have contributed to the evolution of two co-factor families that regulate the same transcription factors through interaction with an overlapping binding site. The loss of the β-strand:α-helix motif dramatically decreases the affinity of Vg/VGLL1-3 (“hot spot” loss) while in Yki/YAP it prevents the ability to compete with Vg/VGLL1-3 and reduces affinity. Therefore, this motif must be preserved in both families. The loss of the Ω-loop generates Vg/VGLL1-3 variants that are still able to bind to Sd/TEAD and to compete with Yki/YAP, while in the latter it abolishes binding (“hot spot” loss). Therefore, the Vg/VGLL1-3 proteins but not the Yki/YAP proteins can lose their Ω-loop. These structural and functional constraints may explain why a β-strand:α-helix or a β-strand:α-helix:Ω-loop motif is present in the Sd/TEAD-binding domain of the Vg/VGLL1-3 proteins while, to our knowledge, only a β-strand:α-helix:Ω-loop motif is found in the Yki/YAP proteins.
Material and methods
Peptides
The synthetic peptides (both N-acetylated and C-amidated) were purchased from Biosynthan (Germany). The purity (> 90%) and the chemical integrity of the peptides were determined by LC–MS from 10 mM stock solutions in 90:10 (v/v) DMSO:water. VGLL285–149, which contains three cysteines (Fig. 1B), was dissolved in 50:50 (v/v) acetontrile:(water + 1 mM TCEP) to avoid oxidation mechanisms induced by DMSO. VGLL285–108 was also prepared in the same solvent. The experiments conducted with VGLL285–149 were done with freshly made solutions (less than 72 h old). The peptide solutions were stored at − 20 °C and for each experiment the solutions were centrifuged to remove potential aggregates; the supernatant was dosed by HPLC and the integrity of the peptide was evaluated by LC–MS. To determine whether acetonitrile affects the output of the biochemical assays, Vg288–337 was also dissolved in 50:50 (v/v) acetonitrile:(water + 1 mM TCEP). The Kd value of this peptide preparation—4.1 ± 0.1 nM (SPR)—is similar to that obtained with a DMSO solution of peptide (3.1 nM; Table 3) showing that the acetonitrile (2% final concentration) does not interfere with our assays.
Cloning, expression and purification of the proteins for the biochemical and biophysical assays
The amino acid sequences of Scalloped (Sd, UniProt P30052, amino acids 223–440), Yorkie (Yki, UniProt Q45VV3, amino acids 30–146), and Vestigial (Vg, UniProt Q26366, amino acids 263–382) from Drosophila melanogaster were back-translated into an Escherichia coli codon-optimized DNA sequence by the GeneArt online ordering tool and synthesized by GeneArt (Thermo Fisher, Switzerland). The DNA fragment encoding Sd223–440 was PCR amplified and inserted into a pET28-derived vector providing an N-terminal His-HRV3C-Avi-tag by HiFi DNA Assembly Cloning (New England Biolabs, Ipswich, MA) according to the instructions of the manufacturer. The mutations to generate Asp276AlaSd and Tyr435HisSd were introduced by the QuikChange II Lightning Site-Directed Mutagenesis kit (Agilent, Santa Clara, CA) according to the manufacturer’s instructions. The DNA fragments encoding Yki30–146 and Vg263–382 were PCR amplified and cloned as described above into a pET derived vector providing an N-terminal His-tag followed by an HRV3C protease site. In a second step, the coding sequences for strep-tagII and rubredoxin were inserted into both constructs between the His-tag and the HRV3C site by HiFi DNA Assembly Cloning. All expression constructs were confirmed by Sanger sequencing. Yki30–146 and Vg263–382 were expressed in E. coli BL21(DE3) cells (Novagen, Madison, WI). For the expression of different Sd proteins, the E. coli BL21(DE3) cells contained in addition a plasmid encoding for biotin ligase BirA (acetyl-CoA carboxylase biotin holoenzyme synthetase). Transformed cells were diluted in LB medium containing 25 µg/ml kanamycin (Invitrogen, Carlsbad, CA) and in addition 34 µg/ml chloramphenicol (Sigma-Aldrich, St Louis, MI) for Sd expression. Pre-cultures were incubated overnight at 37 °C with constant agitation and diluted into fresh medium containing antibiotics and the cultures were grown at 37 °C until OD600 = 0.8. Protein expression was induced by adding 1 mM isopropyl β-D-1-thiogalactopyranoside (AppliChem GmbH, Germany) (and 135 µg/ml biotin (Sigma-Aldrich, St Louis, MI) for Sd expression). The cells were grown overnight at 18 °C, harvested by centrifugation, frozen in dry ice and stored at − 80 °C. All purification steps were carried out at 4–8 °C unless stated otherwise. Frozen cell pellets were thawed on ice and re-suspended in Buffer A (50 mM TRIS·HCl pH 8.0, 1 M NaCl, 2 mM MgCl2, 1 mM Tris(2-carboxyethyl)phosphine (TCEP), 20 mM imidazole, 0.1% (v:v) Tween 20, 0.5 mM biotin (Sigma-Aldrich, St Louis, MI), 5% (v:v) glycerol), cOmplete EDTA free protease inhibitor (1 tablet/50 ml buffer; Roche, Switzerland), and TurboNuclease (20 µl/50 ml buffer; Sigma-Aldrich, St Louis, MI). The cells were disrupted by 3 passes through an EmulsiFlex C3 high pressure homogenizer (Avestin Inc., Canada) at 800–1000 bar and centrifuged at 43,000 × g for 30 min. The cleared supernatant was loaded onto two HisTrap HP 1 mL columns (GE Healthcare, Chicago, IL) mounted in series and equilibrated with Buffer A. The columns were washed with Buffer A followed by Buffer B (50 mM TRIS·HCl pH 8.5, 100 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 20 mM imidazole, 5% (v:v) glycerol). Bound proteins were eluted with a linear gradient of Buffer B containing 250 mM imidazole. The fractions containing Scalloped were pooled and the His6-tag was cleaved off by overnight incubation during dialysis in Buffer B with 1% (w:w) recombinant HRV 3C protease (produced in-house). The dialysed protein was loaded onto two His-Trap HP 1 mL columns (GE Healthcare, Chicago, IL) mounted in series and equilibrated with Buffer B. The flow-through fractions containing the cleaved protein were collected, pooled, and concentrated by centrifugation in Amicon Ultra-15 centrifugal units (MilliporeSigma, Burlington, MA). After determination of the protein concentration by RP-HPLC, a threefold molar equivalent of myristoyl coenzyme A (lithium salt; Sigma-Aldrich, St Louis, MI) was added and incubated at room temperature for 30–60 min. The reaction products were loaded onto a HiLoad 16/600 Superdex 75 prep grade column (GE Healthcare, Chicago, IL) equilibrated with 50 mM TRIS·HCl pH 8.0, 100 mM NaCl, 2 mM MgCl2, 1 mM TCEP, 5% (v:v) glycerol. Protein fractions containing Scalloped were concentrated by centrifugation in Amicon Ultra-15 cartridges (MilliporeSigma, Burlington, MA), aliquoted, snap-frozen in dry ice and stored at − 80 °C. Yki30–146 and Vg263–382 were expressed as described for Scalloped except that E. coli NiCo21 (DE3) cells (New England Biolabs, Ipswich, MA) grown in TBmod plus MOPS medium supplemented with 25 µg/ml kanamycin and no biotin were used. Yki30–146 was purified as described for Scalloped but 300 mM NaCl was used during protease cleavage. For purification of Vg263–382, Buffer B contained during protease cleavage 300 mM NaCl and 0.1% (v/v) CHAPS (Sigma-Aldrich, St Louis, MI). The HiLoad 16/600 Superdex 75 prep grade column (GE Healthcare, Chicago, IL) was either equilibrated as for the other proteins or alternatively equilibrated with 50 mM HEPES, 100 mM KCl, 0.25 mM TCEP, 1 mM EDTA, 0.05% Tween 20, pH 7.4. The purity, quantity and identity of all proteins were determined by RP-HPLC and LC–MS (Fig. S7).
Circular Dichroism, surface plasmon resonance and FRET assay
Representative experiments and methodology are presented on: Fig. S4, Circular Dichroism; Fig. S5, Surface Plasmon Resonance; Fig. S8, TR-FRET assay.
Structural biology
The DNA fragment encoding for wtSd, obtained as described above, was PCR amplified and inserted into a pET28-derived vector providing an N-terminal His-HRV3C-tag by HiFi DNA Assembly Cloning (New England Biolabs, Ipswich, MA) according to the instructions of the manufacturer. The construct encoding for wtSd was expressed in E. coli BL21(DE3) cells (Novagen, Madison, WI) without addition of biotin to the medium and purified according the protocol identical to the one described above for the biotinylated Sd proteins. Crystals of the complex between untagged wtSd and Vg298–337 were grown at 293oK using the sitting drop vapour diffusion method. wtSd (8.3 mg/ml) in 50 mM Tris pH 8.0, 250 mM NaCl, 2 mM MgCl2, 1 mM TCEP and 5% glycerol was pre-incubated with 0.5 mM Vg298–337 (molar ratio Vg298–337/wtSd ~ 1.5). For crystallization, the peptide-protein complex was mixed with an equal volume of the reservoir solution (0.3 µL + 0.3 µL). Initial crystals were obtained using 0.1 M Bis–Tris pH 6.5 and 25% PEG3350 as reservoir solution. Micro seeds were prepared from these conditions and used for further crystallization trials. Diffraction quality crystals were obtained using 0.2 M MgCl2,6H2O, 0.1 M Bis–Tris pH 6.5 and 25% PEG3350 as reservoir solution. Prior to shock cooling in liquid nitrogen, the crystals were soaked for a few seconds in reservoir solution containing 30% glycerol. X-ray diffraction data were collected at the Swiss Light Source (SLS, beamline X10SA) using an Eiger pixel detector. Raw diffraction data from two crystals originating from the same crystallization drop were analysed and processed using the autoPROC47/STARANISO (Tickle et al., Global Phasing Ltd., Cambridge, UK) toolbox. The structure was solved by molecular replacement with PHASER48 using the coordinates of previously solved in-house structures of TEAD4 as the search model. The software programs COOT49 and BUSTER (BUSTER version 2.11.7, www.globalphasing.com) were used for iterative rounds of model building and structure refinement.
Molecular modelling
A model of TEAD4217–434 in complex with VGLL285–149 was constructed by homology. The crystal structure of YAP in complex with TEAD4 (PDB 6GE3) was used as the starting point. The regions corresponding to the α-helix and the linker region of YAP were deleted and the Ω-loop of YAP, YAP85–99, was mutated to mimic the Ω-loop of VGLL2, VGLL2135–149. The crystal structure of TEAD4 in complex with VGLL1 (PDB 5Z2Q) was next superimposed on this partial model. TEAD4 from this structure was deleted and the β-strand:α-helix region of VGLL1, VGLL127–51, was mutated to mimic VGLL285–108. Finally, an arbitrary conformation allowing the α-helix and the Ω-loop sequences to be connected in 3D was given to an amino acid stretch corresponding to the linker region VGLL2109–136. Molecular dynamics simulation of 10 ns with an explicit water solvation model was run using the Desmond module (default parameters) in the molecular modelling package Maestro (Schrödinger Inc. Cambridge, MA). The β-strand:α-helix and Ω-loop regions of VGLL2 were quite stable during the simulation, maintaining their secondary structure. At the end of the simulation, the linker region converged towards a loop conformation with no regular secondary structure that appeared stabilized by an aromatic stacking interaction between residues Tyr115VGLL2 and Phe196VGLL2.
Cellular biology
HEK293FT YAP/TAZ double knockout (ko) cells were established as follows: The sgRNA (CRISPR) targeting sequences for YAP (‘#1’: TGG GGG CTG TGA CGT TCA TC) and WWTR1 (‘#2’: TCC AGC ACC GAC TCG GG and ‘#B’: GTC CAG CAC CGA CTC GTC GG) were cloned into pU6gRNA_CMV-SPyCas9-T2Apuro. 293FT YAP ko cells were created by transfecting pU6gRNA_YAP_1_CMV-SPyCas9-T2Apuro in 293FT cells using Lipofectamine 2000 (Thermo Fisher, Switzerland). Following transient puromycin selection, clones were picked and characterized for (homozygous) absence of YAP at genomic and protein expression level. Selected 293FT YAP ko clones were subsequently co-transfected with pU6gRNA_WWTR1_2_CMV-SPyCas9-T2Apuro and pU6gRNA_WWTR1_B_CMV-SPyCas9-T2Apuro. Following the same selection procedure, the picked clones were characterized for (homozygous) absence of TAZ (WWTR1) at genomic and protein expression level, resulting in verified HEK293FT double YAP/TAZ ko cells. VGLL2 cloning was performed as follows: VGLL2 (RefSeq NM_182645.3) was amplified by standard PCR from a pDONR221-VGLL2 (human codon-optimized) clone, obtained from GeneArt (Germany), using primers containing N-terminal (FLAG)3 epitope. The PCR product was cloned by Gateway reaction into pcDNA3.1 Hygro-DEST (Invitrogen, Carlsbad, CA), according to the manufacturer’s protocol. The pcDNA3.1 Hygro-DEST_VGLL2_Ω_loop mutant was generated by use of the QuikChange Lightning Site-Directed Mutagenesis kit (Stratagene, San Diego, CA), with oligo 5′CCC TTG GAG AGA CTG CAG CTT CTA TCA GGC TCC TGT GCC 3′ (removing amino acid positions 134–149) and the sequence of resulting cDNA clones verified. All other cellular biology tools and methods, namely TEAD4 constructs and co-immunoprecipitations, were essentially as described previously30,50. The inter-experimental reproducibility across the co-IP experiments appeared less robust in the wild-type HEK293FT (i.e. with intact YAP and TAZ) setting, perhaps due to a more complex competition of the various TEAD binding partners (i.e., YAP, TAZ and Vgll2). However, the results obtained in these cells with regard to VGLL2:TEAD4 complex formation and the impact of the Asp272AlaTEAD4 and delΩVGLL2 mutations were similar to the ones obtained with the double HEK293FT YAP/TAZ ko cells.
In vivo biology
Molecular cloning and generation of transgenic fly lines. pUAS-HA:Vg-attb constructs were generated by Gibson Assembly from amplified fragments of the Vg cDNA and the pUASt-attb vector. A GGGS linker was added between HA and Vg ORF to reduced possible positional effect. The resulting constructs (pUAS-HA::Vg, pUAS-HA::VgdelΩ (lacking amino acids 323 to 337), pUAS-HA::Vgdelα:β (lacking amino acids 288 to 311) and pUAS-HA::Vgdelα:β:Ω (lacking amino acids 288 to 337)) were injected into eggs laid by vasa-integrase; attB86Fb flies to establish the transgenic lines. Fly stocks and fly husbandry. en-Gal4 and ey-Gal4 (2nd Chromosome) are described in Flybase. For temporal control of Gal4 function, animals carrying the en-Gal4, UAS-HA::Vg and the tubulin-Gal80ts insertions were kept at 17 °C during development and transferred at 29 °C for 24 h before dissection. Immunostaining and imaging. The primary antibodies used in this study were: 1:300 anti-HA (3F10, Roche, Switzerland) and 1:500 anti-Hth (Gift from N. Azpiazu, Autonomous University of Madrid, Spain). Goat anti-Rabbit Alexa680 and goat anti-Rat Alexa555 secondary antibodies (Thermo Fisher Scientific, Waltham, MA) were employed. The immunostaining protocol was performed as follows: third instar stage larvae were dissected in cold PBS (Cat. number 20012019, Gibco Thermo Fisher Scientific, Waltham, MA) and fixed 30 min in 4% PFA-PBS solution. After washing in PBS and permeabilizing in PBT (0.3% Triton-PBS), samples were blocked in 5% NGS-PBT for 1 h. Samples were then incubated in primary antibody diluted in blocking solution over-night. Next morning, samples were washed in PBT and incubated in secondary antibody diluted in blocking solution for 2 h. After extensive washing first with PBT and then with PBS, imaginal discs were separated from the cuticle and mounted in Vectashield with DAPI (H-1200, Vector Laboratories, Burlingame, CA). All steps were performed at room-temperature except primary antibody incubation that was performed at 4 °C. Wing disc imaging was performed with LSM point confocal 880. The changes in the different eye phenotypical categories were quantified by an unpaired t-test of three independent experiments.
Data availability
The refined coordinates of the Vg:Sd complex structure have been deposited in the Protein Data Bank (www.wwpdb.org) under accession code 6Y20.
References
Ma, S., Meng, Z., Chen, R. & Guan, K. L. The Hippo pathway: biology and pathophysiology. Annu. Rev. Biochem. 88, 577–604. https://doi.org/10.1146/annurev-biochem-013118-111829 (2019).
Wang, Y., Yu, A. & Yu, F. X. The Hippo pathway in tissue homeostasis and regeneration. Protein Cell 8, 349–359 (2017).
Moya, I. M. & Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell. Biol. 20, 211–226 (2018).
Wang, J., Liu, S., Heallen, T. & Martin, J. F. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 15, 672–684 (2018).
Fu, V., Plouffe, S. W. & Guan, K. L. The Hippo pathway in organ development, homeostasis, and regeneration. Curr. Opin. Cell Biol. 49, 99–107 (2017).
Hong, A. W., Meng, Z. & Guan, K. L. The Hippo pathway in intestinal regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 13, 324–337 (2016).
Maugeri-Sacca, M. & De Maria, R. The Hippo pathway in normal development and cancer. Pharmacol. Ther. 186, 60–72 (2018).
Park, J. H., Shin, J. E. & Park, H. W. The role of Hippo pathway in cancer stem cell biology. Mol. Cells 41, 83–92 (2018).
Janse van Rensburg, H. J. & Yang, X. The roles of the Hippo pathway in cancer metastasis. Cell Signal28, 1761–1772 (2016).
Zanconato, F., Cordenonsi, M. & Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 29, 783–803 (2016).
Santucci, M. et al. The Hippo pathway and YAP/TAZ-TEAD protein–protein interaction as targets for regenerative medicine and cancer treatment. J. Med. Chem. 58, 4857–4873 (2015).
Moya, I. M. et al. Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science 366, 1029–1034. https://doi.org/10.1126/science.aaw9886 (2019).
Kakiuchi-Kiyota, S., Schutten, M. M., Zhong, Y., Crawford, J. J. & Dey, A. Safety considerations in the development of Hippo pathway inhibitors in cancers. Front. Cell Dev. Biol. 7, 156. https://doi.org/10.3389/fcell.2019.00156 (2019).
Lin, K. C., Park, H. W. & Guan, K. L. Regulation of the Hippo pathway transcription factor TEAD. Trends Biochem. Sci. 42, 862–872 (2017).
Landin-Malt, A., Benhaddou, A., Zider, A. & Flagiello, D. An evolutionary, structural and functional overview of the mammalian TEAD1 and TEAD2 transcription factors. Gene 591, 292–303 (2016).
Liu, A. M., Wong, K. F., Jiang, X., Qiao, Y. & Luk, J. M. Regulators of mammalian Hippo pathway in cancer. Biochim. Biophys. Acta 1826, 357–364 (2012).
Chen, L. et al. Structural basis of YAP recognition by TEAD4 in the Hippo pathway. Genes Dev. 24, 290–300 (2010).
Li, Z. et al. Structural insights into the YAP and TEAD complex. Genes Dev. 24, 235–240 (2010).
Pobbati, A. V., Chan, S. W., Lee, I., Song, H. & Hong, W. Structural and functional similarity between Vgll1-TEAD and YAP-TEAD complexes. Structure 20, 1135–1140 (2012).
Simon, E., Faucheux, C., Zider, A., Thézé, N. & Thiébaud, P. From vestigial to vestigial-like: the Drosophila gene that has taken wing. Dev. Genes. Evol. 226, 297–315 (2016).
Simmonds, A. J. et al. Molecular interactions between Vestigial and Scalloped promote wing formation in Drosophila. Genes Dev 12, 3815–3820. https://doi.org/10.1101/gad.12.24.3815 (1998).
Ohde, T. et al. Vestigial and scalloped in the ladybird beetle: a conserved function in wing development and a novel function in pupal ecdysis. Insect. Mol. Biol. 18, 571–581 (2009).
Chan, P. Y. et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 12, 282–289 (2016).
Noland, C. L. et al. Palmitoylation of TEAD transcription factors is required for their stability and function in Hippo pathway signalling. Structure 24, 179–186 (2016).
Mesrouze, Y. et al. Effect of the acylation of TEAD4 on its interaction with co-activators YAP and TAZ. Prot. Sci. 26, 2399–2409 (2017).
Hau, J. C. et al. The TEAD4-YAP/TAZ protein–protein interaction: expected similarities and unexpected differences. ChemBioChem 14, 1218–1225 (2013).
Mesrouze, Y. et al. The surprising features of the TEAD4-Vgll1 protein–protein interaction. ChemBioChem 15, 537–542 (2014).
Karlsson, R., Katsamba, P. S., Nordin, H., Pol, E. & Myszka, D. G. Analyzing a kinetic titration series using affinity biosensors. Anal. Biochem. 349, 136–147 (2006).
Mesrouze, Y. et al. Different recognition of TEAD transcription factor by the conserved β-strand:loop:α-helix motif of the TEAD-binding site of YAP and VGLL1. ChemistrySelect 1, 2993–2997 (2016).
Mesrouze, Y. et al. Dissection of the interaction between the intrinsically disordered YAP protein and the transcription factor TEAD. eLife6, e25068 (2017).
Zhang, Z. et al. Structure-based design and synthesis of potent cyclic peptides inhibiting the YAP-TEAD protein–protein interaction. ACS Med. Chem. Lett. 5, 993–998 (2014).
Mesrouze, Y. et al. Adaptation of the bound intrinsically disordered protein YAP to mutations at the YAP:TEAD interface. Prot. Sci. 27, 1810–1820 (2018).
Fossdal, R. et al. A novel TEAD1 mutation is the causative allele in Sveinsson’s chorioretinal atrophy (helicoid peripapillary chorioretinal degeneration). Hum. Mol. Genet. 13, 975–981 (2004).
Bokhovchuk, F. et al. Molecular and structural characterization of a TEAD mutation at the origin of Sveinsson’s chorioretinal atrophy. FEBS J 286, 2381–2398. https://doi.org/10.1111/febs.14817 (2019).
Williams, J. A., Bell, J. B. & Carroll, S. B. Control of Drosophila wing and haltere development by the nuclear vestigial gene product. Genes Dev 5, 2481–2495. https://doi.org/10.1101/gad.5.12b.2481 (1991).
Baena-Lopez, L. A. & Garcia-Bellido, A. Genetic requirements of vestigial in the regulation of Drosophila wing development. Development 130, 197–208. https://doi.org/10.1242/dev.00187 (2003).
Takanaka, Y. & Courey, A. J. SUMO enhances vestigial function during wing morphogenesis. Mech. Dev. 122, 1130–1137. https://doi.org/10.1016/j.mod.2005.05.004 (2005).
Kaan, H. Y. K., Sim, A. Y. L., Tan, S. K. J., Verma, C. & Song, H. Targeting YAP/TAZ-TEAD protein–protein interactions using fragment-based and computational modelling approaches. PLoS ONE 12, e0178381 (2017).
Gibault, F. et al. Toward the discovery of a novel class of YAP-TEAD interaction inhibitors by virtual screening approach targeting the YAP-TEAD protein–protein interface. Cancer 10, E140 (2018).
Dehal, P. & Boore, J. L. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 3, 1700–1708 (2005).
Panopoulou, G. et al. New evidence for genome-wide duplications at the origin of vertebrates using an amphioxus gene set and completed animal genomes. Genome Res. 13, 1056–1066 (2003).
Faucheux, C. et al. Vestigial like gene family expression in Xenopus: common and divergent features with other vertebrates. Int. J. Dev. 54, 1375–1382 (2010).
Huminiecki, L. & Wolfe, K. H. Divergence of spatial gene expression profiles following species-specific gene duplications in human and mouse. Genome Res. 14, 1870–1879 (2004).
Lynch, M. & Katju, V. The altered evolutionary trajectories of gene duplicates. Tends Genet. 20, 544–549 (2004).
Seoighe, C., Johnston, C. R. & Shileds, D. C. Significantly different patterns of amino acid replacement after gene duplication as compared to after speciation. Mol. Biol. Evol. 20, 484–490 (2003).
Figeac, N. et al. VGLL3 operates via TEAD1, TEAD3 and TEAD4 to influence myogenesis in skeletal muscle. J. Cell Sci.132 (2019).
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D 67, 293–302 (2011).
McCoy, A. J. et al. PHASER crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D 60, 2126–2132 (2004).
Bokhovchuk, F. et al. Identification of FAM181A and FAM181B as new interactors with the TEAD transcription factors. Protein Sci. 29, 509–520 (2020).
Acknowledgements
The work of MA and GA was supported by the Kantons Basel-Stadt and Basel-Land and by grants from the SNSF to MA. GA was supported by a “Fellowship for Excellence” of the International PhD Program in Molecular Life Sciences of the Biozentrum, University of Basel.
Author information
Authors and Affiliations
Contributions
Y.M., G.A., F.B., T.M., C.D., F.V., M.M., C.Z., P.F. and R.W. performed research and analysed data. T.V., D.E., P.F., C.S., T.S., M.A. and P.C. supervised the experimental work. M.A. and P.C. wrote the manuscript. P.C. designed research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mesrouze, Y., Aguilar, G., Bokhovchuk, F. et al. A new perspective on the interaction between the Vg/VGLL1-3 proteins and the TEAD transcription factors. Sci Rep 10, 17442 (2020). https://doi.org/10.1038/s41598-020-74584-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-74584-x
This article is cited by
-
Regulation of type I interferon signature by VGLL3 in the fibroblast-like synoviocytes of rheumatoid arthritis patients via targeting the Hippo pathway
Arthritis Research & Therapy (2022)
-
The role of lysine palmitoylation/myristoylation in the function of the TEAD transcription factors
Scientific Reports (2022)
-
RNA-sequencing of myxoinflammatory fibroblastic sarcomas reveals a novel SND1::BRAF fusion and 3 different molecular aberrations with the potential to upregulate the TEAD1 gene including SEC23IP::VGLL3 and TEAD1::MRTFB gene fusions
Virchows Archiv (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
