
Abstract
Global temperature changes have emphasized the need to understand how species adapt to thermal stress across their ranges. Genetic mechanisms may contribute to variation in thermal tolerance, providing evidence for how organisms adapt to local environments. We determine physiological thermal limits and characterize genome-wide transcriptional changes at these limits in bumble bees using laboratory-reared Bombus vosnesenskii workers. We analyze bees reared from latitudinal (35.7–45.7°N) and altitudinal (7–2154 m) extremes of the species’ range to correlate thermal tolerance and gene expression among populations from different climates. We find that critical thermal minima (CTMIN) exhibit strong associations with local minimums at the location of queen origin, while critical thermal maximum (CTMAX) was invariant among populations. Concordant patterns are apparent in gene expression data, with regional differentiation following cold exposure, and expression shifts invariant among populations under high temperatures. Furthermore, we identify several modules of co-expressed genes that tightly correlate with critical thermal limits and temperature at the region of origin. Our results reveal that local adaptation in thermal limits and gene expression may facilitate cold tolerance across a species range, whereas high temperature responses are likely constrained, both of which may have implications for climate change responses of bumble bees.
Similar content being viewed by others

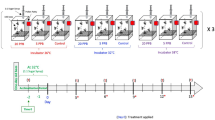

Introduction
Understanding organismal responses to abiotic extremes has become a pressing concern given ongoing and projected changes in environmental conditions1,2. Species with large geographic distributions can encounter substantial environmental heterogeneity3,4 and are excellent models for illuminating the evolution of physiological and genetic variation related to temperature. The degree to which individuals are adapted to regional conditions or tolerate a range of abiotic variation via flexibility or plasticity is of particular interest5, and how selection has shaped range-wide responses to abiotic stress through molecular mechanisms such as gene expression regulation has received increasing attention6,7,8,9,10. Linking physiology and transcriptomics can thus be a valuable approach for teasing apart how species with large ranges adapt to local variation in their environment5.
Gradients in ambient temperature drive numerous biogeographic phenomena5,11, including contemporary range dynamics under global climate change11. Thermal tolerance, or the critical thermal limits (CTLs) of organismal performance at high (CTMAX) and low (CTMIN) temperatures12, is one metric that may be shaped by selection to climate variation5. However, the strength of correlations between climate and CTLs differ at high and low temperatures11,13 due to differences in the relative selective importance of high or low temperature extremes14. For example, studies have revealed that many terrestrial ectotherms show little variation in CTMAX but pronounced differences in CTMIN with geographic temperature gradients14,15,16. The mechanisms underlying this spatial variation in thermal limits will inform species responses to changing temperatures but have rarely been studied (but see17). Shifts in gene expression likely contribute to physiological responses that underly range-wide variation in thermal tolerances, responses that may also be shaped by natural selection and ultimately limit species distributions5,10,18.
Mountain ranges provide a unique opportunity to investigate how organisms respond to thermal extremes, as temperature declines both with increasing latitude and over shorter distances with increasing elevation13,19. Therefore, montane organisms can face similar abiotic extremes at multiple spatial scales, and sampling across latitude and altitude can provide evidence for climate adaptation by separating the effects of space and environment across a species range10,13. The physiology of adaptation to variation in thermal stress with altitude or latitude has been studied both within18,20,21,22 and between12,23,24 species. Studies employing populations from both spatial dimensions remain rare but may be especially likely to reveal how selection shapes variation in thermal tolerance and underlying molecular mechanisms.
Bumble bees (Hymenoptera: Apidae: Bombus) are large-bodied, eusocial bees that are ecologically and economically important pollinators and common in mountainous regions of the Northern Hemisphere25. Bumble bees are facultatively endothermic, capable of elevating body temperatures when necessary (e.g., for foraging, incubating brood, etc.), but able to conserve energy by allowing body temperature to track ambient temperatures. This ability and related morphological, behavioral, and physiological adaptations enable bumble bee activity at low ambient temperatures26, and facilitate their success in cold environments27,28. We thus expect physiological responses of bumble bees to cold are likely to be especially important for shaping their past and present distributions29. However, recent bumble bee declines30 may in part stem from rising temperatures31, suggesting that physiological responses to heat may also determine persistence of bumble bee populations and species. Critically, as responses to thermal extremes may vary among populations particularly for widely distributed species18,32, such population variation in thermal tolerance could profoundly alter predictions of the impacts of changing climates on bumble bees.
To investigate intraspecific variation in functional responses to thermal stress, we measured CTLs and gene expression among Bombus vosnesenskii sourced from populations from southern California (CA) and northern Oregon (OR), USA, near the extremes of the species range (Fig. 1). B. vosnesenskii occurs across broad altitudinal ranges throughout its distribution, and is characterized by weak genetic differentiation (genome-wide FST = 0.003), facilitating identification of strong local effects with little confounding influence from population structure3,33. We employed a common-garden style approach with laboratory colonies reared from queens collected at high and low elevations at both high and low latitudes to separate species-wide responses to thermal stress from population-specific differences that may reflect local adaptation. We measured CTMAX and CTMIN in laboratory-reared B. vosnesenskii workers and employed transcriptomics (RNAseq) to test how gene expression responses to extreme thermal stress correlate with population-specific thermal tolerance and environmental conditions at the population of origin.

Collection location and physiological tests. (A) Queens for laboratory colony rearing were collected from sites near the spatial and bioclimatic extremes of the Bombus vosnesenskii range from relatively low and high elevations in southern CA and northern OR (Table S1). The map was created in ESRI ArcMap 10.8, with state outlines and digital elevation data obtained from https://www.diva-gis.org/Data. The B. vosnesenskii range outline was taken from the species distribution model published perviously3,33. (B) Mean monthly temperatures over the course of a year for queen collecting sites. (C) The CTMIN temperature of laboratory reared workers is strongly correlated with the annual mean temperature of collection sites of the founding queens. The coastal L-CA colony discussed in Results is indicated by a black dot in each panel.
We use this system to address a series of related hypotheses. First, we hypothesize that local adaptation will match critical thermal limits to local environmental conditions7,8,34,35,36, predicting that bees from warmer and colder sites have higher and lower CTLs, respectively. Selection may not be the same at hot and cold extreme temperatures14,23,37, however, which could also produce contrasting population-environment correlations at CTMAX and CTMIN. Alternatively, if variation in thermal tolerance is largely shaped by flexible or plastic individual responses, then thermal limits should be independent of region of origin38,39. Parallel hypotheses can be examined for gene expression. If intraspecific variation in gene expression contributes to population level differences in thermal tolerance, we hypothesize that differential gene expression will vary among populations under thermal stress treatments. If responses to temperature are largely plastic, we expect that treatment (CTMAX vs CTMIN) will have the largest effects on expression, with no effect of environmental temperature at the region of origin. Finally, we aim to identify key molecular processes involved in the bumble bee temperature stress response, and especially in population-specific variation in this response that may provide possible targets of local adaptation.
Results
Bumble bee cold tolerance varies regionally, tracking local temperatures
Laboratory colonies of Bombus vosnesenskii were established from wild queens collected from four regions: low-elevation CA (L-CA), high-elevation CA (H-CA), low-elevation OR (L-OR), and high-elevation OR (H-OR) (Fig. 1A, “SI Appendix”, Table S1), with each region representing a distinct thermal environment, as highlighted by mean monthly temperature over the course of an average year (Fig. 1B). CTMIN of laboratory-reared B. vosnesenskii workers varied significantly with region of origin (ANOVA, F3,8 = 8.6, P = 0.007; Table S2; Dataset S1), with bees from colder regions having reduced CTMIN (Fig. 1C). Across colonies, CTMIN increased significantly with Annual Mean Temperature (AMT) at queen collection localities, with bees from H-OR colonies tolerating temperatures as low as − 7 °C, 12 °C lower than L-CA bees (linear mixed model40, t10 = 5.9, P < 0.001; Fig. 1C; Table S3). CTMAX averaged 51.6 °C (4.4 °C SD) across colonies (N = 13) and did not vary significantly among regions (ANOVA, F3,8 = 3.01, P = 0.1, Table S2) or show a significant association with local temperature (t10 = 5.9, P = 0.2; Table S3, Dataset S1).
Region of origin and temperature both effect expression patterns
We performed RNAseq on bees from the CTMIN and CTMAX experiments plus untreated controls, and first explored the data by determining best fit general linear models for expression patterns for samples grouped by geographic region (L-CA, H-CA, L-OR, H-OR) or treatment (CTMIN, CTMAX, control) with the Akaike Information Criterion (AIC)41. This preliminary analysis suggested that both experimental treatment and region of origin altered expression patterns, with 46% of genes demonstrating a best-fit model incorporating the experimental temperature at CTL (CTTEMP), AMT, or some combination (Table S4, Dataset S2). We then tested specific hypotheses for differential expression at individual genes by specifying contrasts using edgeR42 and limma43 (blocking by colony). We identified significant differential expression in at least one treatment, region, or treatment-by-region contrast in 442 genes (Benjamini–Hochberg44 FDR ≤ 0.05, Dataset S2). Most of these significantly differentially expressed genes (N = 354) had a significant treatment effect, with 296 genes uniquely differentially expressed between all three pairwise treatment contrasts (Fig. 2A; Dataset S2G). Sixty-eight genes demonstrated differential expression due to region of origin, with 55 genes unique to regional comparisons (Fig. 2A; Dataset S2G). Region-specific effects were primarily driven by unique gene expression of H-OR bees following thermal stress (Fig. 2A).

Summary of differential gene expression analysis patterns. (A) Venn diagrams summarizing the total number of genes demonstrating significant FDR-corrected (5%) effects, including (left) genes with treatment, region, or interaction effects, (center) genes differentially expressed among CTMAX, CTMIN, and control treatments for the 296 genes exhibiting treatment effects alone, and (right) 55 genes with region effects alone. See Dataset S2G for details. (B) Network analysis of co-expression with WGCNA grouped genes into 26 modules, six of which (labeled 1–6 on heat map edges) demonstrated a significant relationship (likelihood ratio test, P < 0.05) with critical thermal temperature (CTTEMP), Annual Mean Temperature (AMT), or some combination of the two. The strongest relationship was observed for Module 2 with CTTEMP (black box; Dataset S3). (C) The eigengene value (expression analog) of module 2 genes (top) and expression of the module 2 hub gene, dnaJ protein homolog 1-like (bottom) as functions of Annual Mean Temperature for each treatment.
Bumble bees show a strong treatment effect on gene expression driven by CTMAX
Bees warmed to CTMAX showed the clearest differential expression relative both to control bees and to bees cooled to CTMIN (Fig. 2A). Of the 296 genes unique to the treatment contrasts (i.e. not also associated with region or treatment-by-region interactions; Fig. 2A), 91 were differentially expressed between CTMAX and both CTMIN and control. Genes with increased expression in CTMAX compared to other treatments were significantly enriched in GO terms including cellular response to temperature stimuli, heat shock protein binding, signaling, and plasma membrane structure (Fig. 3A, Dataset S2). Genes with decreased relative expression in CTMAX covered a broader range of GO terms, including RNA polymerase II (RNAP II) transcription and gene expression, metabolic pathways and processes, and transmembrane transporter activity (Fig. 3A, Dataset S2). Comparisons between CTMAX and CTMIN revealed altered expression of genes in several relevant GO categories that did not overlap with comparisons between CTMAX and control, including neurogenesis, nervous system and muscle development, response to heat, and transmembrane transport activity (Dataset S2). In contrast to these strong expression differences in response to CTMAX, only one gene (uncharacterized, LOC105681062) was differentially expressed between control and CTMIN treatment bees across colonies (Dataset S2G) and less stringent FDR correction (10%) identified only three additional genes (Fig. S1), suggesting a subtler species-wide (i.e. not considering population effects) genomic response to cooling.

Gene Ontology enrichment analysis of edgeR/limma and WCGNA results. Significantly enriched gene ontology terms identified in edgeR/limma are listed on the rows for different contrasts, with information on the WCGNA modules (1–6) that are also enriched for that term. Each panel has a different main factor for the contrast (top label), with GO enrichment in genes with decreased or increased expression in the main factor relative to the contrast factor (bottom label). (A) Treatment comparisons (control and CTMIN) against CTMAX treatment, presenting full GO analysis results (Dataset S2) reduced with ReViGO and further filtered to include common terms from other thermal stress studies for the purposes of presentation clarity. (B) Region comparisons (L-CA, H-CA, and L-OR) against H-OR bees, with terms reduced as in (A). (C) Regional CTMIN comparisons (L-CAMIN, H-CAMIN, and L-ORMIN) against H-ORMIN bees. GO terms were summarized with ReViGO but did not require further filtering for presentation clarity. Complete sets of GO terms for all edgeR/Limma and WCGNA results are provided in Datasets S1 and S2, respectively.
Bees from the coldest sites have pronounced region-specific gene expression
Bees from the coldest sites (H-OR) showed the broadest thermal tolerance range, driven by strongly reduced CTMIN (Fig. 1C). Of the 55 genes that were differentially expressed solely due to population of origin (Fig. 2), 53 involved pairwise comparisons with H-OR (Dataset S2). These included increased expression in H-OR of genes related to DNA-templated transcription regulation and muscle and synapse development and activity, as well as reduced expression of genes related to transmembrane transport (calcium and zinc) and biosynthetic pathways such as ribosome biogenesis (Fig. 3B).
Although 91 genes were significantly differentially expressed in at least one region-by-treatment contrast, most of these were also significant in at least one main effect contrast (Fig. 2A). To clarify discussion and address our main hypotheses about regional local adaptation under thermal stress, we focus primarily on contrasts between regions for each treatment. Control bees did not show differential gene expression related to region of origin. However, bees from different regions differed in their responses to being cooled to CTMIN or heated to CTMAX. Only five genes had regional effects under CTMAX, all of which were only differentially expressed between H-OR vs. L-OR. The greatest number of genes (N = 14) exhibiting within-treatment population of origin effects were in CTMIN treatment bees, all involving contrasts with H-ORMIN (14 genes with L-ORMIN, two with L-CAMIN, and one with H-CAMIN). Most (13 of 14) were downregulated in H-ORMIN relative to L-ORMIN and are involved in GO processes such as protein folding and stabilization, heat shock (and Hsp90) protein and chaperone binding, nuclear pore transport, and translation regulation. Upregulated GO terms for H-ORMIN related to cell membrane composition/flotillins.
Notably, relatively few genes and GO terms were detected as significant between H-OR and L-CA bees, despite the large differences in CTMIN (Figs. 2A, 3B,C). One possible explanation is that one L-CA colony came from a coastal locality with unusually low annual temperature variation and correspondingly low CTMIN relative to other L-CA bees (Fig. 1B,C). Exclusion of this colony from analyses would have reduced statistical power for categorical group comparisons, and so we instead opted to take into account colony-specific environmental and physiological information through WCGNA gene expression module analysis.
Linking region of origin, physiology, and gene expression with gene expression modules
To test how gene expression is shaped by both source environmental conditions and physiological responses of individual bees under thermal tolerance treatments, we employed network analysis with WGCNA45 to reduce the complex gene-by-gene patterns into covarying expression modules. Genes clustered into 26 modules, 13 of which correlated significantly with predictors of interest in initial analyses (Dataset S3). The most significant association in initial univariate correlation analysis was between Module 2 and CTTEMP (RCT = 0.74, P = 1.8 × 10–11). Likelihood regression analysis of the eigengene values (i.e. expression) of environmentally correlated modules identified six modules that were significantly best fit by a linear model that included AMT, CTTEMP, or some combination of the two, and we focus the remainder of our discussion on these (Dataset S3).
Module 2 produced the strongest relationship between expression and explanatory variables of all modules [best fit by the model eigengene ~ 1 + AMT + CTTEMP + (1|Colony) (LRT: P = 2.53 × 10–12)] and demonstrated the greatest overlap with the edgeR/limma42,43 gene-by-gene analyses (85 of 111 genes). The major effect on Module 2 expression was CTTEMP, but region of origin (AMT) effects were clearly apparent in control and CTMIN treatments, with patterns of expression at CTMIN and CTMAX that mirror the physiology data (Fig. 2C). Module 2 was enriched in genes relating to response to stress and stimulus (including cold), transcription regulation activity (particularly gene silencing), and various components of muscle, and contained many of the major significant genes that were differentially expressed among treatments (e.g., heat shock genes) (Dataset S3). The hub-gene was LOC100743914 (Dataset S3), a dnaJ homolog and putative small heat shock protein, which was differentially expressed in 16 gene-by-gene edgeR contrasts (Dataset S2).
Five other modules were best fit by significant non-null models (0.020 ≤ P ≤ 0.049). Module 1 was best fit by a full factorial interaction model [eigengene ~ 1 + AMT + CTTEMP + AMT * CTTEMP + (1|Colony)], and genes in this module (Datasets S1 and S2) were involved in neurogenesis, metabolism, DNA transcription, regulation of growth, and general cell cycle processes (Fig. 3; Dataset S3). Two of the remaining modules were best fit by CTTEMP only models, with highest eigengene expression values for Module 3 and Module 4 genes observed in CTMIN and CTMAX bees respectively. GO terms in Modules 3 and 4 supports results from other analyses, showing increased expression in CTMIN bees related to various catabolic and metabolic processes, gene expression, and transmembrane transporter activity (Fig. 3). Modules 5 and 6 were best fit by AMT only models, with Module 6 in particular containing genes with GO terms related to muscle development, cellular differentiation, protein binding, and cell-to-cell signaling (Fig. 3, Dataset S3C).
Discussion
This study demonstrates that variation in the local abiotic niche may drive intraspecific variation in physiology and associated molecular responses when bumble bees are exposed to thermal stress, and that population-dependent responses differ with heat versus cold. As predicted, laboratory-reared Bombus vosnesenskii worker bees from different geographic regions demonstrate strong population-level differences in cold tolerance (CTMIN) and gene expression responses at low temperatures. The correlation between CTMIN and source locality AMT, and the identification of co-expressed gene sets (e.g., Module 2) with parallel responses to AMT and CTTEMP, suggest the potential for local adaptation in physiological and gene expression responses. In contrast, although the largest gene expression responses are produced by warming to upper thermal limits, gene expression under heat stress is conserved across populations, which mirrors a lack of interpopulation variation in CTMAX itself. Although direct evidence for selection on CTLs and gene expression will require further work, these data suggest that if local environment shapes thermal tolerance of bumble bees, it may be responses to low temperatures that are most malleable, and that such variation may be in part associated with gene expression. Such results are consistent with the importance of cold adaptation for the evolution of Bombus diversity above the species level27,28, while also indicating potential constraints for adaptation to increasing temperatures in the warmest parts of species ranges31,46.
The importance of temperature extremes as drivers of thermal tolerance evolution is well-known14, and several studies indicate that physiological cold tolerance thresholds in terrestrial ectotherms can change along spatial-environmental gradients while upper thresholds are constant14,23,24,47,48. Our data extend these observations to facultative endotherms and show complementary patterns for associated molecular processes. A possible explanation for the lack of variation in gene expression and CTMAX is that bumble bees experience high thoracic temperatures during flight regardless of ambient conditions26, and have therefore evolved to deal with uniformly high body temperatures. Bumble bees also rely on behavioral thermoregulation to mediate stress at high ambient temperatures (e.g., limiting foraging activity)49,50,51. The heat shock response is clearly robust based on gene expression data, largely relating to processes including unfolded protein binding, signaling, regulation of transcription and gene expression, metabolic pathways and processes, and transmembrane transporter activity52,53,54. However, limited variation in the point at which this response is initiated across populations indicates there may be physiological or molecular constraints on the evolution of heat tolerance55,56.
Variation among source populations in gene expression at CTMIN suggests several mechanisms underlying the physiological response to cold stress that may be shaped by local adaptation. The onset of chill coma at CTMIN produces spasms that are likely symptomatic of failure at the neuromuscular junction or locally in the muscle57, and differentially expressed genes involved in neurogenesis and muscle structure and function are detected in both treatment and region analyses (Dataset S3C). Failure of the neuromuscular junction can involve changes in ion transport channels that regulate membrane potential57. Calcium regulation is especially important for the cold tolerance response58 and, for example, H-OR bees demonstrated decreased expression in a homolog of calumenin (LOC100743248), a gene known to play a role in thermal stress tolerance54,59. Differences in membrane composition, rigidity, and permeability are also important for the cold shock response, and increased expression of flotillin-1 (LOC100740847) in H-OR bees60 reflects the potential importance of cell membrane adaptations among B. vosnesenskii populations58,60,61,62.
One class of genes, the small heat shock proteins (sHSPs), was involved in responses to both high temperatures and population-specific responses to cold. Geographic variation in expression of HSPs is known to influence adaptation for thermal tolerance across species ranges8,18,22,63,64. The importance of sHSPs in B. vosnesenskii was clearest in the WGCNA module analysis; for example, the hub gene of Module 2 (Fig. 2) is the sHSP dnaJ, and this module contains numerous HSPs that were overrepresented in various treatment and region contrasts (e.g., LOC100742443-protein lethal(2)essential for life-like is differentially expressed in 14 different contrasts including control vs. CTMAX, CTMAX vs. CTMIN, and H-OR vs. L-OR). The consistent regional expression patterns of BAG domain-containing protein Samui-like (another Module 2 gene), especially involving H-OR bees, is also of interest because this gene interacts with HSPs in the cold stress response and maintenance of muscle membrane potential, and has previously been detected as a key player in cold stress for other bees65,66. Intraspecific variation in both constitutive and inducible expression of heat shock and related proteins is thus one mechanism that may underly adaptation to hot and cold thermal extremes in Bombus populations.
Beyond the extreme temperature responses revealed here, there are several avenues for future work. First, there is evidence that bumble bees can show signs of heat stress at ambient temperatures as low as 36 °C67, significantly lower than CTMAX in B. vosnesenskii. Our assays reflect physiological limitations68 at extreme temperatures; but behavioral and ecological responses of bees to less extreme temperatures may also vary among populations and should be investigated. Second, our results suggest that cold tolerance may be more responsive to local selective pressures than heat tolerance, but worker bees are typically active during periods where ambient temperatures as low as the observed CTMIN values are uncommon. Worker CTMIN temperatures are closer to those that might be experienced by queens during overwintering or nest founding, which are likely strong targets of selection69. Our results show some overlap with differentially expressed genes observed in diapausing/post-diapausing B. terrestris queens and some Megachile66,70, and future research on old stress in queens and links to worker physiology would be particularly informative for understanding true targets of selection for cold tolerance. A related caveat is that while experimental workers were reared in the lab and never experienced field conditions that would alter their physiology via plasticity or acclimation, queens did overwinter in the field, which could contribute to maternal effects relating to region of origin71,72. Such effects might be avoided by rearing second-generation colonies, but this proved unsuccessful for B. vosnesenskii and is a technical challenge that must be overcome. Third, we cannot exclude the possibility that the regional correlation between CTMIN and differential gene expression may reflect different extremes of cold stress exposure, owing to the different temperatures at which populations enter chill coma. Unfortunately, because of such regional differences in CTMIN it is impossible to evaluate gene expression patterns in all bees for the same critical physiological threshold. Based on our results here, future studies might thus be designed to address related questions by, for example, comparing gene expression among populations at the same temperatures through time during the cooling process. Finally, we studied thoracic muscle given its importance in bumble bee thermoregulation and flight26,50,73, however, head and abdominal tissues may show different levels of sensitivity to heat or cold exposure74. Ultimately, additional experiments will be necessary for untangling contributions to cold tolerance from genetic adaptation, plasticity, maternal effects, and roles of different tissues.
In conclusion, we have demonstrated that bumble bees exhibit conservation in physiological upper thermal tolerances and gene expression, and interpopulation variation in lower thermal tolerances and gene expression that match conditions in their local niche. Our results provide one possible mechanism that could link global climate change to bumble bee range contractions, which appears to result from erosion in southern and low-lying regions combined with failure to colonize north of existing range margins31. Populations in warmer regions may experience environmental temperatures which exceed their inflexible upper thermal limits, while poleward or upslope range expansions could be limited if cold extremes at available sites exceed CTMIN of dispersers. These conclusions have implications for bumble bee conservation, suggesting that managers may need to consider future changes in temperature extremes as well as means24,75, and should also include evaluation of population-specific phenotypes and genomic variation76. Additional work to characterize physiology and functional genomics at the spatial and abiotic extremes of Bombus species ranges must remain a priority if we are to understand the potential for adaptation to changing temperatures.
Methods
Queen collection and thermal physiology experiments
Queens were transported from collecting sites to the USDA Pollinating Insect Research Unit (PIRU) (Logan, UT USA) for colony initiation (“SI Methods”). Colonies with > 20 workers (N = 13, 3–4 per region) were transported to the University of Wyoming (Laramie, WY USA) for experiments, where they were maintained at 25 °C and 12:12 h L:D cycles. All experiments used workers that developed under these common laboratory conditions from wild caught queens. While conditions in lab are different from those experienced in the field for all populations, these conditions are standardized across colonies. Experiments using workers raised in the lab prevents individual plasticity arising from field exposure, but queens are wild, so maternal effects could be present. Unfortunately, it was not possible to raise a sufficient number of second-generation colonies from lab-reared queens. Environmental data for each collection site was extracted from WorldClim v2 (0.5 min resolution)77.
Physiological tests were conducted as part of a larger study of Bombus thermal physiology using validated techniques for measuring critical thermal limits68. Workers were arbitrarily selected, placed in individual 2-dram clear glass vials, and randomly assigned to control (room temperature, 22 °C), upper (CTMAX) or lower (CTMIN) temperature treatments. Vials and a thermocouple were placed in machined wells in a solid aluminum block attached to an insulated thermoelectric plate. For CTMIN and CTMAX, bees were held at 25 °C for 15 min before decreasing or increasing the temperature at 0.25 °C min−1, respectively78,79,80. At CTMIN, bees exhibit characteristic signs of “chill coma”, while CTMAX is indicated by uncontrollable muscle spasms68. Upon reaching thermal limits, bees were immediately trisected between body sections using a sterile scalpel, placed in RNAlater (Qiagen), and stored at − 80 °C. We used repeated measures ANOVA and linear mixed effects models40 (degrees of freedom and P values determined with lmerTest81) to test for regional differences in CTMIN and CTMAX (“SI Methods”; Dataset S1), as well as correlation with various BioClim77 variables characterizing the environmental variation in thermal conditions, including Annual Mean Temperature, Minimum Temperature of the Coldest Month, Maximum Temperature of the Warmest Month, and Mean Diurnal Range (Fig. S2; Table S3). Although CTLs can be shaped by extremes and variability of local climate, we focus on Annual Mean Temperature as a metric to characterize the average climate at each collecting site because it is strongly correlated with the other environmental variables of interest and CTL patterns were similar across multiple variables (Fig. S2).
RNA extraction, sequencing, and bioinformatics
Samples for RNAseq were arbitrarily ordered to avoid batch effects. Whole RNA was extracted from thoracic muscle (E.Z.N.A. Total RNA Kit II; Omega Bio-tek, Inc.) because of the role of musculature for thermogenesis and thermal tolerance50,73,82 and the importance of homogenous tissues in RNAseq83. We analyzed one control bee and two bees from each CT treatment per colony, except in one case (N = 60 bees from 13 colonies; SI Table S1).
Each bee was sequenced via poly-A amplified directional 50 bp paired-end RNAseq to ~ 20 M reads (± 2.1 M SE) on a HiSeq 2500 (Illumina, Inc) by HudsonAlpha Institute for Biotechnology (Huntsville, AL) (data available on the Sequence Read Archive SAMN13234508-13234603). Reads were processed using TrimGalore v0.4.284,85 and aligned to the Bombus impatiens genome v2.0 (BIMP2.086,87) with TopHat v2.1.188 (stranded, maximum three mismatches permitted, intron length distance 20–50,000 bp, mate inner distance 400 ± 30 bp). Gene ontologies for BIMP2.0 were assigned using a reciprocal best hit approach89 from the Drosophila melanogaster genome. Using NCBI BLAST + 2.9.090, blast databases were created from the protein sequences of both BIMP2.0 and D. melanogaster assembly Release 6 plus ISO1 MT. Bombus impatiens genes which were identified as a single D. melanogaster gene’s best scoring hit in reciprocal subject-query blastP analyses (e-value threshold ≤ 10–5) were assigned the GO term(s) associated with its fly ortholog in Ensembl 201891 using biomartr v0.9.092. In total, 7088 genes had D. melanogaster orthologs, and 6645 of these were assigned GO terms.
Gene expression and gene ontology enrichment analyses
RNAseq analyses used edgeR v3.22.341,42 and limma v3.36.243. Raw counts were transformed to counts per million (CPM), retaining genes with ≥ 1 CPM in ≥ 2 samples. Analyses were blocked by colony with limma duplicateCorrelation. Analysis was then conducted in three stages: (1) model-based sub-setting using AIC; (2) individual gene-by-gene differential expression analyses; (3) gene co-expression module analyses.
We first used limma selectModel to determine a best fit general linear model for each gene from alternative nested models (Region, Treatment, Region + Treatment, or Full model with an interaction) with both the default results, as well as the model with the smallest number of parameters (“simplest model”) with an AIC score within 2 of the minimum AIC41 (Table S4, Dataset S2A).
Second, we performed individual gene-by-gene analyses with a contrasts approach in edgeR to test for region, treatment, and region-by-treatment effects on gene expression. Significance was assessed at a False Discovery Rate (FDR) ≤ 0.05, using Benjamini–Hochberg adjusted P-values44 of all genes (N = 9328) and contrasts on a per-factor basis (e.g. three pairwise contrasts between treatments; six pairwise contrasts between region levels; 66 region-by-treatment pairwise contrasts for the interaction term).
Finally, to identify clusters of genes with co-expression patterns (modules) and correlate expression with individual-specific physiological results and environmental data (versus regional groupings of queen collection sites used above), data were assigned to expression modules using weighted gene co-expression network analysis in WGCNA v1.64.145 following best practices and previous studies18,93. As predictors, we used Annual Mean Temperature (AMT) at queen collection sites to capture effects of local environment, and CTTEMP (i.e. experimental CTMIN or CTMAX, controls = 22 °C) at which each bee was sampled to capture treatment effects. We retained modules with significant responses to AMT and/or CTTEMP (P ≤ 0.05 from function corPvalueStudent). For each filtered module, we used linear mixed-effects models (lmer40) to identify a best fit model (colony as a random effect; AMT and CTTEMP as fixed effects, including additive and interaction terms) for gene expression (WGCNA eigengene value) (“SI Methods”).
Most genes were expressed in two or more samples (80%; 9328/11,600), including most GO-annotated genes (95%; 6327/6645). GO enrichment analyses were performed with topGO v2.30.094 using classic Fisher’s Exact Test with default settings95 for biological process (BP), molecular function (MF), and cellular component (CC). We consider enrichment significant at P ≤ 0.05, as we were primarily interested in examining functional information for genes that were already stringently selected following conservative FDR correction, and GO enrichment P values do not conform to distributions typically required for FDR correction96. For ease of visualizing GO terms in presented results, GO lists were simplified using ReViGO97 (allowed similarity = small), and then filtered for terms (with parent and child terms) previously identified as enriched in thermal stress studies in other organisms58,98,99,100. However, complete unfiltered GO results can be found in Datasets S1E and S2C, and intermediate datasets and R code can be found on DRYAD (https://doi.org/10.5061/dryad.tmpg4f4wx).
References
Hoffmann, A. A. & Willi, Y. Detecting genetic responses to environmental change. Nat. Rev. Genet. 9, 421–432 (2008).
Hoffmann, A. A. & Sgrò, C. M. Climate change and evolutionary adaptation. Nature 470, 479–485 (2011).
Jackson, J. M. et al. Distance, elevation and environment as drivers of diversity and divergence in bumble bees across latitude and altitude. Mol. Ecol. 27, 2926–2942 (2018).
Lowe, W. H. & McPeek, M. A. Is dispersal neutral?. Trends Ecol. Evol. 29, 444–450 (2014).
Somero, G. N. Linking biogeography to physiology: Evolutionary and acclimatory adjustments of thermal limits. Front. Zool. 2, 1 (2005).
Savolainen, O., Lascoux, M. & Merilä, J. Ecological genomics of local adaptation. Nat. Rev. Genet. 14, 807–820 (2013).
McKenzie, D. J., Estivales, G., Svendsen, J. C., Steffensen, J. F. & Agnèse, J.-F. Local sdaptation to altitude underlies divergent thermal physiology in tropical killifishes of the genus Aphyosemion. PLoS ONE 8, e54345 (2013).
Fangue, N. A., Hofmeister, M. & Schulte, P. M. Intraspecific variation in thermal tolerance and heat shock protein gene expression in common killifish, Fundulus heteroclitus. J. Exp. Biol. 209, 2859–2872 (2006).
Dunning, L. T., Dennis, A. B., Sinclair, B. J., Newcomb, R. D. & Buckley, T. R. Divergent transcriptional responses to low temperature among populations of alpine and lowland species of New Zealand stick insects (Micrarchus). Mol. Ecol. 23, 2712–2726 (2014).
Keller, I., Alexander, J. M., Holderegger, R. & Edwards, P. J. Widespread phenotypic and genetic divergence along altitudinal gradients in animals. J. Evol. Biol. 26, 2527–2543 (2013).
Sunday, J. M., Bates, A. E. & Dulvy, N. K. Thermal tolerance and the global redistribution of animals. Nat. Clim. Chang. 2, 686–690 (2012).
Sheldon, K. S. & Tewksbury, J. J. The impact of seasonality in temperature on thermal tolerance and elevational range size. Ecology 95, 2134–2143 (2014).
Dillon, M. E. & Lozier, J. D. Adaptation to the abiotic environment in insects: the influence of variability on ecophysiology and evolutionary genomics. Curr. Opin. Insect Sci. 36, 131–139 (2019).
Sunday, J. et al. Thermal tolerance patterns across latitude and elevation. Philos. Trans. R Soc. B Biol. Sci. 374, 20190036 (2019).
Gaston, K. J. et al. Macrophysiology: A conceptual reunification. Am. Nat. 174, 595–612 (2009).
MacLean, H. J. et al. Evolution and plasticity of thermal performance: an analysis of variation in thermal tolerance and fitness in 22 Drosophila species. Philos. Trans. R Soc. B Biol. Sci. 374, 20180548 (2019).
Andersen, M. K. & Overgaard, J. Maintenance of hindgut reabsorption during cold exposure is a key adaptation for Drosophila cold tolerance. J. Exp. Biol. 223, jeb213934 (2020).
Campbell-Staton, S. C., Bare, A., Losos, J. B., Edwards, S. V. & Cheviron, Z. A. Physiological and regulatory underpinnings of geographic variation in reptilian cold tolerance across a latitudinal cline. Mol. Ecol. 27, 2243–2255 (2018).
Dillon, M. E., Frazier, M. R. & Dudley, R. Into thin air: Physiology and evolution of alpine insects. Integr. Comp. Biol. 46, 49–61 (2006).
Stillman, J. H. & Tagmount, A. Seasonal and latitudinal acclimatization of cardiac transcriptome responses to thermal stress in porcelain crabs, Petrolisthes cinctipes. Mol. Ecol. 18, 4206–4226 (2009).
Scott, G. R., Elogio, T. S., Lui, M. A., Storz, J. F. & Cheviron, Z. A. Adaptive modifications of muscle phenotype in high-altitude deer mice are associated with evolved changes in gene regulation. Mol. Biol. Evol. 32, 1962–1976 (2015).
Schoville, S. D., Barreto, F. S., Moy, G. W., Wolff, A. & Burton, R. S. Investigating the molecular basis of local adaptation to thermal stress: Population differences in gene expression across the transcriptome of the copepod Tigriopus californicus. BMC Evol. Biol. 12, 170 (2012).
Kellermann, V. et al. Upper thermal limits of Drosophila are linked to species distributions and strongly constrained phylogenetically. Proc. Natl. Acad. Sci. 109, 16228–16233 (2012).
Sunday, J. M. et al. Thermal-safety margins and the necessity of thermoregulatory behavior across latitude and elevation. Proc. Natl. Acad. Sci. 111, 5610–5615 (2014).
Cameron, S. A., Hines, H. M. & Williams, P. H. A comprehensive phylogeny of the bumble bees (Bombus). Biol. J. Linn. Soc. 91, 161–188 (2007).
Heinrich, B. Heat exchange in relation to blood flow between thorax and abdomen in bumblebees. J. Exp. Biol. 64, 561–585 (1976).
Hines, H. M. Historical biogeography, divergence times, and diversification patterns of bumble bees (Hymenoptera: Apidae: Bombus). Syst. Biol. 57, 58–75 (2008).
Heinrich, B. Bumblebee Economics (Harvard University Press, Cambridge, Massachusetts, London, 1979).
Williams, P. H., Lobo, J. M. & Meseguer, A. S. Bumblebees take the high road: Climatically integrative biogeography shows that escape from Tibet, not Tibetan uplift, is associated with divergences of present-day Mendacibombus. Ecography 41, 461–477 (2018).
Cameron, S. A. et al. Patterns of widespread decline in North American bumble bees. Proc. Natl. Acad. Sci. USA 108, 662–667 (2011).
Kerr, J. T. et al. Climate change impacts on bumblebees converge across continents. Science 349, 177–180 (2015).
Eliason, E. J. et al. Differences in thermal tolerance among sockeye salmon populations. Science 332, 109–112 (2011).
Jackson, J. M. et al. Local adaptation across a complex bioclimatic landscape in two montane bumble bee species. Mol. Ecol. 29, 920–939 (2020).
Hargreaves, A. L. & Eckert, C. G. Local adaptation primes cold-edge populations for range expansion but not warming-induced range shifts. Ecol. Lett. 22, 78–88 (2019).
Kellermann, V. & van Heerwaarden, B. Terrestrial insects and climate change: Adaptive responses in key traits. Physiol. Entomol. 44, 99–115 (2019).
Andersen, J. L. et al. How to assess Drosophila cold tolerance: Chill coma temperature and lower lethal temperature are the best predictors of cold distribution limits. Funct. Ecol. 29, 55–65 (2015).
Lonsdale, D. J. & Levinton, J. S. Latitudinal differentiation in copepod growth: An adaptation to temperature. Ecology 66, 1397–1407 (1985).
King, N. G., McKeown, N. J., Smale, D. A. & Moore, P. J. The importance of phenotypic plasticity and local adaptation in driving intraspecific variability in thermal niches of marine macrophytes. Ecography 41, 1469–1484 (2018).
Rivière, B. Phenotypic plasticity and population-level variation in thermal physiology of the bumblebee Bombus impatiens. MSc Thesis, U. of Ottowa (2012).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B 57, 289–300 (1995).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Sirois-Delisle, C. & Kerr, J. T. Climate change-driven range losses among bumblebee species are poised to accelerate. Sci. Rep. 8, 14464 (2018).
Muñoz, M. M. & Bodensteiner, B. L. Janzen’s Hypothesis meets the Bogert Effect: Connecting climate variation, thermoregulatory behavior, and rates of physiological evolution. Integr. Org. Biol. 1, oby002 (2019).
Kellermann, V., van Heerwaarden, B., Sgro, C. M. & Hoffmann, A. A. Fundamental evolutionary limits in ecological traits drive Drosophila species distributions. Science 325, 1244–1246 (2009).
Heinrich, B. Thermoregulation in bumblebees. J. Comp. Physiol. B 96, 155–166 (1975).
Heinrich, B., Esch, H., Plath, O. & Entomologist, H. Thermoregulation in bees. Am. Sci. 82, 164–170 (1994).
Vogt, F. D. Thermoregulation in bumblebee colonies. I. Thermoregulatory versus brood-maintenance behaviors during acute changes in ambient temperature. Physiol. Zool. 59, 55–59 (1986).
Chidawanyika, F. & Terblanche, J. S. Rapid thermal responses and thermal tolerance in adult codling moth Cydia pomonella (Lepidoptera: Tortricidae). J. Insect Physiol. 57, 108–117 (2011).
Sørensen, J. G., Nielsen, M. M., Kruhøffer, M., Justesen, J. & Loeschcke, V. Full genome gene expression analysis of the heat stress response in Drosophila melanogaster. Cell Stress Chaperones 10, 312–328 (2005).
Ruiz-Jones, L. J. & Palumbi, S. R. Tidal heat pulses on a reef trigger a fine-tuned transcriptional response in corals to maintain homeostasis. Sci. Adv. 3, e1601298 (2017).
Araújo, M. B. et al. Heat freezes niche evolution. Ecol. Lett. 16, 1206–1219 (2013).
Hoffmann, A. A., Chown, S. L. & Clusella-Trullas, S. Upper thermal limits in terrestrial ectotherms: How constrained are they?. Funct. Ecol. 27, 934–949 (2013).
Overgaard, J., Sørensen, J. G., Petersen, S. O., Loeschcke, V. & Holmstrup, M. Changes in membrane lipid composition following rapid cold hardening in Drosophila melanogaster. J. Insect Physiol. 51, 1173–1182 (2005).
Overgaard, J. & MacMillan, H. A. The integrative physiology of insect chill tolerance. Annu. Rev. Physiol. 79, 187–208 (2017).
DeSalvo, M. K. et al. Differential gene expression during thermal stress and bleaching in the Caribbean coral Montastraea faveolata. Mol. Ecol. 17, 3952–3971 (2008).
Lee, R. E., Damodaran, K., Yi, S.-X. & Lorigan, G. A. Rapid cold-hardening increases membrane fluidity and cold tolerance of insect cells. Cryobiology 52, 459–463 (2006).
Brankatschk, M. et al. A temperature-dependent switch in feeding preference improves Drosophila development and survival in the cold. Dev. Cell 46, 781-793.e4 (2018).
Chen, S., Bawa, D., Besshoh, S., Gurd, J. W. & Brown, I. R. Association of heat shock proteins and neuronal membrane components with lipid rafts from the rat brain. J. Neurosci. Res. 81, 522–529 (2005).
Rako, L., Blacket, M. J., McKechnie, S. W. & Hoffman, A. A. Candidate genes and thermal phenotypes: identifying ecologically important genetic variation for thermotolerance in the Australian Drosophila melanogaster cline. Mol. Ecol. 16, 2948–2957 (2007).
Sørensen, J. G. Application of heat shock protein expression for detecting natural adaptation and exposure to stress in natural populations. Curr. Zool. 56, 703–713 (2010).
Melicher, D. et al. Immediate transcriptional response to a temperature pulse under a fluctuating thermal regime. Integr. Comp. Biol. 59, 320–337 (2019).
Yocum, G. D. et al. Environmental history impacts gene expression during diapause development in the alfalfa leafcutting bee, Megachile rotundata. J. Exp. Biol. 221, jeb173443 (2018).
Couvillon, M. J., Fitzpatrick, G. & Dornhaus, A. Ambient air temperature does not predict whether small or large workers forage in bumble bees (Bombus impatiens). Psyche 2010, 31–33 (2010).
Oyen, K. J. & Dillon, M. E. Critical thermal limits of bumble bees (Bombus impatiens) are marked by stereotypical behaviors and are unchanged by acclimation, age, or feeding status. J. Exp. Biol. 221, jeb.165589 (2018).
Woodard, S. H. et al. Molecular tools and bumble bees: Revealing hidden details of ecology and evolution in a model system. Mol. Ecol. 24, 2916–2936 (2015).
Amsalem, E., Galbraith, D. A., Cnaani, J., Teal, P. E. A. & Grozinger, C. M. Conservation and modification of genetic and physiological toolkits underpinning diapause in bumble bee queens. Mol. Ecol. 24, 5596–5615 (2015).
Massamba-N’Siala, G., Prevedelli, D. & Simonini, R. Trans-generational plasticity in physiological thermal tolerance is modulated by maternal pre-reproductive environment in the polychaete, Ophryotrocha labronica. J. Exp. Biol. 217, 2004–2012 (2014).
Lockwood, B. L., Julick, C. R. & Montooth, K. L. Maternal loading of a small heat shock protein increases embryo thermal tolerance in Drosophila melanogaster. J. Exp. Biol. 220, 4492–4501 (2017).
Esch, H., Goller, F. & Heinrich, B. How do bees shiver?. Naturwissenschaften 78, 325–328 (1991).
Chowanski, S. et al. The physiological role of fat body and muscle tissues in response to cold stress in the tropical cockroach Gromphadorhina coquereliana. PLoS One. 12, e0173100 (2017).
Stott, P. How climate change affects extreme weather events. Science 352, 1517–1518 (2016).
Lozier, J. D. et al. Relocation risky for bumblebee colonies. Science 350, 286–287 (2015).
Hijmans, R. J., Cameron, S. E., Parra, J. L., Jones, P. G. & Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 25, 1965–1978 (2005).
Oyen, K. J., Giri, S. & Dillon, M. E. Altitudinal variation in bumble bee (Bombus) critical thermal limits. J. Therm. Biol. 59, 52–57 (2016).
Terblanche, J. S. et al. Ecologically relevant measures of tolerance to potentially lethal temperatures. J. Exp. Biol. 214, 3713–3725 (2011).
Jørgensen, L. B., Robertson, R. M. & Overgaard, J. Neural dysfunction correlates with heat coma and CTMAX in Drosophila but does not set the boundaries for heat stress survival. J. Exp. Biol. 223, jeb218750 (2020).
Kuznetsova, A., Brockhoff, P. B. & Christensen, R. H. B. lmerTest Package: Tests in linear mixed effects models. J. Stat. Softw. 82, 1–26 (2017).
Andersen, M. K. & Overgaard, J. The central nervous system and muscular system play different roles for chill coma onset and recovery in insects. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 233, 10–16 (2019).
Johnson, B. R., Atallah, J. & Plachetzki, D. C. The importance of tissue specificity for RNA-seq: Highlighting the errors of composite structure extractions. BMC Genom. 14, 1 (2013).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Krueger, F. & Felix Krueger. Trim Galore. https://github.com/FelixKrueger/TrimGalore (2015).
National Center for Biotechnology Information. Bombus impatiens 2.0. Genome (1988). ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/188/095/GCF_000188095.1_BIMP_2.0/GCF_000188095.1_BIMP_2.0_rna.fna.gz. (Accessed 5 November 2015)
Sadd, B. B. M. et al. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 16, 1–32 (2015).
Kim, D. et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Moreno-Hagelsieb, G. & Latimer, K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 24, 319–324 (2008).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Res. 46, D754–D761 (2018).
Drost, H.-G. & Paszkowski, J. Biomartr: Genomic data retrieval with R. Bioinformatics 33, btw821 (2017).
Velotta, J. P., Jones, J., Wolf, C. J. & Cheviron, Z. A. Transcriptomic plasticity in brown adipose tissue contributes to an enhanced capacity for nonshivering thermogenesis in deer mice. Mol. Ecol. 25, 2870–2886 (2016).
Alexa, A. & Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology v.2.36.0.a (R package, 2019).
Rivals, I., Personnaz, L., Taing, L. & Potier, M.-C. Enrichment or depletion of a GO category within a class of genes: Which test?. Bioinformatics 23, 401–407 (2007).
Gould, B. A., Chen, Y. & Lowry, D. B. Gene regulatory divergence between locally adapted ecotypes in their native habitats. Mol. Ecol. 27, 4174–4188 (2018).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6, e21800 (2011).
Xiong, Y. et al. Comparative transcriptome analysis reveals differentially expressed genes in the Asian citrus psyllid (Diaphorina citri) upon heat shock. Comp. Biochem. Physiol. D 30, 256–261 (2019).
Jia, Y. et al. Comparative transcriptome analysis reveals adaptive evolution of Notopterygium incisum and Notopterygium franchetii, two high-alpine herbal species endemic to China. Molecules 22, 1–17 (2017).
King, A. M. & MacRae, T. H. Insect heat shock proteins during stress and diapause. Annu. Rev. Entomol. 60, 59–75 (2015).
Acknowledgements
This work was funded by the University of Alabama College of Arts and Sciences, the National Science Foundation (DEB-1457645/1457659 and EF-1921585 to JDL, MED and JPS), and the US Department of Agriculture (2018-67012-28040 to MLP). We would also like to thank the National Parks Service for access to Sequoia National Park, the National Forest Service for access to Willamette and Mt. Hood National Forests, and the state of Oregon for access to Memaloose State Park.
Author information
Authors and Affiliations
Contributions
M.L.P. performed RNAseq and bioinformatics, and K.J.O. conducted physiology assays. J.D.H. and J.P.S. conducted queen sampling and colony rearing. J.M.J. assisted with GIS and preliminary field work. J.D.L., M.E.D., and J.P.S. conceived of the study, and M.L.P. and J.D.L. wrote much of the paper with input, editing, and other intellectual contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pimsler, M.L., Oyen, K.J., Herndon, J.D. et al. Biogeographic parallels in thermal tolerance and gene expression variation under temperature stress in a widespread bumble bee. Sci Rep 10, 17063 (2020). https://doi.org/10.1038/s41598-020-73391-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73391-8
This article is cited by
-
Cryptic genotypic and phenotypic diversity in parapatric bumble bee populations associated with minimum cold temperatures
Biodiversity and Conservation (2024)
-
Cuticular Hydrocarbon Profiles of Himalayan Bumble Bees (Hymenoptera: Bombus Latreille) are Species-Specific and Show Elevational Variation
Journal of Chemical Ecology (2024)
-
Genome-wide DNA methylation patterns in bumble bee (Bombus vosnesenskii) populations from spatial-environmental range extremes
Scientific Reports (2023)
-
Effects of imidacloprid on survival and nest development in the neo-tropical bumblebee Bombus ephippiatus
Apidologie (2022)
-
Body mass and sex, not local climate, drive differences in chill coma recovery times in common garden reared bumble bees
Journal of Comparative Physiology B (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
