
Abstract
Pure sensory polyneuropathy of genetic origin is rare in childhood and hence important to document the clinical and genetic etiologies from single or multi-center studies. This study focuses on a retrospective chart-review of neurological examinations and genetic and electrodiagnostic data of confirmed sensory polyneuropathy in subjects at a tertiary-care Children’s Hospital from 2013 to 2019. Twenty subjects were identified and included. Neurological examination and electrodiagnostic testing showed gait-difficulties, absent tendon reflexes, decreased joint-position, positive Romberg’s test and large fiber sensory polyneuropathy on sensory nerve conduction studies in all patients associated with lower-extremity spasticity (6), cardiac abnormalities or cardiomyopathy (5), developmental delay (4), scoliosis (3), epilepsy (3) and hearing-difficulties (2). Confirmation of genetic diagnosis in correlation with clinical presentation was obtained in all cases (COX20 n = 2, HADHA n = 2, POLG n = 1, FXN n = 4, ATXN2 n = 3, ATM n = 3, GAN n = 2, SPG7 n = 1, ZFYVE26 n = 1, FH n = 1). Our single-center study shows genetic sensory polyneuropathies associated with progressive neurodegenerative disorders such as mitochondrial ataxia, Friedreich ataxia, spinocerebellar ataxia type 2, ataxia telangiectasia, spastic paraplegia, giant axonal neuropathy, and fumarate hydratase deficiency. We also present our cohort data in light of clinical features reported for each gene-specific disease subtype in the literature and highlight the importance of genetic testing in the relevant clinical context of electrophysiological findings of peripheral sensory polyneuropathy.
Similar content being viewed by others

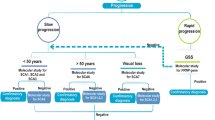

Introduction
Sensory polyneuropathy is rare in children1 with a broad differential diagnosis that includes both acquired and genetic etiologies2. Exposure to medications or supplements, including multiple chemotherapy agents such as cisplatin, carboplatin, and pyridoxine may induce a large fiber predominant sensory polyneuropathy. Trauma, compression, injury during surgical procedures, infections (Leprosy, Human Immunodeficiency Virus), vitamin B12 deficiency, diabetes mellitus and the rare Miller Fisher variant of Guillain–Barré syndrome (GBS) also present with definitive sensory dysfunction3. Genetic etiologies cause a heterogeneous group of progressive neurodegenerative disorders, but a paucity of literature on detailed clinical presentation, electrodiagnostic, and genetic-workup for children’s sensory polyneuropathy necessitates further characterization. In this study, we describe a single center pediatric electrodiagnostic (EDX) laboratory experience identifying pure sensory polyneuropathy on nerve conduction study (NCS) with detailed clinical features and genetic etiologies and compared each subtype findings to available English literature on rare inherited sensory polyneuropathies.
Results
Twelve hundred sixty one pediatric EDX studies were performed from 2013 to 2019. One hundred fifty seven studies (12%) were classified as sensory motor polyneuropathy and 24 studies (2%) had pure sensory polyneuropathy without gender preference. Further review of the subjects with pure sensory polyneuropathy on NCS revealed 20 children (12 girls, 8 boys, mean age of 13.6 ± 4.1 years) with confirmed genetic diagnosis with clinical correlation. The remaining 4 children had acquired sensory polyneuropathy i.e. Miller Fisher variant GBS, diabetes mellitus and were excluded from this study.
The study cohort presented with gait difficulties and on examination had sensory ataxia 100% (n = 20), spasticity 30% (n = 6), global developmental delay 20% (n = 4), cardiac defects or cardiomyopathy 25% (n = 5), scoliosis 15% (n = 3), epilepsy 15% (n = 3), and hearing difficulties 10% (n = 2). Significant family histories of neuromuscular illness or parental consanguinity were absent. Study subjects had genetic confirmation with pathogenic variants in mitochondrial genes [30%: COX20 (OMIM#614698) n = 2, HADHA (OMIM#600890) n = 2, POLG (OMIM#174763) n = 1, FH (OMIM# 136850) n = 1], Friedreich-ataxia (21%: FXN (OMIM#606829) n = 4), spinocerebellar ataxia type-2 (15%: ATXN2 (OMIM#601517) n = 3), ataxia-telangiectasia (15%: ATM (OMIM#607585) n = 3), giant axonal neuropathy (10%: GAN (OMIM# 605379), n = 2), and 5% each in spastic paraplegia 7 (SPG7 (OMIM#602783) n = 1) and spastic paraplegia 15 (ZFYVE26 (OMIM# 612012) n = 1). Subjects on an average had a 4.9 ± 3.3 years delay from symptom-onset to genetic confirmation. Clinical presentation with genetic variant and review of literature are presented in Tables 1 and 2. Below we give detailed genotypic and phenotypic features of subtypes of genetic childhood polyneuropathies identified in this study.
Mitochondrial sensory polyneuropathy subtypes
POLG-related polyneuropathy
20-year-old girl with identification of POLG variants [c.2243G>C (p.W784S)]; [c.3609_3612dupAACT] showed progressive sensory ataxia, intractable generalized epilepsy, magnetic resonance imaging (MRI)-confirmed cerebellum atrophy, and speech delays. Although the c.2243G>C (pW784S) is a variant of uncertain significance (VUS), clinical correlation and identification of a second pathogenic variant suggested that the POLG gene in this case is disease causing. The patient showed normal muscle strength but had nystagmus, ataxia, and dysarthria as well as absent reflexes and died at age 21 years due to liver failure.
HADHA-related mitochondrial trifunctional protein (MTP) deficiency
A 14-year-old female showed recurrent rhabdomyolysis, retinitis pigmentosa, mitral valve insufficiency, seizures with episodic weakness and gait abnormalities. The patient demonstrated tightened heel cords, decreased lower-extremity reflexes, absent sensory nerve action potentials (SNAPs). Pathogenic variants in HADHA gene [c.1528G>C (p.E510Q)]; [c.1620 + 2_162 + 0delTAAGG] were identified in this patient confirming molecular diagnosis.
A 5 year-old boy with history of early onset motor delay and walking difficulty first presented to pediatric neuromuscular clinics and electrodiagnostic (EDX) laboratory. He had bilateral hand tremors, waddling gait, and absent reflexes in lower extremities. EDX confirmed sensory polyneuropathy. Soon after, he was admitted to a cardiac intensive care unit with a diagnosis of severe cardiomyopathy with cardiac failure. Exome sequencing revealed a clinically correlated disease-causing variant in HADHA gene [(c.1418C>A (p.Ala473Asp)] in this patient, and after cardiac transplantation, motor strength in lower extremities improved. Genetic testing on its own was not strictly confirmatory, as a second variant in the HADHA gene was not identified; however, clinical correlation of genetic testing results suggested HADHA as the most likely disease-causing gene.
COX20-related polyneuropathy
An 18-year-old girl with sensory neuropathy, ataxia, distal extremity weakness, spasticity, and hearing loss presented farsightedness and esotropia at 3 years of age and developed gait instability by age four. The patient also presented chronic gastrointestinal problems with a strong family history of biopsy-confirmed Celiac disease on the maternal side with negative results in brain and spine MRI, Charcot-Marie-Tooth and Friedreich ataxia-specific genetic-testing. Her 10-year-old younger brother, delivered as twin B at 36 weeks of gestation with a brief respiratory distress showed speech and gross motor delays. The patient sat up at 12 months, walked at 18 months and could run by age four at which the onset of alternating esotropia, strabismus, and gait ataxia were noted. At 5 years of age, hypotonia and hyperreflexia were recorded in this patient and subsequent EDX revealed sensory polyneuropathy. Whole exome sequencing (WES) revealed pathogenic variants in the COX20 gene for both siblings: [c.157 + 3G>C (IVS2 + 3G>C)]; [c.41A>G (p.Lys14Arg)]; and [c.340G>A (p.Gly114Ser)]. c.157 + 3G>C variant was paternally inherited, and the p.Lys14Arg and p.Gly114Ser variants were maternally inherited.
FH-related fumarase deficiency
A 10-month-old boy with fumarate hydratase deficiency presented with global developmental delays, seizures, low muscle tone and absent tendon reflexes on neurological examination. His work up showed absent SNAPs on NCS, abnormal electroencephalogram (EEG) and cerebral atrophy on the brain MRI. The patient also had strabismus with some visual impairments, fatigue and poor weight gain, and anemia. In addition, some cardiac abnormalities such as pulmonary artery hypertension, and pulmonary stenosis, as well as urinary tract infection were also present. The patient harbored FH gene variants: [c.697C>T (p.R233C)]; [c.1431_1433dup].
Spinocerebellar ataxia (SCA)
ATXN2-related spinocerebellar ataxia type 2
Identical 17-year-old twin brothers showed progressive ataxia, myoclonic jerks, feeding difficulty, and weight loss with an onset age of 10 years. The brothers also showed bradykinesia, spasticity, decreased pinprick, vibration, and feet proprioception, as well as dysmetria, absent reflexes in lower extremities and wide-based ataxic gait. Symptoms worsened with need for gastrostomy tube, anti-seizure medications and benzodiazepines. Brain MRIs showed cerebellar atrophy and NCS had absent SNAPs for both brothers. The older twin had genetic confirmation and harbored the pathogenic variant [c.1564_1565delGA (p.Glu522Ilefs43)] in ATXN2.
A 14-year-old girl with ataxia, dysphagia, and developmental delay with onset age of 4 years demonstrated dysarthria, increased tone in extremities and axial hypotonia, decreased vibration and proprioception, ataxia and dysmetria, and absent tendon reflexes. MRI brain showed diffuse cerebral and cerebellar volume loss. She had absent SNAPs on NCS and her genetic testing confirmed predicted pathogenic variant in ATXN2 gene.
Ataxia telangiectasia
ATM-related ataxia telangiectasia
Sisters aged 11 and 12 years at the time of examination with a single ATM gene pathogenic variant [c.1564_1565delGA (p.Glu522Ilefs43)] showed progressive ataxia, myoclonus, bulbar conjunctiva and skin telangiectasia, immunodeficiency on chronic intravenous immunoglobulins and recurrent skin infections. Even though a second variant was not identified, clinical correlation of genetic testing suggested the ATM gene was disease causing. On examination, patients demonstrated nystagmus, dysarthria, ataxia, absent tendon reflexes and absent SNAPs.
A 3-year-old female with normal early development with rolling and sitting at age 3 and 6 months respectively but later developing balance problems, drooling and fine motor problems and in-toeing of gait, was found to harbor a heterozygous pathogenic variant in ATM gene [c.4544dup (p.Asn1515Lysfs*16)], in addition to two VUSs, [c.7397C>A (p.Ala2466Glu)] and [c.7502A>G (p.Asn2501Ser)]. No seizures, headaches or focal weakness were noted in this patient, but frequent ear infections and difficulty in maintaining sleep were noted. No family history of neuromuscular or balance problems was found. The genetic testing results, taken together with the clinical suspicion for the disease, were suggestive of a diagnosis of Ataxia Telangiectasia.
Friedreich's ataxia
FXN-related Friedreich ataxia
A 16 year old with confirmed genetic diagnosis of 899 GAA-repeats of FXN showed progressive gait instability, scoliosis, and absent SNAPs. The patient presented with good muscle strength but needed walking assistance with decreased vibration in her extremities with dysmetria, absent tendon reflexes and bilateral Babinski.
Three other children were evaluated with similar presentation including cardiomyopathy and attention deficit hyperactivity disorder (ADHD) with 800–1250 range FXN GAA triplet repeats among whom one patient also harbored one pathogenic missense variant [c.317 T>C (p.Leu106Ser)] in FXN gene.
Spastic paraplegia (SPG)
ZFYVE26-related spastic paraplegia
A 16-year-old girl showed progressive ataxia, seizures, scoliosis, diffuse cerebellar atrophy, and hearing loss. This patient showed low muscle tone, absent reflexes in lower extremities, ataxia, and absent SNAPs. The patient harbored VUSs in ZFYVE26 gene [c.2300G>A (p.R767H)]; [c.2799C>T (p.L933 =)] that are likely disease-causing variants in ZFYVE26 based on clinical correlation suggesting SPG15 (OMIM# 612,012).
SPG7-related spastic paraplegia
A 17-year-old boy with scoliosis and gait abnormalities showed decreased lower extremities strength and reflexes with contractures, decreased vibration and temperature sensation. EDX showed absent SNAPs. MRI of brain/spine showed significant scoliosis. Genetic testing pointed towards SPG7 gene with a likely pathogenic variant [exon 1: c.1A>G] with a possible autosomal dominant inheritance confirming diagnosis of SPG7 (OMIM# 602,783) in correlation with clinical data.
Giant axonal neuropathy
GAN-related giant axonal neuropathy-1
A 6-year-old boy showed progressive gait abnormality, hammer toes, high arched feet bilaterally with 4/5 strength (MRC scale) in distal feet muscles, absent distal tendon reflexes in the lower extremities and intact in upper extremities. EMG findings were indicative of inherited moderate to severe sensory predominant polyneuropathy. MRI of brain showed evidence of symmetric signal abnormalities primarily involving the dentate nuclei of the cerebellum, deep cerebellar and cerebral white matter. Diminished N-acetylaspartate peak in MR spectroscopy suggested nonspecific neuronal dysfunction appearance but possible metabolic disorder such as a mitochondrial abnormality or an aminoaciduria among others. The patient was found to harbor a homozygous pathogenic variant [c.851 + 1G>A] in the GAN gene. Taken together, the clinical features, genetic results, and correlation with published evidence4 confirmed a giant axonal neuropathy-1.
Another 10-year-old girl harboring two pathogenic variants in a compound heterozygous manner in the GAN gene [c.805C>T (p.Arg269Trp)]; [c.732delT (p.Ile244MetfsX33)] showed wide based, high steppage gait with sensory ataxia with positive Rhomberg’s signs. The wide-based gait was worse with eyes closed or in the dark. Truncal ataxia was greater than appendicular ataxia. The subject showed decreased muscle bulk in the bilateral lower extremities and distal hands without any scapular winging and fast disease progression towards lower limb immobilization. Sensory evaluation revealed decreased vibration in joint position on pin prick in bilateral feet and absent deep tendon reflexes and flexor plantar responses. The patient also showed vocal cord paralysis, episodes of tachycardia, difficulty breathing, poor cough reflex, and difficulty in swallowing. Taken together, definitive molecular diagnosis confirmed giant axonal neuropathy-1.
Discussion
In our study, we retrospectively analyzed pure sensory polyneuropathy cases of genetic origin seen over a period of 7 years at a tertiary care Children’s hospital electrodiagnostic laboratory (Table 1) and performed an English literature search for each gene-specific disease type (Table 2). Although limited by the heterogeneity of different sequencing tests performed at physician discretion, thorough analyses of a single-center experience of childhood genetic sensory polyneuropathy are needed to understand the etiologies and variabilities of each disease subtype.
In our study, sensory polyneuropathy of genetic origin accounted for less than 2% of all pediatric electrodiagnostics performed and affected young children without gender preference. Average delay to genetic confirmation was 5 years. All children showed gait problems secondary to large fiber predominant sensory polyneuropathy with loss of joint position and positive Rhomberg, spasticity with or without cerebellar component and varying degrees of learning difficulties, scoliosis, epilepsy, swallowing difficulty, hearing loss and cardiomyopathy as co-morbidities. All study subjects had no response in SNAP’s. Therefore, we were unable to assess and quantify disease severity variabilities based on nerve conduction velocity (NCV) in NCS. However, on clinical examination the severity of neurodegeneration varied. In all subjects, we identified length-dependent abnormality with absence of sensory modalities like reduced vibration sense, joint position and reduced or absent tendon reflexes to be present in bilateral distal lower extremities more than the upper extremities. Clinically, however, on NCS both upper and lower extremities showed no response.
Mitochondrial genes were the most common genetic etiology in our cohort. Up to 1/3rd of mitochondrial disease patients may have a peripheral neuropathy but are often undiagnosed due to the severity of other symptoms82. In this report, we describe siblings with a novel COX20-associated disorder. COX20 codes for a protein that is involved in the assembly and stability of mitochondrial complex IV, the final component of the respiratory chain. Clinical features include childhood-onset progressive cerebellar ataxia, sensory neuropathy, hypotonia, areflexia, dysarthria. The natural history of this condition is emerging recently albeit from small number of cases27,28,29,30,83. Interestingly, the two siblings even with similar genotypes showed some heterogeneity in clinical presentations, such as only one of them showing initial respiratory distress, which agrees with the established mechanism of respiratory chain assembly intermediate accumulation causing reduced respiratory capacity in the absence of COX20 protein as shown in HEK293 cells84. Thus, it is possible that other genetic modifiers not identified in this study could drive the phenotypic differences. We also did not observe the severe cognitive or intellectual disabilities of COX20-related diseases typically reported in literature. We also identified a POLG-associated female patient showing ataxia, seizures, speech and motor delays, ophthalmic abnormalities, and ultimately succumbing to liver problems at age 20 years, a feature seen in POLG-associated Alper-Huttenlocher syndrome in neonates or children85. Two unrelated patients in our cohort were diagnosed with mitochondrial trifunctional protein (HADHA) deficiency. The majority of neuropathy cases described includes sensorimotor polyneuropathy with pure sensory neuropathy being a less represented subset of HADHA-associated neuropathies86,87,88. Their symptoms correlated well with that of long-chain 3-hydroxyl-CoA dehydrogenase deficiency (LCHADD) characterized by early-onset cardiomyopathy, hypoglycemia, neuropathy, pigmentary retinopathy, and sudden death—although the patients in our cohort survived. Previously, it was suggested that children born of a mother with severe fatty liver pregnancy should be genetically tested for LCHAD or MTP89. Interestingly, the patient with two identified HADHA variants showed cardiac aortic valve insufficiency but not cardiomyopathy per se as in patients with one identified HADHA variant. Moreover, the two HADHA patients show clinical heterogeneity that needs to be accounted for, such as patient# 2 who showed rhabdomyolysis, ophthalmic defects and seizures unlike patient# 3. Further testing on patient# 3 such as genome sequencing to identify possible deletion or duplication or deep-intronic variant that exome sequencing may not find needs to be done to identify the second HADHA variant. We also identified a fumarate hydratase (fumarase) deficiency case which showed severe weakness, generalized severe hypotonia, focal epilepsy and infantile spasms and developmental delay. Fumarase is an enzyme involved in the mitochondrial Kreb’s cycle, converting fumarate to malate and hence this subtype should be included in the mitochondrial sensory polyneuropathy group as it causes generalized muscle hypotonic and brain and developmental disorders. Interestingly, the patient showed cardiac symptoms such as pulmonary stenosis and pulmonary artery hypertension, and urinary tract infection which are generally not observed in fumarase deficient children, suggesting broader clinical spectrum of cardiac comorbidities.
Sensory polyneuropathy is well-described Friedreich ataxias (FA) feature90 although the precise pathogenesis is controversial and considered secondary to mitochondrial metabolism dysfunction91. We identified four FA cases with patients# 14 and 16 harboring the largest FXN trinucleotide repeat length and showing more severe phenotypes including cardiomyopathy, scoliosis, bladder dysfunction and developmental delay as was previously suggested61,64. It is also interesting to note that patient# 14, with a single allele repeat and a missense variant, showed slightly milder phenotypes, similar to previous findings66,67,68. Peripheral neuropathy is a predominating symptom in spinocerebellar ataxia 2 (SCA2) patients with>80% having abnormal nerve function. Spinocerebellar ataxias may range from motor or sensory neuropathies to a combination of both. Other studies have demonstrated pure sensory neuropathy being most prevalent among SCA392, but it was absent from our cohort. Ataxia telangiectasia is an autosomal recessive disorder affecting the neuromuscular- and the immune-system characterized by progressive cerebellar ataxia, oculomotor apraxia, choreoathetosis, oculocutaneous telangiectasia, frequent infections, and increased cancer risk with hematological malignancy being the most common93. Spastic paraplegias are a heterogeneous group of slowly progressive motor neuron disorders which presents frequently with lower limb spasticity and weakness94. Moreover, the SCA2 siblings age of disease onset and diagnosis were at 3 weeks which is, to our knowledge, the youngest cases reported so far being even younger than previously identified youngest case at 3 months95. Unlike reported symptoms in literature, the two siblings harboring ATXN2 pathogenic variants show signs of epilepsy and anti-seizure medication had to be given (patients 7 and 8 in Table 1). It is likely that ATXN2 aberration causes a brain developmental issue as reflected in the developmental delay in patient# 9 (Table 1) as well.
We identified two cases of spastic paraplegia (SPG), specifically SPG15 (ZFYVE26) and SPG7 (SPG7), with SPG15 showing a bigger and broader phenotypic onslaught compared to SPG7 in the form of seizures, hearing loss, and cerebellar atrophy. Among the giant axonal neuropathy (GAN) cases, we identified uncommon features not found in the literature survey, such as tachycardia and breathing difficulty in particular for patient# 20, making the clinical presentation of GAN broader.
Overall, pathophysiological follow up of childhood sensory polyneuropathies are critical as it could potentially be a marker for later onset neurodegenerative diseases. Our study shows the importance of peripheral nerve degeneration in sensory ataxia and peripheral nerve involvement in rare genetic polyneuropathies. Electrodiagnostic findings of peripheral sensory polyneuropathy in the appropriate clinical context mandates further genetic testing and subsequent clinical correlation. A genetic diagnosis is important as it provides a unifying diagnosis to coordinate care, prognostic and anticipatory guidance for clinicians (cardiology referral for suspect mitochondrial and Friedreich ataxia patients) and families (breast cancer risk in ATM carrier mothers), recurrence risks for family planning purposes, and possible additional treatment options.
Patients and methods
A parent or a legal guardian of all pediatric participants in this study provided written informed consent and the study was approved by the Children’s Healthcare of Atlanta (CHOA) and Emory University (EU) Ethics Committee and Institutional Review Board (CHOA IRB 13-151 and EU IRB00075815). All methods were carried out in accordance with relevant IRB guidelines and regulations. A retrospective chart review of the pediatric electrodiagnostic database at a tertiary care Children’s Hospital from 2013 to 2019 was performed. Children with absent sensory nerve action potentials (SNAPs) on nerve conduction studies (NCS) with remaining of the study being unremarkable were included. Clinical neuromuscular examination findings, genetic testing results and the clinical course of the above mentioned subjects were reviewed.
Board-certified pediatric neurologists performed the clinical examination and electrodiagnostic studies. Clinical examination focused on gait, motor strength, tendon reflexes, sensory modalities (pinprick, joint position, vibration and temperature) and Babinski and Romberg signs. SNAPs were recorded through antidromic electrode stimulation of sural, superficial fibular and plantar nerves using 15 mm disposable surface disc electrodes (part No. 019-415200; Natus Neurology, Middleton, WI) and on median and ulnar nerves (digits II and IV, respectively) using standard ring electrodes (part No. 9013S0332; Natus Neurology, Middleton, WI). Limb temperatures were kept between 28 and 32 °C and all data was collected using the Synergy software program (Natus Medical Incorporated, San Carlos, CA).
Genetic testing requests included PMP 22 deletion/ duplication, Friedreich ataxia trinucleotide repeats, spinocerebellar ataxia and whole exome sequencing (WES) or hereditary neuropathy gene-panel sequencing for patients with pure sensory polyneuropathy. Genetically confirmed cases (based on pathogenic variant identification and/or clinical correlation of the identified variants) were reviewed and variant pathogenicity was determined following American College of Medical Genetics and Genomics (ACMG) guidelines96. Disease-causing genes in cases of identification of variants of uncertain significance (VUSs) were determined by interpretation of most likely genetic cause based on clinical data correlation with genotype. Thereafter, an attempt was made to search and review similar published English literature cases.
Ethics standards
All informed consents were obtained and the study was performed according to Institutional Review Board protocol. We, the authors, confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Data availability
De-identified datasets will be available to other researchers from corresponding authors on reasonable request after publication as per Institutional Review Board protocol.
Abbreviations
- EDX:
-
Electrodiagnostic
- SNAPs:
-
Sensory nerve action potentials
- OMIM:
-
Online Mendelian inheritance of man (https://www.omim.org/)
- COX20 :
-
Cytochrome C oxidase assembly factor
- POLG :
-
Polymerase gamma
- HADHA :
-
Hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase
- FXN :
-
Frataxin
- ATXN2 :
-
Ataxin-2
- ATM :
-
Ataxia-telangiectasia mutated
- ZFYVE26 :
-
Zinc finger FYVE-type containing 26
- FH :
-
Fumarate hydratase
- SPG:
-
Spastic paraplegia
- SCA:
-
Spinocerebellar ataxia
- GAN:
-
Giant axonal neuropathy
References
Sladky, J. T. Neuropathy in childhood. Semin. Neurol. 7, 67–75. https://doi.org/10.1055/s-2008-1041407 (1987).
Mancuso, M. et al. “Mitochondrial neuropathies”: a survey from the large cohort of the Italian Network. Neuromusc. Disord. 26, 272–276. https://doi.org/10.1016/j.nmd.2016.02.008 (2016).
Yang, J. et al. Pure sensory Guillain-Barre syndrome: a case report and review of the literature. Exp. Ther. Med. 8, 1397–1401. https://doi.org/10.3892/etm.2014.1955 (2014).
Garg, M., Kulkarni, S. D., Hegde, A. U., Desai, M. & Sayed, R. J. Giant axonal neuropathy: clinical, radiological, and genetic features. Ann. Indian Acad. Neurol. 21, 304–308. https://doi.org/10.4103/aian.AIAN_82_18 (2018).
Tang, S. et al. Mitochondrial DNA polymerase gamma mutations: an ever expanding molecular and clinical spectrum. J. Med. Genet. 48, 669–681. https://doi.org/10.1136/jmedgenet-2011-100222 (2011).
Van Goethem, G., Dermaut, B., Lofgren, A., Martin, J. J. & Van Broeckhoven, C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 28, 211–212. https://doi.org/10.1038/90034 (2001).
Kollberg, G. et al. Low frequency of mtDNA point mutations in patients with PEO associated with POLG1 mutations. Eur. J. Hum. Genet. 13, 463–469. https://doi.org/10.1038/sj.ejhg.5201341 (2005).
Hudson, G. et al. POLG1, C10ORF2, and ANT1 mutations are uncommon in sporadic progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Neurology 66, 1439–1441. https://doi.org/10.1212/01.wnl.0000210486.32196.24 (2006).
Van Goethem, G. et al. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromusc. Disord. 13, 133–142. https://doi.org/10.1016/s0960-8966(02)00216-x (2003).
Winterthun, S. et al. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology 64, 1204–1208. https://doi.org/10.1212/01.WNL.0000156516.77696.5A (2005).
Gonzalez-Vioque, E. et al. Association of novel POLG mutations and multiple mitochondrial DNA deletions with variable clinical phenotypes in a Spanish population. Arch. Neurol. 63, 107–111. https://doi.org/10.1001/archneur.63.1.107 (2006).
Naviaux, R. K. & Nguyen, K. V. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann. Neurol. 55, 706–712. https://doi.org/10.1002/ana.20079 (2004).
Naviaux, R. K. & Nguyen, K. V. POLG mutations associated with Alpers syndrome and mitochondrial DNA depletion. Ann. Neurol. 58, 491. https://doi.org/10.1002/ana.20544 (2005).
Davidzon, G. et al. POLG mutations and Alpers syndrome. Ann. Neurol. 57, 921–923. https://doi.org/10.1002/ana.20498 (2005).
Ferrari, G. et al. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-gammaA. Brain 128, 723–731. https://doi.org/10.1093/brain/awh410 (2005).
Milone, M. & Massie, R. Polymerase gamma 1 mutations: clinical correlations. Neurologist 16, 84–91. https://doi.org/10.1097/NRL.0b013e3181c78a89 (2010).
Vissing, J. et al. Multiple mtDNA deletions with features of MNGIE. Neurology 59, 926–929. https://doi.org/10.1212/wnl.59.6.926 (2002).
Giordano, C. et al. Fatal congenital myopathy and gastrointestinal pseudo-obstruction due to POLG1 mutations. Neurology 72, 1103–1105. https://doi.org/10.1212/01.wnl.0000345002.47396.e1 (2009).
Phillips, J. et al. POLG mutations presenting as Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 24, 213–218. https://doi.org/10.1111/jns.12313 (2019).
Ibdah, J. A. et al. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N. Engl. J. Med. 340, 1723–1731. https://doi.org/10.1056/NEJM199906033402204 (1999).
Brackett, J. C. et al. Two alpha subunit donor splice site mutations cause human trifunctional protein deficiency. J. Clin. Investig. 95, 2076–2082. https://doi.org/10.1172/JCI117894 (1995).
Sims, H. F. et al. The molecular basis of pediatric long chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with maternal acute fatty liver of pregnancy. Proc. Natl. Acad. Sci. USA 92, 841–845. https://doi.org/10.1073/pnas.92.3.841 (1995).
Diebold, I. et al. HADHA and HADHB gene associated phenotypes: identification of rare variants in a patient cohort by Next Generation Sequencing. Mol. Cell Probes 44, 14–20. https://doi.org/10.1016/j.mcp.2019.01.003 (2019).
Bo, R. et al. Clinical and molecular investigation of 14 Japanese patients with complete TFP deficiency: a comparison with Caucasian cases. J. Hum. Genet. 62, 809–814. https://doi.org/10.1038/jhg.2017.52 (2017).
Kwiatkowska, J. et al. Clinical course and cardiovascular outcomes in patients with the long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Cardiol. J. 24, 101–104. https://doi.org/10.5603/CJ.2017.0009 (2017).
Llorca-Cardenosa, A., Catala-Mora, J., Garcia-Cazorla, A., Meavilla, S. & Castejon-Ponce, E. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: a case report. Arch. Soc. Esp. Oftalmol. 91, 236–239. https://doi.org/10.1016/j.oftal.2016.01.006 (2016).
Szklarczyk, R. et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum. Mol. Genet. 22, 656–667. https://doi.org/10.1093/hmg/dds473 (2013).
Doss, S. et al. Recessive dystonia-ataxia syndrome in a Turkish family caused by a COX20 (FAM36A) mutation. J. Neurol. 261, 207–212. https://doi.org/10.1007/s00415-013-7177-7 (2014).
Otero, M. G. et al. Novel pathogenic COX20 variants causing dysarthria, ataxia, and sensory neuropathy. Ann. Clin. Transl. Neurol. 6, 154–160. https://doi.org/10.1002/acn3.661 (2019).
Xu, H. et al. Observation of novel COX20 mutations related to autosomal recessive axonal neuropathy and static encephalopathy. Hum. Genet. 138, 749–756. https://doi.org/10.1007/s00439-019-02026-4 (2019).
Kerrigan, J. F., Aleck, K. A., Tarby, T. J., Bird, C. R. & Heidenreich, R. A. Fumaric aciduria: clinical and imaging features. Ann. Neurol. 47, 583–588 (2000).
Mroch, A. R., Laudenschlager, M. & Flanagan, J. D. Detection of a novel FH whole gene deletion in the propositus leading to subsequent prenatal diagnosis in a sibship with fumarase deficiency. Am. J. Med. Genet. A 158A, 155–158. https://doi.org/10.1002/ajmg.a.34344 (2012).
Bourgeron, T. et al. Mutation of the fumarase gene in two siblings with progressive encephalopathy and fumarase deficiency. J. Clin. Invest. 93, 2514–2518. https://doi.org/10.1172/JCI117261 (1994).
Zinn, A. B., Kerr, D. S. & Hoppel, C. L. Fumarase deficiency: a new cause of mitochondrial encephalomyopathy. N. Engl. J. Med. 315, 469–475. https://doi.org/10.1056/NEJM198608213150801 (1986).
Petrova-Benedict, R., Robinson, B. H., Stacey, T. E., Mistry, J. & Chalmers, R. A. Deficient fumarase activity in an infant with fumaricacidemia and its distribution between the different forms of the enzyme seen on isoelectric focusing. Am. J. Hum. Genet. 40, 257–266 (1987).
Snodgrass, P. J. Fumarase deficiency. N. Engl. J. Med. 316, 345–346 (1987).
Gellera, C. et al. Fumarase deficiency is an autosomal recessive encephalopathy affecting both the mitochondrial and the cytosolic enzymes. Neurology 40, 495–499. https://doi.org/10.1212/wnl.40.3_part_1.495 (1990).
Vara, R. et al. Fumarase deficiency associated with noninflammatory biliary atresia. J. Pediatr. Gastroenterol. Nutr. 58, e32-34. https://doi.org/10.1097/MPG.0b013e31826f7526 (2014).
Prasad, C., Napier, M. P., Rupar, C. A. & Prasad, C. Fumarase deficiency: a rare disorder on the crossroads of clinical and metabolic genetics, neurology and cancer. Clin. Dysmorphol. 26, 117–120. https://doi.org/10.1097/MCD.0000000000000148 (2017).
Ottolenghi, C. et al. Clinical and biochemical heterogeneity associated with fumarase deficiency. Hum. Mutat. 32, 1046–1052. https://doi.org/10.1002/humu.21534 (2011).
Deschauer, M. et al. Molecular and biochemical investigations in fumarase deficiency. Mol. Genet. Metab. 88, 146–152. https://doi.org/10.1016/j.ymgme.2006.01.007 (2006).
Maradin, M. et al. Fumaric aciduria: mild phenotype in a 8-year-old girl with novel mutations. J. Inherit. Metab. Dis. 29, 683. https://doi.org/10.1007/s10545-006-0321-0 (2006).
Remes, A. M., Rantala, H., Hiltunen, J. K., Leisti, J. & Ruokonen, A. Fumarase deficiency: two siblings with enlarged cerebral ventricles and polyhydramnios in utero. Pediatrics 89, 730–734 (1992).
Pulst, S. M. et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 14, 269–276. https://doi.org/10.1038/ng1196-269 (1996).
Cancel, G. et al. Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families. Hum. Mol. Genet. 6, 709–715. https://doi.org/10.1093/hmg/6.5.709 (1997).
Choudhry, S., Mukerji, M., Srivastava, A. K., Jain, S. & Brahmachari, S. K. CAG repeat instability at SCA2 locus: anchoring CAA interruptions and linked single nucleotide polymorphisms. Hum. Mol. Genet. 10, 2437–2446. https://doi.org/10.1093/hmg/10.21.2437 (2001).
Kasinathan, A., Saini, A. G., Sankhyan, N. & Singhi, P. Spinocerebellar ataxia type 2 with onset at toddlerhood. J. Clin. Neuromusc. Dis. 18, 244–245. https://doi.org/10.1097/CND.0000000000000161 (2017).
Figueroa, K. P. et al. Genetic variance in the spinocerebellar ataxia type 2 (ATXN2) gene in children with severe early onset obesity. PLoS ONE 4, e8280. https://doi.org/10.1371/journal.pone.0008280 (2009).
Gatti, R. A. Localizing the genes for ataxia-telangiectasia: a human model for inherited cancer susceptibility. Adv. Cancer Res. 56, 77–104. https://doi.org/10.1016/s0065-230x(08)60478-9 (1991).
Sedgwick, R. P. & Boder, E. Progressive ataxia in childhood with particular reference to ataxia-telangiectasia. Neurology 10, 705–715. https://doi.org/10.1212/wnl.10.7.705 (1960).
Byrd, P. J. et al. Mutations revealed by sequencing the 5’ half of the gene for ataxia telangiectasia. Hum. Mol. Genet. 5, 145–149. https://doi.org/10.1093/hmg/5.1.145 (1996).
Gilad, S. et al. Predominance of null mutations in ataxia-telangiectasia. Hum. Mol. Genet. 5, 433–439. https://doi.org/10.1093/hmg/5.4.433 (1996).
Taylor, A. M. Ataxia telangiectasia genes and predisposition to leukaemia, lymphoma and breast cancer. Br. J. Cancer 66, 5–9. https://doi.org/10.1038/bjc.1992.208 (1992).
Harding, A. E. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 104, 589–620. https://doi.org/10.1093/brain/104.3.589 (1981).
Finocchiaro, G., Baio, G., Micossi, P., Pozza, G. & di Donato, S. Glucose metabolism alterations in Friedreich’s ataxia. Neurology 38, 1292–1296. https://doi.org/10.1212/wnl.38.8.1292 (1988).
Campuzano, V. et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427. https://doi.org/10.1126/science.271.5254.1423 (1996).
Galea, C. A. et al. Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 79, 485–495. https://doi.org/10.1002/ana.24595 (2016).
Delatycki, M. B. et al. G130V, a common FRDA point mutation, appears to have arisen from a common founder. Hum. Genet. 105, 343–346. https://doi.org/10.1007/s004399900142 (1999).
De Castro, M. et al. Genotype and phenotype analysis of Friedreich’s ataxia compound heterozygous patients. Hum. Genet. 106, 86–92. https://doi.org/10.1007/s004399900201 (2000).
Schols, L. et al. Genetic background of apparently idiopathic sporadic cerebellar ataxia. Hum. Genet. 107, 132–137. https://doi.org/10.1007/s004390000346 (2000).
Filla, A. et al. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 59, 554–560 (1996).
Durr, A. et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N. Engl. J. Med. 335, 1169–1175. https://doi.org/10.1056/NEJM199610173351601 (1996).
Bidichandani, S. I., Ashizawa, T. & Patel, P. I. Atypical Friedreich ataxia caused by compound heterozygosity for a novel missense mutation and the GAA triplet-repeat expansion. Am. J. Hum. Genet. 60, 1251–1256 (1997).
Monros, E. et al. Phenotype correlation and intergenerational dynamics of the Friedreich ataxia GAA trinucleotide repeat. Am. J. Hum. Genet. 61, 101–110. https://doi.org/10.1086/513887 (1997).
Delatycki, M. B. et al. Clinical and genetic study of Friedreich ataxia in an Australian population. Am. J. Med. Genet. 87, 168–174. https://doi.org/10.1002/(sici)1096-8628(19991119)87:2%3c168::aid-ajmg8%3e3.0.co;2-2 (1999).
Clark, E., Strawser, C., Schadt, K. & Lynch, D. R. Identification of a novel missense mutation in Friedreich’s ataxia-FXN(W) (168R). Ann. Clin. Transl. Neurol. 6, 812–816. https://doi.org/10.1002/acn3.728 (2019).
Ygland, E. et al. Atypical Friedreich ataxia in patients with FXN p.R165P point mutation or comorbid hemochromatosis. Parkinsonism Relat. Disord. 20, 919–923. https://doi.org/10.1016/j.parkreldis.2014.04.018 (2014).
Clark, E., Butler, J. S., Isaacs, C. J., Napierala, M. & Lynch, D. R. Selected missense mutations impair frataxin processing in Friedreich ataxia. Ann. Clin. Transl. Neurol. 4, 575–584. https://doi.org/10.1002/acn3.433 (2017).
Kara, E. et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 139, 1904–1918. https://doi.org/10.1093/brain/aww111 (2016).
Boukhris, A. et al. Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin. Genet. 75, 527–536. https://doi.org/10.1111/j.1399-0004.2009.01176.x (2009).
Schule, R. et al. Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J. Neurol. Neurosurg. Psychiatry 80, 1402–1404. https://doi.org/10.1136/jnnp.2008.167528 (2009).
Pensato, V. et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 137, 1907–1920. https://doi.org/10.1093/brain/awu121 (2014).
Goizet, C. et al. SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology 73, 1111–1119. https://doi.org/10.1212/WNL.0b013e3181bacf59 (2009).
Hewamadduma, C. A. et al. Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations. Neurol. Genet. 4, e279. https://doi.org/10.1212/NXG.0000000000000279 (2018).
Choquet, K. et al. SPG7 mutations explain a significant proportion of French Canadian spastic ataxia cases. Eur. J. Hum. Genet. 24, 1016–1021. https://doi.org/10.1038/ejhg.2015.240 (2016).
Kuhlenbaumer, G. et al. Giant axonal neuropathy (GAN): case report and two novel mutations in the gigaxonin gene. Neurology 58, 1273–1276. https://doi.org/10.1212/wnl.58.8.1273 (2002).
Bruno, C. et al. Clinical and molecular findings in patients with giant axonal neuropathy (GAN). Neurology 62, 13–16. https://doi.org/10.1212/01.wnl.0000101676.41505.a7 (2004).
Zemmouri, R. et al. Charcot-Marie-Tooth 2-like presentation of an Algerian family with giant axonal neuropathy. Neuromusc. Disord. 10, 592–598. https://doi.org/10.1016/s0960-8966(00)00141-3 (2000).
Tazir, M. et al. Phenotypic variability in giant axonal neuropathy. Neuromusc. Disord. 19, 270–274. https://doi.org/10.1016/j.nmd.2009.01.011 (2009).
Buysse, K. et al. Giant axonal neuropathy caused by compound heterozygosity for a maternally inherited microdeletion and a paternal mutation within the GAN gene. Am. J. Med. Genet. A 152A, 2802–2804. https://doi.org/10.1002/ajmg.a.33508 (2010).
Normendez-Martinez, M. I. et al. Two novel mutations in the GAN gene causing giant axonal neuropathy. World J. Pediatr. 14, 298–304. https://doi.org/10.1007/s12519-018-0140-z (2018).
Finsterer, J. Mitochondrial neuropathy. Clin. Neurol. Neurosurg. 107, 181–186. https://doi.org/10.1016/j.clineuro.2004.07.001 (2005).
Lorenzi, I. et al. The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis. Biochim. Biophys. Acta https://doi.org/10.1016/j.bbamcr.2017.11.010 (2018).
Bourens, M., Boulet, A., Leary, S. C. & Barrientos, A. Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum. Mol. Genet. 23, 2901–2913. https://doi.org/10.1093/hmg/ddu003 (2014).
Rahman, S. & Copeland, W. C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 15, 40–52. https://doi.org/10.1038/s41582-018-0101-0 (2019).
Spiekerkoetter, U. et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J. Inherit. Metab. Dis. 32, 488–497. https://doi.org/10.1007/s10545-009-1125-9 (2009).
Hayes, B., Lynch, B., O’Keefe, M., Monavari, A. A. & Treacy, E. P. Long chain fatty acid oxidation defects in children: importance of detection and treatment options. Iran. J. Med. Sci. 176, 189–192. https://doi.org/10.1007/s11845-007-0025-y (2007).
Olpin, S. E. et al. Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J. Inherit. Metab. Dis. 28, 533–544. https://doi.org/10.1007/s10545-005-0533-8 (2005).
Isaacs, J. D. Jr. et al. Maternal acute fatty liver of pregnancy associated with fetal trifunctional protein deficiency: molecular characterization of a novel maternal mutant allele. Pediatr. Res. 40, 393–398. https://doi.org/10.1203/00006450-199609000-00005 (1996).
Morral, J. A., Davis, A. N., Qian, J., Gelman, B. B. & Koeppen, A. H. Pathology and pathogenesis of sensory neuropathy in Friedreich’s ataxia. Acta Neuropathol. 120, 97–108. https://doi.org/10.1007/s00401-010-0675-0 (2010).
Mulroy, E. et al. Peripheral nerve ultrasound in Friedreich ataxia. Muscle Nerve 57, 852–856. https://doi.org/10.1002/mus.26012 (2018).
Yadav, R. et al. Electrophysiological evaluation of spinocerebellar ataxias 1, 2 and 3. J. Neurol. Sci. 312, 142–145. https://doi.org/10.1016/j.jns.2011.07.051 (2012).
van Os, N. J. H. et al. Ataxia-telangiectasia: recommendations for multidisciplinary treatment. Dev. Med. Child. Neurol. 59, 680–689. https://doi.org/10.1111/dmcn.13424 (2017).
Salinas, S., Proukakis, C., Crosby, A. & Warner, T. T. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 7, 1127–1138. https://doi.org/10.1016/S1474-4422(08)70258-8 (2008).
Mao, R. et al. Childhood-onset ataxia: testing for large CAG-repeats in SCA2 and SCA7. Am. J. Med. Genet. 110, 338–345. https://doi.org/10.1002/ajmg.10467 (2002).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Acknowledgements
We thank the patients and their parents for their support and participation in this study. We thank L. Gloster for the final editing and proofreading of the manuscript.
Funding
This study was supported by Muscular Dystrophy Association (MDA) Development Grant and Children’s Healthcare of Atlanta pilot grant to Dr. Chakravorty.
Author information
Authors and Affiliations
Contributions
S.C. and S.V. designed the study, curated and analyzed data, and wrote the initial draft of the manuscript. R.R.L. and S.V. performed the clinical examination and NCS. Genetic testing results, genetically confirmed cases and clinical correlation of variants was performed and reviewed by experienced clinical geneticist and researcher S.C. and student researcher M.J. E., and board-certified medical genetic counselor R.L. All authors contributed to the retrospective curation of data and revising of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chakravorty, S., Logan, R., Elson, M.J. et al. Expanding the genotype–phenotype correlation of childhood sensory polyneuropathy of genetic origin. Sci Rep 10, 16184 (2020). https://doi.org/10.1038/s41598-020-73219-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73219-5
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
