
Abstract
Fighting smart diseases requires smart vaccines. Novel ways to present protective immunogenic peptide epitopes to human immune systems are needed. Herein, we focus on Self Assembling Protein Nanoparticles (SAPNs) as scaffolds/platforms for vaccine delivery that produce strong immune responses against Toxoplasma gondii in HLA supermotif, transgenic mice. Herein, we present a useful platform to present peptides that elicit CD4+, CD8+ T and B cell immune responses in a core architecture, formed by flagellin, administered in combination with TLR4 ligand-emulsion (GLA-SE) adjuvant. We demonstrate protection of HLA-A*11:01, HLA-A*02:01, and HLA-B*07:02 mice against toxoplasmosis by (i) this novel chimeric polypeptide, containing epitopes that elicit CD8+ T cells, CD4+ T helper cells, and IgG2b antibodies, and (ii) adjuvant activation of innate immune TLR4 and TLR5 pathways. HLA-A*11:01, HLA-A*02:01, and HLA-B*07:02q11 transgenic mouse splenocytes with peptides demonstrated predicted genetic restrictions. This creates a new paradigm-shifting vaccine approach to prevent toxoplasmosis, extendable to other diseases.
Similar content being viewed by others


Introduction
Toxoplasma gondii causes serious human illness. This apicomplexan, obligate, intracellular, parasite can infect all human cells, especially those of the brain and eye. Immunocompromised individuals, the fetus and newborn infant are most severely affected1. Severe active infection can cause death, respiratory failure as well as harm other organs, even in those without known immune compromise. Encephalitis and ophthalmologic disease cause substantial morbidity. Although anti-parasitic medicines, i.e., pyrimethamine and sulfadiazine are effective against tachyzoites, they do not eradicate dormant, encysted parasite forms1,2. Thus, one of the aspects of our study, is to address the gap of a substantial need to develop a potent and safe vaccine. This work builds on our foundation of novel work using a bioinformatics/immunosense/empiric approach with human cells and HLA transgenic mice3,4,5,6,7. We defined panels of octamer/nonamer peptides that bind to major HLA Class I supermotifs. IFN-γ producing CD8+ T cells specific to these peptides were detected in individuals with three Human Leukocyte Antigen (HLA) supermotifs (HLA supertype A03, A02, and B07) present in large proportions of the world population. We used peptides that bind to a subset of these HLA alleles to immunize HLA supermotif transgenic mice as a proof of principle3,4,5,7. These pooled peptides when given with the TLR4 agonist adjuvant, GLA-SE (created by The Infectious Diseases Research Institute, IDRI) are able to produce protective memory CD8+ T lymphocytes that reduce the amounts of T. gondii in specific HLA transgenic mice. Therefore, we utilized GLA-SE as one of our adjuvants8,9,10. GLA-SE has an excellent pre-clinical track record and is in development through human clinical trials including for malignancies, viral, protozoan, and bacterial infections. Our previous work has demonstrated a superior effect of GLA-SE compared to ALUM6,11, when added to peptides eliciting cell mediated responses (CD8+ and CD4+ T lymphocytes) that protect against Toxoplasma. Our hypothesis is to create a safe and potent vaccine based on our preliminary immunosense and Self Assembling Protein Nanoparticle (SAPN) data, both with Toxoplasma infections and malaria11,12,13. We then embarked on engineering the peptides into SAPNs. These proteins serve as vaccine core platforms and have the ability to present immunogenic pathogen fragments to the host’s immune system (patent US8575110 B213,14,15). These include CD8+, CD4+ T-, and B cells to promote strong cellular and humoral responses12,16,17. Because of their size and shape, they reach and are processed in follicular dendritic cells. They are flexible in design, and easy to produce rapidly. In addition, they do not present the risks of live attenuated vaccine strains. In our recent work (Prototype 1, Fig. 1a), we have engineered a SAPN that contains the T. gondii B07 binding epitope of the dense granule protein GRA720–28 (LPQFATAAT) and an universal CD4+ T cell eliciting epitope (PADRE)12. Immunization of HLA-B*07:02 mice with these SAPNs, induced strong CD8+ T cell-dependent protective immunity against Type I and Type II T. gondii parasites, although protection is not complete. These findings highlight the development of a safe and effective T cell epitope-based toxoplasmosis vaccine. Furthermore, we developed a novel type of SAPN, called Prototype 2, that contains five HLA-A*11:01-restricted CD8+ T cell epitopes (Fig. 1a). This Prototype 2 incorporates the TLR5 agonist flagellin as a a scaffold and as an immunopotentiator to make a self-adjuvanting SAPN11 (patent application EP1415060018). This multiepitope construct has been shown to induce IFN-γ and protect against type II parasites in HLA-A*11:01 mice, demonstrating the ability of the SAPNs to improve vaccine potency of CD8-based immunization approaches11.

(a) Prototype constructs used for SAPN studies. The core particle of different SAPN constructs used for vaccine studies against toxoplasmosis was composed of the pentameric (green) and trimeric (blue) coiled coils. Attached to the core are the TLR5 agonist flagellin (purple) and the B cell epitope MIC1 (red) and depending on the particular construct the CD8+ epitopes (orange) and CD4+ epitopes (magenta) are either engineered into flagellin or the trimeric coiled coil or attached to the N-terminal end of the protein chain. Prototype 1 (P1)12 incorporates the T. gondii B07 epitope LPQFATAAT (GRA720–28) and an universal CD4+ T-cell epitope (derived from PADRE) into the SAPN. Prototype 2 (P2)11 incorporates five HLA-A*11:01-restricted CD8+ epitopes KSFKDILPK(SAG1224-232), STFWPCLLR(SAG2C13-21), AVVSLLRLLK(GRA589-98), SSAYVFSVK(SRS52A250-258), and AMLTAFFLR(GRA6164-172), and the universal CD4+ T-cell epitope. All are integrated into a flagellin sequence as a component of the nanoparticle to make it a self-adjuvanted SAPN. Prototype 3 (P3) (design construct of the current study), in addition to the five HLA-A*11:01-restricted CD8+ epitopes and PADRE, the B cell MIC1 protein, 4 HLA-A*02:01-restricted CD8+ epitopes, and one HLA-B*07:01-restricted CD8+ epitope are attached to the N-terminal end of the protein chain. (b) Gene cloning of nanoparticle proteins. The polypeptide was cloned between the NdeI/EcoRI restriction sites in pET23b vector (Novagen) and yield the following sequence (see more details in the “Materials and methods”).
Here we present the designs and immunological profiling of a novel SAPN containing five CD8+ T cell eliciting HLA-A*11:01 binding protective epitopes, one CD8+ HLA-B*07:02, four CD8+ HLA- A*02:01, a pan-allelic CD4 epitope, and a MIC1 B cell epitope to yield the SAPN prototype called “ToxAll” (prototype 3, Fig. 1a). In addition to the introduction of several epitopes from three HLA supermotifs, another new aspect of our recent SAPN presented herein is the introduction of MIC1 as a B cell epitope (Fig. 1b). MIC1 was chosen because of it’s involvement in the interactions of the parasite with its host cell receptor during the early stages of host cell attachment, invasion, virulence and pathogenicity19. Hence, our study of ToxAll demonstrated the ability to elicit antibodies against the B cell epitope MIC1 in a humoral immune response and at the same time it delivered the specific HLA supermotif -restricted CD8+ and CD4+ epitopes to elicit a cellular immune response.
Results
Generation of SAPN nanoparticles
Herein, we develop tools to improve our vaccine platform using a structure-based “plug and play” approach to the SAPN. We incorporated five CD8+ HLA-A*11:01 protective epitopes, one CD8+ HLA-B*07:02, four CD8+ HLA-A*02:01, a pan-allelic CD4+ epitope and a MIC1 B-cell epitope to yield the SAPN prototype called “ToxAll”. Figure 1b represents the sequence of different epitopes separated by proteasomal cleavage sites. We used the properties of trimeric and pentameric coiled coils sequences to plug and assemble the protective epitopes into a particle (Fig. 2a). HLA-A*11:01 CD8+ epitopes (gold) are engineered into flagellin. The CD4+ T cell eliciting epitope is engineered into the trimeric coiled coil. HLA-*A02:01, HLA-B*07:02, and CD4+ T cell eliciting epitopes are attached to the N-terminal end of the protein chain, while B-cell eliciting epitope, MIC1 appears to be hidden in the assembled particle. In a first step of our design, we attached the MIC1 protein to the N-terminal end of the protein chain of the SAPN (Fig. 2a). This MIC1 protein is presented on the tip of the pentameric coiled coil and displayed in a repetitive manner on the surface of the SAPN. The surface of the MIC1 protein that interacts with the cell receptor was exposed on the surface of the nanoparticles, thus inducing production of antibodies against the interaction site of MIC1. In a second step, we combined MIC1 SAPN with the prototype that carries the five HLA-A*11:01 epitopes, one HLA-B*07:02 and four HLA-A*02:01 T. gondii CD8+ T cell eliciting epitopes, the CD4+ T cell eliciting epitope PADRE intercalated into the flagellin portion of the design (Fig. 2a).

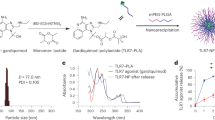
(a) Computer Model of ToxAll. The core particle is composed of the pentameric and trimeric coiled coils. They are shown in green and blue, respectively. Attached to the trimeric coiled coil is the TLR5 agonist flagellin (purple) while the B cell epitope MIC1 (red) is attached to the pentameric coiled coil. The HLA-A*11:01 CD8+ epitopes (gold) are engineered into flagellin, the CD4+ epitope is engineered into the trimeric coiled coil, the HLA-A*02:01 and HLA-B*07:01 CD8+ epitopes are attached to the N-terminal end of the protein chain. While the B-cell epitope MIC1 appears to be hidden in the assembled particle, the structures of the pentamer and trimer reveal that there is ample void space that allows antibodies to bind to MIC1 in the assembled structure. (b) Biochemical and Biophysical Analysis. Part (A) SDS-PAGE of the purified construct. Part (B) Transmission electron microscopy of ToxAll showing relatively uniform and non-aggregating nanoparticles. The bar corresponds to 100 nm. Part (C) DLS size distribution analysis of ToxAll showing an average nanoparticle size of ~ 40 nm.
The ToxAll construct was expressed in E. coli. It was purified and folded to form nanoparticles as described in the “Materials and methods”. Figure 2b shows the molecular size of the protein ~ 75 kDa in Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with a uniform and non aggregate distribution of particles ~ 40 nm in diameter.
SAPN that contains three HLA-A*02:01, one HLA-B*07:02, five HLA-A*11:01 CD8+ T cell eliciting epitopes, and the pan-allelic CD4+ T cell eliciting epitope PADRE, confers protection against Toxoplasma
The mice were immunized with either ToxAll with GLA-SE, Empty-SAPN with GLA-SE, or saline following the design presented in Fig. 3a. Immunogenicity was compared using magnitude of IFN-γ secretion from mouse splenocytes of HLA-A*11:01, HLA-A*02:01, and HLA-B*07:02. Figures 3, 4 and 5 show ELISpot experiment with IFN-γ spot formation from splenocytes, which were tested using a pool of HLA-A*11:01 or HLA-A*02:01 or HLA-B*07:02 restricted CD8+ T cell epitopes with or without PADRE and some mismatched HLA Class I restricted peptides as controls. Our data show that ToxAll elicits CD8+ T cells that respond to HLA-A*02:01, HLA-A*11:01, and HLA-B*07:02 binding constituent peptides and can present such antigens in a MHC restricted manner and CD4+ T cells that respond to PADRE. To test the properties of this SAPN in vivo, mice were challenged two weeks after the last immunization with ToxAll using 2000 Me49-Fluc, type II strain, T. gondii. Twenty-one days after the challenge, brains from these mice were removed and imaged using a Xenogen in vivo imaging system. As shown in Figs. 3d, 4c, and 5c, numbers of luciferase expressing parasites in brains of HLA-A*11:01, HLA-A*02:01, and HLA-B*07:02 transgenic mice immunized with ToxAll + GLA-SE were less (p < 0.05) in mice that received Empty-SAPN or PBS as controls. These correlate with the reduction of number of cysts per brain (Figs. 3f, 4e, and 5e). For experiments with two groups, between-group comparisons were made using the nonparametric Mann–Whitney test. For experiments with three or more groups, data were initially compared by the nonparametric Kruskal–Wallis test. If results from that were statistically significant, then relevant pairwise comparisons were made using GraphPad Prism 7 software (GraphPad Software, San Diego, CA). Results are expressed as the mean ± SD and considered statistically significant at p < 0.05.

(a) Experimental design of Immunization of HLA-A*11:01 (and other) transgenic mice. (b, c) IFN-γ- producing T cells were measured by ELISpot assay. Mouse splenocytes were stimulated with pooled HLA-A*11:01 restricted CD8+ T cell epitopes (A03; HLA-A03 supertype allele of HLA-A*11:01) with or without PADRE. In some experiments as in the photograph of the well in b HLA-A*02:01 restricted CD8+ T cell peptides were used. There were 2 mice of the 6 in the Tox-All GLA-SE group that had splenocytes that had some response to these HLA-A*02:01 restricted peptides in contrast to the well shown in (b) (data not shown in photograph or graph of ELISpot data). There were 3 wells of splenocytes for each mouse. A mean value was determined for each mouse. (b,c) Present the data of the IFN-γ spot formation from 2 separate experiments combined. The numbers of mice were as shown in the figure with a total n = 4–6 mice per each group of controls or peptides utilized in vitro. Each symbol represents IFN-γ secretion for one mouse. The horizontal line represents the mean of these IFN-γ spot formation determinations. (d,e) Luciferase expression was reduced in the brains of HLA-A*11:01 mice that had been immunized with ToxAll plus GLA-SE. This was measured after challenge with T. gondii ME49-Fluc (Type II) expressing luciferase. Mice (n = 5 per experiment) were immunized, and challenged intraperitoneally 14 days after the last boost, with 2000 Type II (Me49-Fluc) parasites. Imaging was performed with the in vivo IVIS imaging system ( Xenogen, Alameda, CA) with surviving mice. The experiment was performed twice beginning with 5 mice per group. The numbers of mice that survived to the 21st day when parasite burden as luminescence or (f) cyst number in brain were determined. (f) enumeration of cysts in mouse brain. Brains from these mice were collected and resuspended in saline and tissue cysts were counted using an optical microscope.

Protective immunity against toxoplasmosis induced by 4 pooled HLA-A*02:01 restricted CD8+ epitopes in HLA-A*02:01 mice. (a,b) IFN-γ producing T cells were measured by ELISpot assay,in the same manner as described in the legend of Fig. 3. Splenocytes from ToxAll plus GLASE and EMPTY SAPN plus GLA-SE immunized mice were stimulated with a pool of 4 HLA-A*02:01 restricted CD8+ T cell peptides in the presence or absence of PADRE, or PADRE alone. PBS group was used as a control. There was a trend toward a small response of splenocytes to HLA-A*02.01 restricted CD8 + T cell epitopes compared to PBS but it did not achieve statistical significance. The response in both groups with PADRE was significant. (c,d) T. gondii luciferase expression in brains of HLA-A*02:01 immunized mice with ToxAll plus GLA-SE was significantly reduced compared to group mice control (PBS). (e) Brains show the reduction of number of cysts in HLA-A*02:01 immunized mice compared to the control group.

ToxAll elicits B07 epitope (GRA720–28, LPQFATAAT) specific immune response in HLA-B*07:02 mice. (a,b) show IFN-γ -producing T cells from splenocytes of HLA-B*07:02 mice immunized with ToxAll plus GLA-SE compared to Empty plus GLA-SE adjuvant. Data show the specificity of splenocytes from HLA-B*07:02 immunized mice with adjuvanted ToxAll to B07 epitope and not to the pool of HLA-A*02:01 restricted CD8+ peptides. The absence of any cross reactivity of HLA haplotypes demonstrates the specificity of that peptide HLA interaction and absence of any contribution of the heterologous combination. (c,d) Luciferase expression from brains of HLA-B*07:02 mice was significantly reduced in ToxAll immunized mice compared to control Empty-SAPN. (d) This reduction in luminescence for the ToxAll SAPN immunized transgenic mice correlates well with the reduction of number of cysts per brain counted shown in e (p = 1.0 because of one outlier in the control group with a much higher number of cysts but the trend show substantial reduction in cyst numbers for all mice by immunization with the ToxAll SAPN administered with GLA-SE).
ToxAll induces humoral responses
To determine whether ToxAll + GLA-SE elicits humoral responses by production of MIC1 antibodies, western blot analysis with sera, from immunized HLA-A*11:01, HLA-A*02:01, and HLA-B*07:02 transgenic mice was performed using total soluble antigen (STAg) fraction T. gondii from ME49 strains. As shown in Fig. 6a, the sera of the 3 immunized transgenic mice reacted only with the 60–70 kDa bands of STAg. The identity of these proteins were identified as MIC1 proteins by mass spectrometry analysis (data not shown). We performed also Immuno Fluorescence Assay (IFA) using sera from MIC1 immunized transgenic mice with infected fibroblasts and as a control, an anti MIC1 antibody. In comparison to the MIC1 antibodies (from a MIC1 complete sequence, used as a control), our data with the SAPN immune sera do not show a staining at the surface of the parasite, as described earlier20. This is consistent with the size of MIC1 plugged in ToxAll (Fig. 6b).

Antibody production. (a) Western blot analysis using protein extracts from ME49 strains with three sera from immunized HLA-A*11:01, HLA-A*02:01 mice, and HLA-B*07:02 that recognize MIC1 proteins. (b) Immuno Fluorescence Assay (IFA) using sera containing MIC1 from immunized transgenic mice in infected fibroblasts right panel. In comparison to MIC1 antibodies (from a MIC1 complete sequence used as a control), our data from the SAPN immunized mice with the truncated MIC sequence, do not show a staining at the surface of the parasite. This is consistent with the size of MIC1 plugged in ToxAll. (c) Detection of the levels of T. gondii specific antibodies and their subclass IgG1 and IgG2b. Serum samples were collected 10 days after the last immunization and antibodies level were tested by ELISA. Humoral responses were assessed by reading the mean of the optical density at 450 nm (OD450) ± SD values.
Quantitation of levels of T. gondii specific antibodies and their subclass IgG1 and IgG2b
In order to determine the relative levels of IgG1 and IgG2a subclass reacting with T. gondii antigens, serum samples were collected from both immunized and non-immunized mice 10 days after the last booster immunization, and detected by ELISA. Compared with the control groups, there are higher levels of IgG and IgG2b antibodies in the sera from immunized mice (Fig. 6c). However, the level of IgG1 antibodies was not changed from immunized mice compared to the control group (Fig. 6c).
Interaction of SAPN with lysosomal enzymes
To understand the way in which the ToxAll processing and presentation is able to deliver T cell immune responses and protection described above, we analyzed in vitro bone marrow dendritic cells (BMDDCs) by electron microscopy. BMDDCs from HLA-A*02:01 transgenic mice were incubated for twenty-four hours with either ToxAll alone, ToxAll with GLA-SE or GLA-SE alone. Results indicate that while GLA-SE is processed at the plasma membrane (consistent with the interaction of the adjuvant to TLR4) (Fig. 7a), SAPNs plus GLA-SE interacts with lysosomal enzymes (Fig. 7c). There is a remarkable, homogeneous distribution and loading of antigenic peptides within the lysosome when SAPNs were added to the adjuvant and emulsion (Fig. 7c).

TEM images. After 24 h co-culture of bone marrow- derived dendritic cells (BMDDCs) from HLA-A*02:01 transgenic mice incubated with (a) GLA-SE alone, (b) ToxAll alone or (c) ToxAll with GLA-SE. GLA-SE present in particles mostly close to cell membranes with a size of ~ 100 nm (blue arrows). When incubated with ToxAll alone, lysosome of BMDDCs didn’t show any processing at 24 h. ToxAll plus GLA-SE interacts with lysosomal enzymes and shows a remarkably homogeneous distribution of particles in the lysosome. The red arrows indicate clusters of gold-encapsulated SAPN.
Discussion
Herein, we engineered the multi-epitope toxoplasmosis nanovaccine ToxAll based on the SAPN concept. It contains five HLA-A*11:01 restricted CD8+ T cell eliciting epitopes, one HLA-B*07:02 restricted CD8+ T cell eliciting epitope, four HLA-A*02:01 restricted CD8+ T cell eliciting epitope, pan-allelic CD4+ T cell eliciting epitope for MHC II haplotypes, the TLR5 agonist flagellin, as well as the fully folded protein domain of MIC1 as a B cell epitope. This forms a SAPN that induced a combination of humoral immune stimulation, as well as cell mediated immune responses.
This construct is built based on Prototype 2 (Please see Fig. 1), which incorporates the TLR5 agonist flagellin as a component of the nanoparticle to make it a self-adjuvanting SAPN11. We found earlier that Prototype 2 elicits CD8+ T cells that respond to HLA-A*11:01 binding constituent peptides, and CD4+ T cells that respond to PADRE. This particular SAPN architecture contains TLR5 agonist flagellin as a scaffold and as an immunopotentiator (patent application EP1415060018) and these SAPN tritiate a robust innate immune response. This innate immune response is elicited through activation of the TLR5 pathway11. In the current construct, we combine HLA A02 and B07 supertypes from our earlier work3,4,5,6 to make a chimeric novel polypeptide with HLA-A*11:01 restricted CD8+ and a new B cell epitope microneme protein, MIC1. We also used our CD4+ T cell eliciting universal, but not parasite specific epitope, PADRE. We attached hydrophobic CD8+ T cell eliciting epitopes at the N-terminal end of the novel chimeric SAPN protein sequence so it would be located in the interior of the protein shell of the particle. Such location is preferred as it is predicted to avoid hydrophobic aggregation between particles in solution, similar to the hydrophobic core of correctly folded proteins. We had earlier unsuccessfully attempted to create linked HLA-A*02:01 restricted peptides as linear constructs as we did with HLA-A*11:01 peptides6 (data not shown).
Even though this final SAPN design looks rather complicated, herein we have successfully engineered a SAPN that combines MIC1, CD8+, CD4+ -T cell eliciting epitopes and flagellin in one single protein chain. This provides a framework that could be utilized to create other similar chimeric polypeptides including other immunogenic peptides and proteins which perhaps have potential to be multiplexed. This could broaden human population coverage still further and to protect against other organisms or diseases where similar immune responses are critical. We then could test them in these or other HLA transgenic mice where genetic restriction for those haplotypes would be operative.
All epitopes were flanked at the carboxy-terminus by N/KAAA spacers. These spacers lead to immunogenic processing that is optimal6. In separate earlier studies, we found that processing and presentation of five HLA-A*11:01 epitopes assembled with N/K alanine linkers in a single polypeptide (called LO poly-epitope) stimulated human cells occur with higher efficiency compared to individual peptides6, and conferred greater protection than the individual peptides did, or did a different combination of peptides with another linker (GPGPG)6. In the HLA-B*07:02, absence of any cross reactivity of HLA-A*02 haplotype peptide while HLA-B*07:02 is present demonstrates the specificity of that peptide HLA- B*07:02 interaction and absence of any contribution of the heterologous combination (Fig. 5). We had demonstrated earlier6 and again herein in Fig. 3 that the construct without its contents called “empty” herein had no difference from PBS in immunogenicity measured as stimulation in vitro by peptides.
The data show that PADRE clearly elicits a very robust response as an universal CD4+ T cell eliciting peptide known to stimulate CD4+ T cells strongly. Even with this strong response there is no parasite specific response, nor would one be expected, as PADRE is not present in T. gondii. PADRE is helpful in priming the initial response but there would be no recall in a natural infection. Thus, one component lacking in the final design is a Toxoplasma- epitope specific for HLA Class II alleles to elicit CD4+ T cells. We have embarked recently on the selection of these candidates from T. gondii that will be part of a final construct (data not shown).
These findings are important conceptually, especially because they show why this chimeric construct that we are building is in total more than the sum of its parts and this is proven in the work herein. This is significant for breadth of population coverage and for specificity of response matching peptide binding data and haplotype specific immune responses.
The results herein and in our earlier work have shown some variations in responsiveness even to the same peptides in terms of robustness at different times. In Figs. 3, 4, and 5, the scale of the vertical axis that shows very robust response to PADRE, makes these differences appear more pronounced. Although the response to PADRE is very robust and is important for stimulating IL2 production, which stimulates CD8+ T cells during the prime and boost immunizations, PADRE is not a T. gondii derived peptide. Since PADRE is not homologous, it will not drive a memory recall response relevant to T. gondii infection.
Additionally, the data with PADRE are important in the proof of principle that an epitope that elicits CD4+ T cells can be presented in this SAPN vaccine and elicits a strong response. The responses to the epitopes that elicit CD8+ T cells were not always robust compared to PADRE. Nonetheless, the genetic restriction demonstrated is critical for this proof of principle study. We had noted earlier that the linkers and arrangement of the peptides in the HLA-A*11:01 construct made the response greater than the 5 peptides separately in a linear construct and we retained that arrangement6. We had been unable to make a linear construct that folded the HLA-A*02:01 bound peptides. We selected an arrangement that allowed those hydrophobic HLA-A*02:01 epitopes to be on the inside face of the SAPN. Even more important than the magnitude of response that can vary or be made stronger by additional copies or linkers6, peptides bound by each HLA haplotype do indeed elicit a response specifically for that haplotype. This is the proof that homologous peptide present in the SAPN elicits the appropriate HLA-restricted T cell response, although in this study the HLA-02 restricted response is modest, along with the MIC protein being able to elicit antibody.
MIC1 has a tandemly duplicated domain related to thrombospondin-1-like domain of thrombospondin that binds to or alters host cell receptors21. Homology of these MIC1 containing domains were found in P. falciparum circumsporozoite protein (PfCSP). When these domains of PfCSP were used in SAPNs they elicited antibodies and protected against P. falciparum, suggesting that MIC1 may be useful in protection against T. gondii. A SAPN vaccine has been created for malaria. This vaccine for this disease caused by the related apicomplexan plasmodial parasites, was created to immunize with PfCSP derived epitopes eliciting T and B cells13,22. Moreover, the crystal structure of MIC1 protein has been solved23. This crystal structure allows the design of specific vaccine components that will induce immune responses that interfere with/impair parasite interactions with host cells; i.e. antibody raised against the surface of MIC1 that interacts with the host receptor will impair parasite interactions dependent on MIC1 with host cells. We have placed MIC1 in our SAPN in order to make an antibody with potential to contribute to blocking the interaction of the parasite with the host24. MIC proteins recently were used as recombinant vaccines that conferred some protection25. Although ToxAll induces humoral responses, sera containing MIC1 from immunized HLA-A*11:01, HLA-A*02:01, and HLA-B*07:02 transgenic mice do not block parasite invasion into fibroblasts (data not shown). MIC1, MIC4 and MIC6 complex has been shown to be critical in invasion26. Thus an antibody to these MIC proteins might contribute to blocking invasion of the host cells as did antibodies critical to invasion such SAG127, and AMA-RON28.
Interestingly, ToxAll plus GLA-SE interacts with lysosomal enzymes (Fig. 7c). There is a remarkable, homogeneous distribution and accumulation of antigenic peptides within the lysozome when SAPNs were added to the adjuvant and emulsion. The results for this localization and pattern are consistent with a similar study performed earlier with Plasmodium falciparum29. It will be important to study the specific kinetics of the processing of ToxAll through BMDDCs. Specifically, and in addition with TEM, confocal microscopy could be examined to support the cleavage of the peptides within the ToxAll SAPN processing during the twenty-four hours. This would contrast efficiency of processing of ToxAll + GLA-SE with ToxAll alone in different organelles (e.g. endoplasmic reticulum, endosome/lysosome).
Additionally, our SAPN contains flagellin. It is expected that our new SAPN design, with flagellin in its scaffold will retain TLR5 activity. In ongoing separate work using another immunization platform30, we included the sequence for ToxAll in a RNA replicon. In this system we found that flagellin encoded in this replicon construct does stimulate TLR5.
Herein, challenges with Me49 type II parasites in HLA-A*11:01, HLA-A*02:01, and HLA-B*07:02 mice demonstrated reduction (87%) from a mean of 1550 to 200 cysts per brain. This correlates with our previous studies that showed also reduction of cysts in brains of mice6,11,12, but not as robustly as our ToxAll. However, none of these vaccine regimens provided complete protection. More peptides will be needed for sufficient population coverage, and possibly multiple copies with these peptides, or to elicit a still more robust response. Nonetheless, these experiments demonstrate that the overall structure can present peptides and proteins to the appropriate compartments to elicit a protective immune response. These critical concepts are illustrated by the data in Figs. 3, 4, 5 and 6 and shape the approaches in our next steps in development of a vaccine.
Conclusion
To our knowledge, the ToxAll presented here represents the first SAPN for the delivery of CD4+, CD8+ T cell eliciting and B cell eliciting epitopes with testing in different HLA transgenic mice.
First, we investigated the folding and the size of the SAPNs by DLS size distribution and electron microscopy analysis. We assessed the functionality of the SAPNs by mixing them with GLA-SE adjuvant. GLA-SE has been recently used extensively in human clinical trials. GLA-SE is one of the oil-in-water (o/w) emulsions developed at IDRI. Emulsion droplets are ~ 100 nm in diameter and are stable for years. This IDRI emulsion formulation is similar to formulations already approved in Europe for influenza vaccines, MF59 and AS03, but in addition contains TLR4 ligand GLA. Emulsions like these effectively and safely induce immune responses to influenza antigens, including enabling dose sparing31,32,33. The mice were immunized with either ToxAll with the adjuvant GLA-SE, Empty-SAPN with GLA-SE, GLA-SE only, or PBS. First, we used PBS as a control in these studies in HLA-A*11:01 mice. Immunogenicity was compared using the magnitude of IFN-γ production by mouse splenocytes ex vivo. We showed that IFN-γ secretion is high in mice immunized with ToxAll plus GLA-SE stimulated by either a pool of peptides or PADRE. To evaluate whether immunization with ToxAll confers any protection against the Type II strain of T. gondii (Me49-Fluc), brains of immunized mice were imaged using a Xenogen in vivo imaging system. We showed that immunized HLA-A*11:01 transgenic mice that received ToxAll had significantly reduced parasite numbers in their brains compared to mice that received the control Empty-SAPN, adjuvant alone, or PBS. This is consistent with the reduction of cysts in the mice immunized with ToxAll (p < 0.05). Thus, our multi-epitope ToxAll elicited protection quantitated as reduction of parasite cyst burden and enhanced survival when administered with GLA-SE laying the foundation for creating a novel type of nanovaccine, to reduce or eliminate initial T. gondii parasite burden.
Understanding the mechanisms whereby CD4+ and CD8+ T cell, and B cell restricted epitopes are associated with protection of HLA-A*11:01, HLA-A*02:01, and HLA-B*07:02 mice against toxoplasmosis, underline the role of the ToxAll scaffold in the rational design of a T cell- epitope based vaccine strategy. This represents a novel approach to generate host neutralizing antibodies against different parasites and in other diseases.
Materials and methods
Cloning ToxAll gene
NdeI/EcoRI restriction sites were used to clone the polypeptide in pET23b vector (Novagen). ToxAll has the sequence shown in Fig. 1b as follows: A his-tag sequence (black); 4 HLA-A*02:01 restricted CD8+ epitopes FMGVLVNSL (GRA629-37), ITMGSLFFV (SRS52A12-20), FMIVSISLV (SAG2X351-360), GLAAAVVAV(SPA82-90) (gold); one HLA-B*07:02, LPQFATAAT (GRA720-28) (gold); the B cell epitope microneme protein 1 (MIC1) of T. gondii p89 (red); the pentameric coiled coil (green); within the trimeric coiled coil (blue), pan-allelic CD4+ epitope string (magenta); five HLA-A*11:01 restricted CD8+ T cell epitopes represented in (gold) AVVSLLRLLK(GRA589-98), AMLTAFFLR(GRA6164-172), KSFKDILPK(SAG1224-232), STFWPCLLR(SAG2C13-21), SSAYVFSVK(SRS52A250-258). These epitopes were placed into the domain D2 and D3 of the flagellin sequence((purple) of Salmonella enterica. This flagellin has the structure that has pdb-code 3V4734. This is from RCSB protein data bank.
A control construct, designed as empty vector contains all the peptide sequences except HLA restricted CD8+, CD4+ eliciting and MIC1 epitopes.
Protein expression, purification, refolding, and analysis of the nanoparticle polypeptide
The construct called ToxAll was expressed in E. coli BL21 (DE3) cells as described in our previous work 3,5. Expression clones were grown at 37 °C in Luria broth media with ampicillin. Protein expression was induced by the Isopropyl β-D-1-thiogalacto-pyranoside (IPTG) to an OD600 of ~ 0.8. After induction at ~ 4 h, cells were removed from 37 °C and centrifuged at 4000×g. Cell pellet was stored at − 80 °C. The cell pellet was thawed on ice, suspended in a lysis buffer containing 9 M urea, 10 mM Tris pH 8, 100 mM NaH2PO4, 20 mM imidazole, and 0.2 mM of the reducing agent Tris-2-carboxyethyl phosphine (TCEP), sonicated, and then centrifuged at 30,500×g for 45 min to clear the lysate. The His-tagged Recombinant protein was purified by using nickel-affinity chromatography followed by Q-Sepharose. The eluate ToxAll protein was first rebuffered in denaturant conditions to the following conditions: 8 M urea, pH 8.5, 20 mM Tris, 50 mM NaCl, 5% Glycerol, 5 mM TCEP, and then refolded in buffer composed of pH 7.5, 20 mM Tris, 50 mM NaCl, and 5% Glycerol. After refolding, the final protein concentration was 0.05 mg/mL using Bradford method. The shape and size of the SAPNs was analyzed using Transmission Electron Microscopy (TEM) and Dynamic Light Scattering (DLS).
Immunizations of mice and quantitation of parasite burden in brains of mice as numbers of cysts after challenge with type II parasites
The mice used in this study were female HLA-A*11:01, HLA-A*02:01 and HLA-B*07:02 transgenic mice. HLA-A*1101/Kb, HLA-A*0201/Kb and HLA-B*0702/Kb were produced originally at Pharmexa-Epimmune (San Diego, CA) on a C57BL/6 (for HLA-A*11:01 and HLA-B*02:01) and C57BL/6 × Balb/C (for HLA-B*07:02) background, embryo-rederived at JAX laboratories and now are bred at the University of Chicago and JAX laboratories. To evaluate the immunogenicity of the SAPNs, transgenic mice were inoculated intramuscularly (i.m.) with 20 μg SAPNs emulsified in 5 μg of GLA-SE three times at two week intervals. For challenge studies, 5 mice per group were immunized, challenged intraperitoneally 14 days after the last boost with 2000 Type II (Me49-Fluc) parasites. They were imaged 21 days after challenge. This imaging was performed using an in vivo imaging system. This system is made by IVIS (Xenogen, Alameda, CA). The experiment was performed twice with 5 mice per group. To proceed, Mice were injected retro-orbitally with luciferin. Specifically, 200 μl of D-luciferin was injected. Mice were anesthetized in an O2-rich induction chamber. Anesthesia was 2% isoflurane. They were imaged 12 min later. Mice brains were analyzed ex-vivo for imaging. Assessment of photonic emissions was performed using software called Living image® 2.20.1 produced by Xenogen. This experiment was performed twice with 5 mice in each group. Mice were euthanized at 21 days after challenge. Brains were collected and resuspended in 1 ml of saline (0.85% NaCl) and tissue cysts were counted in 50 μl of homogenate using optical microscope. The obtained number was multiplied by 20 to obtain the tissue cysts per mouse brain. Dolichos biflorus agglutinin was used in parallel for the confirmation of cysts.
ELISpot assay on murine splenocytes
Harvested spleens were pressed through a screen. This was a 70 μm screen . This created a single-cell suspension. Then, this suspension was depleted of erythrocytes by using AKC lysis buffer. The AKC buffer contained 10 mM KHCO3, 160 mM NH4Cl, and 100 mM EDTA. Following washing splenocytes twice using Hank’s Balanced Salt Solution (HBSS) splenocytes were resuspended in complete RPMI medium. RPMI-1640 was supplemented with 2 mM L-GlutaMax. We then performed an IFN-γ enzyme linked immunospot (ELISpot) assay. For this assayα we used α-mouse IFN-γ mAb (AN18). We also used biotinylated α-mouse IFN-γmAb (R4–6A2)4,6,11. The reagents and antibodies that we used for the ELISpot assay were from Mabtech, Inc (Cincinnati, OH). 2.5–5 × 105 splenocytes were placed per well . We used at least three replicate wells for each of the conditions. Our results are expressed as the numbers of SFCs (spot forming cells) per 106 mouse splenocytes. There were 3 mice per group, with 3 determinations per mouse with each experiment performed two separate times (n = 6 in total).
Measurement of humoral IgG subclass responses
Ten days after the second immunization, we measured the isotype and antibody to MIC1. Levels of serum antigen specific to IgG1 and IgG2b antibodies were determined by ELISA, as described earlier35. Briefly, 50 µg of tachyzoite Antigen Lysate (TLA) in 50 mM carbonate buffer (pH 9.6) was adsorbed overnight onto 96 well plates (Corning Incorporated, Corning, NY). The plates were blocked with a blocking buffer composed of 1% of milk in saline and 0.05% Tween 20 (PBST) and incubated with the mouse serum (diluted with PBS 1:25) for 2 h at 37 °C. Then, wells were washed three times with PBST and individual anti mouse IgG-IgG1 and IgG2b conjugated to horseradish peroxidase (HRP) (Sigma-Aldrich) sera was added for 1 h at 37 °C. 3,3′, 5, 5′-tetramethylbenzidine (TMB) from (Sigma-Aldrich) was used to detect the peroxidase activity. After blocking the experiment with 2 M H2SO4, the optical density at 450 nM (OD = 450 nm) was recorded using a microplate fluorimeter.
Immunoblot analysis of MIC1 reacting with the serum from immunized mice was performed using tachyzoite Antigen Lysate (TLA) in 3 mice per group. This experiment was performed twice. In brief, 50 µg of TLA were suspended in sodium dodecyl 10% sulfate polyacrylamide gel electrophoresis (SDS-PAGE), electro-blotted to a nitrocellulose membrane and probed with serum from immunized mice diluted 1/50 in PBS-T containing 1% BSA, for 1 h, at 24 °C. Reacting antibodies were detected using 1/5000 peroxidase-conjugated goat anti-mouse IgG (Sigma), for 1 h, at 24 °C and reactions were visualized with DAB substrate kit (Pierce). For IFA, we used sera from mice and anti MIC1 antibody kindly provided by Dr. M. Lebrun, France.
Electron microscopy
To better understand the properties of ToxAll for the delivery of immunogenic epitopes, we analyzed by electron microscopy the processing and presentation of ToxAll in bone marrow derived dendritic cells (BMDDCs). BMDDCs were cultured overnight with 20 µg of SAPN in the presence or absence of 5 μg of GLA-SE. The cells were harvested and spun at 300 × g for 10 min. The cell pellet was placed into planchettes (Ted Pella), cryo-fixed using a High-Pressure Freezing (HPF) Machine, and placed immediately into cryo-tubes containing a frozen cocktail of 0.1% uranyl acetate and 0.25% glutaraldehyde in anhydrous acetone. Samples were frozen, washed with acetone, infiltrated with Lowicryl resin, and mounted on 300 mesh copper grids (EMS). Images were collected on an electron microscope operated at 120 kV (Tecnai Spirit; FEI) with a 2 K ultra scan digital camera (Gatan).
Statistical analysis
For each assay, groups include untreated or mock treated controls. For experiments with two groups, between-group comparisons were made using the nonparametric Mann–Whitney test. For experiments with three or more groups, data were initially compared by the nonparametric Kruskal–Wallis test. If results from that were statistically significant, then relevant pairwise comparisons were made using GraphPad Prism 7 software (GraphPad Software, San Diego, CA). Results are expressed as the mean ± SD and considered statistically significant at p < 0.05.
Ethics approval
All methods were carried out in accordance with relevant guidelines and regulations. Animal experiments were performed with the review and approval of the Institutional Care and Committee at the University of Chicago (AICUC# 71734).
Data availability
All data will be shared.
References
McLeod R, V. T. C., Boyer K. in Nelson Textbook of Pediatrics, 20th Edition Ch. 290, 1723–1732 (Elsevier, New York, 2015).
McLeod, R. et al. Prematurity and severity are associated with Toxoplasma gondii alleles (NCCCTS, 1981–2009). Clin. Infect Dis. 54, 1595–1605. https://doi.org/10.1093/cid/cis258 (2012).
Tan, T. G. et al. Identification of T. gondii epitopes, adjuvants, and host genetic factors that influence protection of mice and humans. Vaccine 28, 3977–3989. https://doi.org/10.1016/j.vaccine.2010.03.028 (2010).
Cong, H. et al. Human immunome, bioinformatic analyses using HLA supermotifs and the parasite genome, binding assays, studies of human T cell responses, and immunization of HLA-A*1101 transgenic mice including novel adjuvants provide a foundation for HLA-A03 restricted CD8+T cell epitope based, adjuvanted vaccine protective against Toxoplasma gondii. Immunome Res. 6, 12. https://doi.org/10.1186/1745-7580-6-12 (2010).
Cong, H. et al. Towards an immunosense vaccine to prevent toxoplasmosis: protective Toxoplasma gondii epitopes restricted by HLA-A*0201. Vaccine 29, 754–762. https://doi.org/10.1016/j.vaccine.2010.11.015 (2010).
El Bissati, K. et al. Adjuvanted multi-epitope vaccines protect HLA-A*11:01 transgenic mice against Toxoplasma gondii. JCI Insight 1, e85955. https://doi.org/10.1172/jci.insight.85955 (2016).
Henriquez, F. L., Woods, S., Cong, H., McLeod, R. & Roberts, C. W. Immunogenetics of Toxoplasma gondii informs vaccine design. Trends Parasitol. 26, 550–555. https://doi.org/10.1016/j.pt.2010.06.004 (2010).
Anderson, R. C. et al. Physicochemical characterization and biological activity of synthetic TLR4 agonist formulations. Colloids Surf. B Biointerfaces 75, 123–132. https://doi.org/10.1016/j.colsurfb.2009.08.022 (2010).
Baldwin, S. L. et al. Enhanced humoral and Type1 cellular immune responses with Fluzone adjuvanted with a synthetic TLR4 agonist formulated in an emulsion. Vaccine 27, 5956–5963. https://doi.org/10.1016/j.vaccine.2009.07.081 (2009).
Fox, C. B., Baldwin, S. L., Vedvick, T. S., Angov, E. & Reed, S. G. Effects on immunogenicity by formulations of emulsion-based adjuvants for malaria vaccines. Clin Vaccine Immunol 19, 1633–1640. https://doi.org/10.1128/CVI.00235-12 (2012).
El Bissati, K. et al. Protein nanovaccine confers robust immunity against Toxoplasma. NPJ Vaccines 2, 24. https://doi.org/10.1038/s41541-017-0024-6 (2017).
El Bissati, K. et al. Effectiveness of a novel immunogenic nanoparticle platform for Toxoplasma peptide vaccine in HLA transgenic mice. Vaccine 32, 3243–3248. https://doi.org/10.1016/j.vaccine.2014.03.092 (2014).
Kaba, S. A. et al. A nonadjuvanted polypeptide nanoparticle vaccine confers long-lasting protection against rodent malaria. J. Immunol. 183, 7268–7277. https://doi.org/10.4049/jimmunol.0901957 (2009).
Burkhard, P. Peptidic nanoparticles as drug delivery and antigen display systems. US8575110B2 (2004).
Raman, S., Machaidze, G., Lustig, A., Aebi, U. & Burkhard, P. Structure-based design of peptides that self-assemble into regular polyhedral nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2, 95–102. https://doi.org/10.1016/j.nano.2006.04.007 (2006).
Burkhard, P. Self-assembling peptide nanoparticles useful as vaccines. US 8546337 (2008).
Kaba, S. A. et al. Protective antibody and CD8+ T-cell responses to the Plasmodium falciparum circumsporozoite protein induced by a nanoparticle vaccine. PLoS ONE https://doi.org/10.1371/journal.pone.0048304 (2012).
Raman, S. K., Machaidze, G., Lustig, A., Aebi, U. & Burkhard, P. Flagellin-containing protein nanoparticles as a vaccine platform. J. Mol. Biol. https://doi.org/10.1016/j.nano.2006.04.007 (2014).
Cerede, O. et al. Synergistic role of micronemal proteins in Toxoplasma gondii virulence. J. Exp. Med. 201, 453–463. https://doi.org/10.1084/jem.20041672 (2005).
Lourenco, E. V. et al. Toxoplasma gondii micronemal protein MIC1 is a lactose-binding lectin. Glycobiology 11, 541–547. https://doi.org/10.1093/glycob/11.7.541 (2001).
Nussenzweig, V. & Nussenzweig, R. S. Rationale for the development of an engineered sporozoite malaria vaccine. Adv. Immunol. 45, 283–334. https://doi.org/10.1016/s0065-2776(08)60695-1 (1989).
Seth, L. et al. Development of a self-assembling protein nanoparticle vaccine targeting Plasmodium falciparum Circumsporozoite Protein delivered in three Army Liposome Formulation adjuvants. Vaccine 35(41), 5448–5454.
Garnett, J. A. et al. Detailed insights from microarray and crystallographic studies into carbohydrate recognition by microneme protein 1 (MIC1) of Toxoplasma gondii. Protein Sci. 18, 1935–1947. https://doi.org/10.1002/pro.204 (2009).
Blader, I. J. & Koshy, A. A. Toxoplasma gondii development of its replicative niche: in its host cell and beyond. Eukaryot Cell 13, 965–976. https://doi.org/10.1128/EC.00081-14 (2014).
Pinzan, C. F. et al. Vaccination with recombinant microneme proteins confers protection against experimental toxoplasmosis in mice. PLoS ONE 10, e0143087. https://doi.org/10.1371/journal.pone.0143087 (2015).
Reiss, M. et al. Identification and characterization of an escorter for two secretory adhesins in Toxoplasma gondii. J. Cell Biol. 152, 563–578. https://doi.org/10.1083/jcb.152.3.563 (2001).
Mineo, J. R. et al. Antibodies to Toxoplasma gondii major surface protein (SAG-1, P30) inhibit infection of host cells and are produced in murine intestine after peroral infection. J. Immunol. 150, 3951–3964 (1993).
Lamarque, M. H. et al. Plasticity and redundancy among AMA-RON pairs ensure host cell entry of Toxoplasma parasites. Nat. Commun. 5, 4098. https://doi.org/10.1038/ncomms5098 (2014).
McCoy, M. E. et al. Mechanisms of protective immune responses induced by the Plasmodium falciparum circumsporozoite protein-based, self-assembling protein nanoparticle vaccine. Malar J. 12, 136. https://doi.org/10.1186/1475-2875-12-136 (2013).
Chahal, J. S. et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 113, E4133-4142. https://doi.org/10.1073/pnas.1600299113 (2016).
Li, Y. et al. Characterization of antibody responses elicited by human immunodeficiency virus type 1 primary isolate trimeric and monomeric envelope glycoproteins in selected adjuvants. J. Virol. 80, 1414–1426. https://doi.org/10.1128/JVI.80.3.1414-1426.2006 (2006).
Nitayaphan, S. et al. A phase I/II trial of HIV SF2 gp120/MF59 vaccine in seronegative thais. AFRIMS-RIHES Vaccine Evaluation Group. Armed Forces Research Institute of Medical Sciences and the Research Institute for Health Sciences. Vaccine 18, 1448–1455. https://doi.org/10.1016/s0264-410x(99)00421-1 (2000).
Reed, S. G., Fox, C. B. & Carter, D. Emulsion-based vaccine adjuvants. In Future Medicine. https://doi.org/10.2217/9781780840604 (2012).
Babapoor, S. et al. A novel vaccine using nanoparticle platform to present immunogenic M2e against avian influenza infection. Influenza Res. Treat. 2011, 126794. https://doi.org/10.1155/2011/126794 (2011).
Meng, M. et al. Evaluation of protective immune responses induced by DNA vaccines encoding Toxoplasma gondii surface antigen 1 (SAG1) and 14-3-3 protein in BALB/c mice. Parasite Vectors 5, 273. https://doi.org/10.1186/1756-3305-5-273 (2012).
Acknowledgements
We gratefully acknowledge the support of the Mann-Cornwell family, Rodriguez, Morel, Engel, Rooney–Alden, Pritzker, Langel, Drago, Mussilami, Quinn, and Rosenthal families for their support of this work. This work was also funded by the National Institutes of Health, Grant numbers R01 AI027530, R01 AI071319, U01 AI077887, and U01 AI082180 from NIH NIAID DMID (to RMc). The research was also supported by the Toxoplasmosis Research Institute, the Knights Templar Eye Foundation and the Institute of translational Medicine Core facility program at the University of Chicago (to KE). We thank Dr. Jeff Alexander, Dr. John Sidney, and Dr. Alessandro Sette for their helpful discussions. We thank also Dr. Maryse Lebrun for providing antibody to MIC1. We thank Kristen Wroblewski for guidance with the statistical analyses. We thank Christopher R. Weber for assistance with finalizing artwork.
Author information
Authors and Affiliations
Contributions
K.E., P.B., and R.M. designed the research. K.E., Y.Z., S.P., S.R., C.K., A.E., A.M.E., J.L., and P.B. performed the research. K.E., Y.Z., S.R., J.L., P.B., and R.M. analyzed the data. K.E., P.B., and R.M. wrote the paper. All authors read and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
P.B. has an interest in Alpha-O Peptides. This is a company with a focus on SAPNs. This company has patents or patents pending on relevant technology. Patent applications pertinent to this work have been filed. Otherwise, the authors have no competing interests. Funding sources did mot influence the decision to submit the paper, nor in the design, collection of data, not in the analysis and writing the report.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
El Bissati, K., Zhou, Y., Paulillo, S.M. et al. Engineering and characterization of a novel Self Assembling Protein for Toxoplasma peptide vaccine in HLA-A*11:01, HLA-A*02:01 and HLA-B*07:02 transgenic mice. Sci Rep 10, 16984 (2020). https://doi.org/10.1038/s41598-020-73210-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73210-0
This article is cited by
-
Toxoplasmosis vaccines: what we have and where to go?
npj Vaccines (2022)
-
Building Programs to Eradicate Toxoplasmosis Part I: Introduction and Overview
Current Pediatrics Reports (2022)
-
Building Programs to Eradicate Toxoplasmosis Part IV: Understanding and Development of Public Health Strategies and Advances “Take a Village”
Current Pediatrics Reports (2022)
-
Toxoplasma gondii – gegenwärtige Arzneimittel und zukünftige Impfstoffe gegen eine unterschätzte Protozoonose
Der Internist (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
