
Abstract
A thorough understanding of malaria vector species composition and their bionomic characteristics is crucial to devise effective and efficient vector control interventions to reduce malaria transmission. It has been well documented in Africa that malaria interventions in the past decade have resulted in major changes in species composition from endophilic Anopheles gambiae to exophilic An. arabiensis. However, the role of cryptic rare mosquito species in malaria transmission is not well known. This study examined the species composition and distribution, with a particular focus on malaria transmission potential of novel, uncharacterized Anopheles cryptic species in western Kenya. Phylogenetic analysis based on ITS2 and COX1 genes revealed 21 Anopheles mosquito species, including two previously unreported novel species. Unusually high rates of Plasmodium sporozoite infections were detected in An. funestus, An. gambiae and eight cryptic rare species. Plasmodium falciparum, P. malariae and P. ovale sporozoite infections were identified with large proportion of mixed species infections in these vectors. This study, for the first time, reports extensive new Anopheles cryptic species involved in the malaria transmission in western Kenya. These findings underscore the importance of non-common Anopheles species in malaria transmission and the need to target them in routine vector control and surveillance efforts.
Similar content being viewed by others

Introduction
Vector control tools, mainly long-lasting insecticide-treated nets (LLIN), indoor residual spraying (IRS), and larval source management (LSM), are key components of malaria control strategies1. The massive scale-up of LLINs and IRS in recent years for malaria vector control has led to major changes in biting behaviour, host preference, breeding range, vectorial capacity or vector competence of mosquito vector species in Africa2. Vector species-specific differences in ecology and behaviour could substantially affect both malaria transmission and the success of vector control strategies. Accurate identification of malaria vector species and their distribution and bionomics is crucial to devise efficient vector control interventions.
In Africa, most malaria entomological studies focus on the two major vector species complexes Anopheles gambiae sensu lato (s.l.), and An. funestus s.l.3,4,5,6,7,8. Such studies usually rely on morphological mosquito species identification with keys that could result in morphological misidentifications or discarding unexpected or unknown cryptic sibling species (or rare species)9. Cryptic rare species are defined as groups of closely related, but genetically isolated species that are difficult or impossible to distinguish by morphological traits10. They are very common in the Anopheles genus9,11,12,13,14,15,16,17. For examples, in southern Africa, Lobo et al. reported 12 of 18 molecularly identified species (including 7 newly identified cryptic species) carrying Plasmodium sporozoites using PCR. In West Africa, a cryptic subgroup of An. gambiae complex, namely GOUNDRY, which exhibits outdoor resting behaviour, was found to be highly susceptible to Plasmodium falciparum infection18,19. Additionally, a new species namely An. fontenillei in the An. gambiae complex was recently discovered in the forested areas of Gabon, Central Africa20. In the western Kenya highlands, 17 species of Anopheles (including 9 cryptic species) were identified, and nearly half of the cryptic species carried P. falciparum parasite DNA12,14,21. These studies indicated that many cryptic Anopheles species may play an important role in malaria transmission across Africa.
Morphologically indistinguishable cryptic sibling species of mosquitoes are challenges to malaria control programs as a result of incomplete understanding of their biology and their role in malaria transmission. Recent advancements in molecular tools, however, offer unique opportunities for accurate and comprehensive analysis of vector-host-parasite interactions. A number of DNA-based molecular tools have been used to identify cryptic species, including PCR amplification or sequencing of the extrachromosomal mitochondrial DNA (mtDNA) and the ribosomal RNA (rRNA) genes. The rRNA genes have been a preferred target because they are well-studied gene family, and the sequences of certain domains of the genes are very highly conserved among species. Sequencing of rRNA genes has become an important tool for systematic studies of highly diverged taxa11. The intergenic spacer (IGS) and internal transcribed spacer 2 (ITS2) region of rRNA gene have been commonly used for development of PCR-based species diagnostic assays and sequence-based cryptic species identifications9,13,14,16,17,22,23,24,25. The intraspecies ITS2 sequence variation of species in An. gambiae complex ranged from 0.07 to 0.43%, whereas interspecies variation ranged between 0.4 and 1.6%26. Mitochondrial DNA fragments from the cytochrome oxidase subunit 1 (COX1) gene have been used as DNA barcodes to identify mosquito species27,28,29. However, molecular species identifications based on COX1 alone may have challenges for discriminating between closely related species or species within a complex30. Indeed, a recent study indicated that ITS2 provides better resolution than COX1 to differentiate An. arabiensis specimens from other An. gambiae complex specimens in eastern Ethiopia31. Accurate identification of diverse vector species is crucial to devise a tailor-made vector intervention for malaria control based on their specific ecology and behaviour. This is particularly critical as African countries anticipate to achieve malaria elimination from many endemic areas by 203032.
To understand the role of cryptic rare vector species in malaria transmission, the present study examined the composition, distribution, and bionomics of Anopheles species in western Kenya. The study also determined Plasmodium sporozoite infection status of cryptic rare Anopheles species in highland and lowland settings of western Kenya. The multiplex-PCR and species-specific PCR methods were used to identify major vector species and blood meal sources. The ITS2 and the COX1 loci were sequenced and analyzed for cryptic rare Anopheles species. Highly sensitive real-time PCR approach was used to detect Plasmodium infections in mosquito vectors.
Results
Overview of molecular determination of Anopheles species
Out of the 3556 Anopheles mosquitoes, 87.1% (3099/3556) were determined by species-specific PCRs or multiplex-PCRs and sequencing as major species An. gambiae sensu stricto (hereafter referred to as An. gambiae) (1440), An. arabiensis (718), and An. funestus sensu stricto (hereafter referred to as An. funestus) (941) in the five study sites (Fig. 1, Table 1, Supplementary Fig. S1). A subset of 21 randomly selected individuals from each major species identified by PCRs were confirmed by ITS2 sequencing based on similarity (> 98%) to the sequences of anopheline voucher species retrieved from NCBI GenBank database (Supplementary Fig. S2).

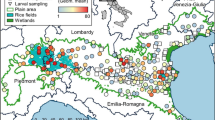
Maps of sampling sites and Anopheles species distribution in western Kenya. (a) distribution of Anopheles major species; (b) distribution of Anopheles rare species. Pie-chart showed the abundance of Anopheles specimens for each site. The maps were generated using ArcGIS Pro 2.6 software. Map source: ESRI, CGIAR, and USGS (available at: www.esri.com).
The remaining 457 collected anophelines (12.9%) were classified into 18 rare species groups based on ITS2 sequence homology. Except for two species groups (An. sp.18 and An. sp.19), the ITS2 sequences of all the species were identified as different species based on their similarity (> 98%) to the sequences of Anopheles voucher species retrieved from NCBI GenBank database (Supplementary Fig. S2). The ITS2 sequences of two species could not match with similarity > 98% threshold to reference anopheline sequences or known vector species in GenBank databases, suggesting the existence of novel cryptic species.
Pairwise comparison of ITS2 sequence similarities of the 21 Anopheles species indicated that except for one pair with 98.5% identity between An. gambiae and An. arabiensis, all pairs showed a similarity of 90% or less with confirmed species classifications (Supplementary Table S1). Phylogenetic tree analysis indicated that the 21 species belong to two different Subgenus (Subgenus Cellia Theobald and Subgenus Anopheles Meigen) in five species series groups, including Myzomyia, Neocellia, Pyretophorus, Cellia, and Myzorhynchus series (Fig. 2, Supplementary Table S2). The two new species An. sp.18 and An. sp.19 as well as An. sp.17 (a recently reported species13) belong to two different series groups, and An. sp.18 belongs to a different Subgenus (Subgenus Anopheles Meigen). The ITS2 sequence of An. sp.9 is homogenous with that of An. theileri (GenBank acc. JN994151) and An. sp. 9 BSL-2014 (GenBank acc. KJ522821)14. The ITS2 sequences obtained in the study are available in GenBank with accession numbers: MT408564-MT408584.

Molecular phylogenetic analysis of ITS2 sequences by Maximum Likelihood method. The phylogenetic tree was constructed using MEGA 7.0 software based on the Kimura 2-parameter model with 1000 bootstrap replicates. Pink filled diamonds showed the major species; red filled circles indicated the novel cryptic species tested positive for Plasmodium infections.
Comparison of morphological and molecular identifications
Of the 3556 mosquitoes tested, 3226 (90.7%) samples with available morphological data were used to evaluate the accuracy of morphological identification as compared to molecular identification. Among the 3226 mosquito samples, 2192 (67.9%) individuals were morphologically identified as An. gambiae s.l., 938 (29.1%) as An. funestus, 94 (2.9%) as An. coustani, and the remaining 2 (0.1%) as An. pharoensis (Table 2). The An. gambiae s.l. complex and An. funestus group (An. funestus, An. cf.rivulorum, and An. leesoni) had a similar percentage of matches (gambiae complex: 85.8%, 1881/2192, funestus group: 85.2%, 799/938) between molecular assay and morphological identification, while only 53.2% of specimens morphologically identified as An. coustani were confirmed by the molecular assay (50/94). Based on molecular assays, An. gambiae s.l. complex had the lowest misidentification (4.3%), followed by An. funestus group (6.8%), while 18.0% (11/61) An. coustani specimens were morphologically misidentified as An. gambiae complex (9) or An. funestus (2). Based on morphological identification, less than 15% of specimens morphologically assigned to An. gambiae complex (14.2%) or An. funestus group (14.8%) were identified as other species, while there were 44 specimens morphologically assigned to An. coustani (46.8%) that were classified by molecular assay into 9 anopheline species, including An. rufipes (17), An. funestus (7), and An. gambiae complex (6). Overall, more than 60% (264/427) of the rare species were morphologically misidentified as An. gambiae s.l. Specifically, nearly 20% (81/427) and 7.2% (31/427) of rare species were misidentified as An. funestus and An. coustani, respectively, whereas only 11.7% (50/427) of the rare species were correctly identified as An. coustani. Altogether, 84.0% (2710/3226) identification alignment was observed between the morphological and molecular analysis (Table 2).
Comparison of Anopheles species distributions and diversity
Overall, the three major species were found in all five study sites but at varying proportions. Anopheles funestus accounted for a large proportion (44.7–98.2%) of species observed throughout the five study sites (Fig. 1A, Table 1). Anopheles gambiae was the predominant species in two highland sites (56.7% in Emutete and 61.3% in Iguhu) and one lowland site (47.73% in Kombewa), whereas An. gambiae was nearly absent in Homa Bay (0.4%), a lowland site, and Kisii (0.6%), a highland site. An. arabiensis, was observed in high proportion in lowland areas (Homa Bay: 70.8% and Kombewa: 24.9%) than in highland areas, which ranged from 1.8% (Emutete) to 5.2% (Iguhu).
Seventeen of 18 rare species were identified in the highland areas, whereas only six rare species were detected in the lowland areas, suggesting that cryptic species might be more related to the sympatric An. gambiae than An. arabiensis. In lowland sites, the most abundant rare anopheline species was An. sp.15 (n = 17), followed by An. rufipes (n = 14) and An. cf.rivulorum (n = 14), whereas multiple rare species (such as An. christyi, An. sp.1, and An. sp.17) were identified in the highlands (Fig. 1B, Table 1).
A significantly higher species diversity was observed in the highland areas than in the lowland areas (Shannon index H, t-test, t = − 6.59, df = 3419, p < 0.001). From 2015 to 2019, the average observed species richness (S) per year was 12.0 ± 0.27 in highland and 6.8 ± 0.25 in lowland (Table 3). In lowland, the highest species abundance (n = 10) was observed in 2018, whereas in highland, the highest species abundance of 15 was detected in 2016 and 2017. A significantly decreased diversity was observed from 2017 to 2018 (Shannon index H, t-test, t = 3.37, df = 340, p < 0.001) and 2018 to 2019 (Shannon index H, t-test, t = 3.83, df = 799, p < 0.001) in lowland. However, in highland, a significant increased diversity was observed from 2016 to 2017 (Shannon index H, t-test, t = − 7.06, df = 833, p < 0.001), then decreased diversity from 2017 to 2018 (Shannon index H, t-test, t = 1.97, df = 787, p < 0.05), and remaining high species richness in 2019 (n = 10) (Table 3).
Molecular determination of Anopheles mosquito host blood meal source
A total of 1,372 blood-fed female mosquitoes from 16 Anopheles species were successfully genotyped for blood meal sources (Table 4). Of these, 41.6% females were identified to have had human blood meals, 53.6% of mosquito blood meals were identified as bovine, whereas the remained 4.8% individuals had blood meals originating from other animals, e.g., goat, pig, and dog. For the major vector species, the highest human blood index was found in An. funestus (0.72), followed by An. gambiae (0.51), whereas very few samples of An. arabiensis had human blood meals (Pearson's Chi-squared test: χ2 = 532.4, df = 8, p < 0.0001). The majority (> 90%) of An. arabiensis blood meals originated from cows (Fig. 3). Similar patterns of blood meal source were observed in the highlands and lowlands. Human blood meal sources were detected in a total of ten rare Anopheles species, including eight rare species from highland and two from lowland. The blood meal source of the other three rare species (An. leesoni, An. maculipalpis, and An. pretoriensis) were identified as bovine (Table 4).

Host blood meal source of Anopheles female mosquitoes in western Kenya. Error bars indicated 95% confidence interval. Bar charts were created in Microsoft Excel 2013 software. Statistical analyses were performed using SAS JMP 14.0 software (SAS Inc., Cary, NC).
Multiplex-qPCR identification of Plasmodium sporozoite infection
Of the total 3,556 female anopheline mosquitoes tested, 348 (9.8%) were positive for Plasmodium sporozoite infections (Table 5). Highest sporozoite rate was observed in An. funestus (17.3%; 95% CI 14.9–19.7%), followed by An. gambiae (9.9%; 95% CI 8.4–11.4%), and An. arabiensis (4.2%; 95% CI 3.6–4.8%). Sporozoite rates were two-fold higher in the lowland than that in highland sites for An. funestus (26.4% vs. 13.1%) and 1.5-fold higher in An. gambiae (14.3% vs. 7.8%). In contrast, An. arabiensis showed a higher sporozoite rate in the highlands (8.3%) compared to the lowlands (3.6%). Surprisingly, Plasmodium sporozoites were also detected in eight out of 18 rare anopheline species, confirming the role of rare cryptic species on malaria transmission in western Kenya (Table 5). Between 2015 and 2019, an increasing trend in the annual sporozoite rate was observed in the three major vector species both in the lowland (Chi-squared test: χ2 = 9.04, df = 1, p < 0.01) and highland (Chi-squared test: χ2 = 8.08, df = 1, p < 0.05) study sites (Fig. 4). From the outdoor collection of mosquito samples in 2016 and 2017, four rare species were found positive for Plasmodium infection in the two highland sites (Iguhu and Emutete) (Supplementary Table S4). No statistically significant difference in Plasmodium infection was detected between indoor and outdoor both in Iguhu (Pearson's Chi-squared test: χ2 = 0.78, df = 1, p = 0.37) and Emutete (Pearson's Chi-squared test: χ2 = 0.07, df = 1, p = 0.78). Three species of Plasmodium were detected with the majority being P. falciparum (87.6%, 305/348), and the remaining were P. malarae and P. ovale (Supplementary Table S5). Mixed species infections were common, accounted for 5.7% (20/348) of the total Plasmodium sporozoite positive samples. No statistically significant difference between major vector species in the component of parasite species was observed both in lowland (Pearson's Chi-squared test: χ2 = 7.35, df = 8, p = 0.49) and highland (Pearson's Chi-squared test: χ2 = 14.84, df = 12, p = 0.25).

Sporozoite rate of major Anopheles mosquito species in western Kenya from 2015 to 2019 (a) highland; (b) lowland. Error bars represented standard errors of sporozoite rates in the tested samples. Bar charts were created in Microsoft Excel 2013 software. Statistical analyses were performed using SAS JMP 14.0 software (SAS Inc., Cary, NC).
Mitochondrial DNA barcode diversity of cryptic species
Eighteen of 21 COX1 sequence groups or species had a 1:1 relationship with the ITS2 sequence groups. Two species, An. sp.18 and An. sp.19 did not show reliable COX1 sequencing results due to failure of the PCR amplification using the universal COX1 barcoding PCR primers, indicating possible sequence variation in primer regions. However, high COX1 sequence variation was observed in the newly discovered species An. sp.17 (Supplementary Fig. S2). Among the 21 tested individuals, which showed identical ITS2 gene sequences, 13 haplotypes (An. sp.17_H01–An. sp.17_H13) of COX1 gene were identified with 17 separating sites. Higher haplotype diversity (Hd = 0.929, Pi = 0.0081) and nucleotide diversity were found in this cryptic species, as compared to An. gambiae (Hd = 0.700, Pi = 0.0029) and An. funestus (Hd = 0.710, Pi = 0.0015) (n = 21 for each species). Phylogenetic tree analysis indicated that at least four haplotype groups with three of them in different clades matching the reference sequences (An. sp.C, An. sp.D, and An. sp.F, respectively) from NCBI GenBank database (Fig. 5, Supplementary Table S3). No matching references in GenBank was found for the clade with haplotypes An. sp.17_H01, An. sp.17_H02, An. sp.17_H04, and An. sp.17_H06. The sequences obtained in the study were deposited in NCBI GenBank with the accession numbers: MT375202-MT375229.

Phylogenetic tree analysis of COX1 haplotypes in the newly discovered Anopheles cryptic species in western Kenya. The phylogenetic tree was constructed using MEGA 7.0 software by the bootstrap method with 1000 replications. The red solid squares indicated predominant haplotypes within species, and solid dark circles represented reference sequences retrieved from GenBank at NCBI. The An. sp.C, An. sp.D, and An. sp.F are anopheline cryptic species reported previously in western Kenya (GenBank accession: MK047666, MK047667, and MK047666).
Discussion
This study, for the first time, reported eight rare Anopheles species with Plasmodium sporozoites and human blood meal sources in western Kenya. Although An. funestus, An. gambiae and An. arabiensis remained as the primary malaria vectors in the region, the presence of 18 cryptic rare Anopheles species—half of them being incriminated as malaria vectors—may have serious implications for malaria control program in western Kenya. Mosquito population modification using the innovative vector control techniques, such as gene drives or Wolbachia transfection, which rely on mating of mosquitoes to spread, will be unlikely to spread into other reproductively isolated cryptic species except the one (s) in which the tool was introduced. Presence of such diversified vector species coupled with Plasmodium-infected outdoor mosquitoes may challenge vector interventions and contribute to malaria transmission stability. Understanding breeding habitats, biting seasonality, resting behaviour and vector competence of these rare vector species is critically required in order to target them in routine malaria control programs.
Phylogenetic tree analysis showed that the seven recently identified and less documented cryptic rare Anopheles species along with An. funestus s.l. complex belong to Myzomyia series group. Among these, four of them (An. sp.1, An. sp.6, An. sp9, and An. sp.17) were positive for human malaria parasites. Similar findings were reported previously that identified new sibling cryptic Anopheles species in western Kenya highlands13,14. However, Plasmodium infections of the rare species An. sp.11 and An. sp.15 are probably the first time being reported in the present study, although these two new species were previously reported without parasite detections14. These two rare species were classified into Cellia and Myzorhynchus series groups, respectively, which encompasses potential vectors, such as An. pharoensis and An. coustani9,24. The present study also for the first time reported two other rare species An. sp.18 and An. sp.19 as novel species, since their sequence data were not found in the GenBank database. However, possibly due to a low number of mosquitoes collected, Plasmodium infections were not detected in these two species, and no host blood meal sources were found for the samples. An unusually high number of malaria sporozoite rates in rare Anopheles species, including An. sp.1 (2/69) and An. sp.17 (3/56), suggested their potential role in malaria transmission in western Kenya. Additionally, detecting Plasmodium infections from outdoor mosquito collections indicates the presence of outdoor transmission that may not be addressed by indoor-based vector interventions. Further study is however required to characterize breeding habitats, vectorial capacities and biting behaviours of these rare species. Future studies should also investigate environmental factors in the highlands of western Kenya that led to presence of a diverse Anopheles species. The potential role of vector competition in the highlands and possible increased availability of breeding sites due to climate change require further study.
A high Plasmodium sporozoite rate (nearly 10% or more) for the primary malaria vector species, An. funestus and An. gambiae, indicated a high level of malaria transmission due to these species in both the highlands and lowlands of western Kenya. Sporozoite rates reported in the present study were over two-fold higher than that of a previous report in western Kenya33, probably due to the use of highly sensitive molecular techniques34 and multiple parasite species examinations in our study. Similar high sporozoite rates were previously reported for An. funestus from elsewhere9. These findings generally confirm that An. funestus and An. gambiae, remain to be the primary malaria vector species in western Kenya. Although these two species are primarily endophagic, the detection of Plasmodium infected mosquitoes outdoors might suggest certain behavioural change in their ecology due to continuous use of indoor-based interventions, as reported in neighboring Tanzania35. Prolonged and wide-spread use of LLINs could, thereby, favour traits such as biting outdoors or early in the evening. In the southwest Pacific, a study found that high levels of malaria transmission have been maintained by An. farauti, a malaria vector that altered its behaviour to blood-feed early in the evening and outdoors and, thereby, avoiding exposure to the insecticides used in IRS36. The potential occurrence of such behavioural change in western Kenya requires further study. On the other hand, high sporozoite rate does not necessarily translate to high malaria transmission since disease transmissibility is affected by a number of genetic and environmental factors that determine vector competence37. The observed high species diversity in the highland areas may be the result of an increase in new clonal niches that potentially conquered new breeding habitats in the highlands. Unlike An. funestus and An. gambiae, An. arabiensis was a dominant vector species in the lowlands of Homa Bay than highlands of western Kenya. This species is a common vector of malaria in the lowlands of East Africa38,39. A low human blood index and low sporozoite rate of An. arabiensis in the lowlands indicate that this species primarily tends to feed on non-human blood meal sources, mainly bovine, as reported in Ethiopia40. Previous study has suggested that climate change may push An. arabiensis to encroach and adapt to the highlands of East Africa41.
This study also highlighted the level of misidentifications and misassignments while using morphological keys for species identification. For instance, there were only four species identified using the standard morphological key42, while molecular assay revealed the presence of 21 species—suggesting that many Anopheles species might be morphologically indistinguishable. The level of misidentification was extremely lower in major species (4–7%) compared to rare species (100%). A number of factors such as level of training and mosquito sample quality determine proper identification of mosquitoes using morphological keys. For instance, morphological identification of An. coustani is not complicated due to its unique and conspicuous white patch on its hind legs42. Yet, our data indicated that a considerable number of this species was misidentified—suggesting the need for additional training to improve the skill of field entomologists. Overall, species identification using morphological key is affected by a number of factors such as: (1) the quality of the specimen (damaged or intact specimen) when collecting mosquitoes using CDC-LT; (2) morphologically indistinguishable sibling mosquito species; (3) low level of skills on use of morphological keys for species identification by field entomologists. The present study indicated that standard morphological identifications were more reliable for the major species than rare species21.
In summary, the present study demonstrated the importance of new Anopheles rare species in malaria transmission in western Kenya. Molecular tools help accurately characterize the Anopheles vector species in malaria endemic areas. For the first time, eight out of 18 rare Anopheles species with Plasmodium sporozoites and human blood source were reported in western Kenya, suggesting their secondary role in malaria transmission. Presence of diversified malaria vector species in the highlands might be the consequence of climate change that potentially created new ecological niches for rare vector species, although this requires further investigations. Future studies need to identify and investigate the breeding habitats, and resting and feeding behaviours of these rare vector species in order to devise appropriate vector control strategies that can target all malaria vector species in the region and advance malaria elimination in Africa.
Methods
Ethics statement
Ethical approval for the study was obtained from the institutional scientific ethical review board of University of California, Irvine, USA and Maseno University, Kenya. Permission was sought from the chief of each study site. Written informed consent was obtained from heads of the households, and individuals who were willing to participate in the study. All methods used in this study were performed in accordance with the relevant guidelines and regulations.
Study sites and sample collections
Malaria vector surveillance was conducted in three highland sites (elevation 1500–2300 m) and two lowland sites (1050–1500 m) in western Kenya (Fig. 1). Malaria transmission in highland regions was traditionally regarded as mesoendemic-hyperendemic, unstable, and limited by low temperature, whereas malaria transmission in lowland regions is holoendemic and year around43,44. Between 2015 and 2019, adult mosquitoes were collected in five study sites: Iguhu, Emutete, and Kisii in highland regions; and Kombewa and Homa Bay in lowland regions. Indoor-resting mosquitoes were collected by CDC light traps (CDC-LT) and pyrethrum spray catches (PSC) in all study sites. Outdoor-resting mosquitoes were collected by CDC-LT, human landing catches (HLC), clay pot sampling (CP), and pit shelters (PS) sampling methods in Iguhu (2016) and Emutete (2017). These study sites and detailed sampling methods of vector surveillance have been described in previous publications45,46,47. Adult female anopheline mosquitoes were identified to species by morphological keys for the Afrotropical Anophelinae42,48. The abdominal status of all individuals was classified into unfed, fed, half gravid and gravid. Mosquito samples were stored individually in 1.5 ml tubes containing desiccated silica gel covered with cotton wool.
DNA extraction and PCR-based determination of the major species
A large subset of morphologically identified mosquitoes (n = 3556) from the collections above were used for DNA extraction, molecular identification of species, and Plasmodium sporozoite infection detections. Female adult mosquitoes were separated individually into head-thoracic portion and abdominal portion for DNA extraction. DNA extraction was performed by using QIAamp 96 DNA QIAcube HT kit on a QIAcube HT 96 automated nucleic acid purification robot (Qiagen, Valencia, CA) according to the manufacturer’s protocol with minor modifications. Specifically, each individual sample was ground in ZR Bashing Bead Lysis tube (2.0 mm beads, Zymo Research Corporation, Irvine, USA) and homogenized in 200 μl lysis solution containing 20 µl of proteinase K (20 mg/ml) using the TissueLyser II system (Qiagen, Hilden, Germany) for 10 min at 30 Hz. The genomic DNA was eluted in a final volume of 100 µl for head-thoracic portion and 200 µl for abdominal portion. For those specimens morphologically identified to An. gambiae s.l. complex, multiplex polymerase chain reaction (Multiplex-PCR) was conducted to distinguish An. gambiae s.s. and An. arabiensis using the species-specific primers (UN/GA/AR) in 28S ribosomal RNA gene according to the method as described by Scott et al.22. For those specimens morphologically identified to An. funestus, single PCR was conducted to confirm species using the species-specific primers (ITS2A/FUN) in the internal transcribed spacer region (ITS2) on the ribosomal DNA as described by Koekemoer et al.49. For the other species and those specimens which failed to amplify by PCR for clear species-specific band or had non-specific or weak amplifications bands50,51 using the species-specific primers, additional two PCR amplifications were performed to further identify morphologically misidentified specimens using the primers UN/GA/AR and ITS2A/FUN, respectively.
DNA sequence-based determination of the rare species
For the specimens which failed to be identified by PCR, and a subset of 21 randomly selected individuals from each major species, additional PCR amplification and DNA sequencing of the ITS2 region of nuclear ribosomal DNA and the COX1 gene were performed using the primer pair ITS2A (TGTGAACTGCAGGACACAT) and ITS2B (TATGCTTAAATTCAGGGGGT) for ITS225, and the primer pair: LCO1490 (GGTCAACAAATCATAAAGATATTGG) and HCO219 (TGATTTTTTGGTCACCCTGAAGTTTA) for COX152. An additional primer pair, 158F (GGTCAACAAATCATAAGGATATTGG) and 890R (GGTCCGAATCCAGGTAAAATTAAA) was designed for COX1 amplification of new species (An. sp.17). The PCR was conducted in a total reaction volume of 17 μl, including 1 μl of DNA template (~ 5 ng), 5 pmol of each primer, and 8.5 μl of DreamTaq Green PCR Master Mix (2X) (Thermo Fisher Scientific, Waltham, MA, USA). The thermocycling protocol consisted of an initial activation step of 3 min at 95 °C, followed by 35 amplification cycles of 30 s at 94 °C, 30 s at 55 °C and 45 s at 72 °C, and a final extension step of 6 min at 72 °C. Five microliters of the amplified fragments were examined by electrophoresis on a 1.5% agarose gel. PCR products showing a clear single band on the agarose gel were purified using an enzymatic PCR clean-up technique: 0.3 μl of Exonuclease I (ExoI, 20U/μl) and 1.7 μl of Shrimp Alkaline Phosphtase (SAP, 1U/μl) and 8.0 μl of PCR products. This mixture was incubated at 37 °C for 30 min, followed by 15 min at 80 °C to inactivate the enzymes, then held at 4 °C until sequencing. The premixed samples containing 3 μl of cleaned PCR product, 3 μl of forward primer (10 μM), and 12 μl of ddH2O were sent to Retrogen Inc (San Diego, CA) for sequencing.
Multiplexed PCR-based blood meal identification
Host blood meal identification of fed mosquitoes was conducted using the multiplexed PCR-based methods as described by Kent et al.29, with minor modifications. Specifically, PCR was conducted in a total reaction volume of 13 μl, including 1 μl of DNA template (~ 5 ng), 5 pmol of each primer (Forward primers: Pig573F, Human741F, Goat894F, Dog368F, Cow121F, and Reverse primer UNREV1025), and 6.5 μl of DreamTaq Green PCR Master Mix (2 ×) (Thermo Fisher Scientific, Waltham, MA, USA). The thermocycling protocol consisted of an initial activation step of 3 min at 95 °C, followed by 35 amplification cycles of 30 s at 94 °C, 30 s at 55 °C and 45 s at 72 °C, and a final extension step of 6 min at 72 °C. Five microliters of the amplified fragments were analyzed by 1.5% agarose gel electrophoresis along with 100 bp DNA ladder.
Multiplexed quantitative PCR (qPCR) assay for Plasmodium infections
The DNA extracted from mosquito head-thoracic portion were used for qPCR identification of Plasmodium sporozoite infection. An multiplexed real-time qPCR assay was performed by using the published species-specific 18 s ribosomal RNA probes and primers for Plasmodium falciparum and P. malariae53 and P. ovale54. Multiplexed qPCR was run on QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific, Carlsbad, CA) in a final volume of 12 µl containing 2 µl of sample DNA, 6 µl of PerfeCTa qPCR ToughMix, Low ROX Master mix (2X) (Quantabio, Beverly, MA), 0.5 µl of each probe (2 µM), 0.4 µl of each forward primers (10 µM), 0.4 µl of each reverse primers (10 µM) and 0.1 µl of double-distilled water. The following temperature profile was applied: hold stage at 50 °C for 2 min and 95 °C for 2 min, followed by 45 cycles of PCR amplification stage at 95 °C for 3 s and 60 °C for 30 s. The standard curve of positive control containing Plasmid DNA (MRA-177 for P. falciprium, MRA-179 for P. malariae, and MRA-180 for P. ovale) from BEI Resources (https://www.beiresources.org) was included in each qPCR plate run with 3 negative controls.
Statistical analysis
The CodonCode Aligner 9.0.1 (CodonCode Corporation, Centerville, MA) was used to check the sequence quality and trim low-quality bases. BioEdit software55 and MView web-based tool56 were used to conduct the alignment of the sequences and to calculate pairwise sequence identity and similarity from multiple sequence alignments. A threshold limit of 98% sequence similarity for ITS2 was used to classify sequences into species groups9. The consensus sequences within group were compared to the NCBI nr/nt database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Sequence groups were assigned into known taxa or voucher specimen based on similarity to a voucher specimen sequence at 98% threshold value. The haplotypes of COX1 sequences were compared to both the NCBI nr/nt database and BOLD database (https://www.barcodinglife.org)29.
Phylogenetic analyses were performed using Maximum Likelihood (ML) algorithms with the General Time Reversible model for ITS2 consensus sequences of species groups and UPGMA with the Kimura 2-parameter model for COX1 haplotypes in the MEGA version 7.057. The tree nodes were evaluated by bootstrap analysis for 1000 replicates and rooted for COX1 haplotypes using a sequence from Aedes aegypti (GenBank: AF390098). The diversity index of Shannon and Simpson, as well as the Simpson’s evenness and dominance index were used to assess Anopheles diversity within and between sites. All these indices were calculated using PAST 4.0 software package58. To compare diversity between highland and lowland, a t-test was used to determine whether they were significantly different59.
Human blood index (HBI) is defined as the proportion of freshly fed mosquitoes containing human blood and calculated as described by Garrett-Jones60. Mixed blood meals were added to the number of each host blood meals when calculating the HBI separately. Sporozoite rate was estimated by the proportion of the number of positive individuals divided by total number of tested individuals with 95% confidence interval61. Statistical analyses were conducted using SAS JMP 14.0 software (SAS Inc., Cary, NC).
References
WHO. Guidelines for Malaria Vector Control (World Health Organization, Geneva, 2020).
WHO. World Malaria Report 2019 (World Health Organization, Geneva, 2019).
Keating, J. et al. Anopheles gambiae s.l. and Anopheles funestus mosquito distributions at 30 villages along the Kenyan coast. J. Med. Entomol. 42, 241–246. https://doi.org/10.1093/jmedent/42.3.241 (2005).
Noutcha, M. A. & Anumdu, C. I. Entomological indices of Anopheles gambiae sensu lato at a rural community in south-west Nigeria. J. Vector Borne Dis. 46, 43–51 (2009).
Sande, S., Zimba, M., Chinwada, P., Masendu, H. T. & Makuwaza, A. Biting behaviour of Anopheles funestus populations in Mutare and Mutasa districts, Manicaland province, Zimbabwe: Implications for the malaria control programme. J. Vector Borne Dis. 53, 118–126 (2016).
Ndenga, B. A. et al. Malaria vectors and their blood-meal sources in an area of high bed net ownership in the western Kenya highlands. Malar. J. 15, 76. https://doi.org/10.1186/s12936-016-1115-y (2016).
Oyewole, I. O. et al. Behaviour and population dynamics of the major anopheline vectors in a malaria endemic area in southern Nigeria. J. Vector Borne Dis. 44, 56–64 (2007).
Okara, R. M. et al. Distribution of the main malaria vectors in Kenya. Malar. J. 9, 69–69. https://doi.org/10.1186/1475-2875-9-69 (2010).
Lobo, N. F. et al. Unexpected diversity of Anopheles species in Eastern Zambia: Implications for evaluating vector behavior and interventions using molecular tools. Sci. Rep. 5, 17952. https://doi.org/10.1038/srep17952 (2015).
Bickford, D. et al. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 22, 148–155. https://doi.org/10.1016/j.tree.2006.11.004 (2007).
Collins, F. H. & Paskewitz, S. M. A review of the use of ribosomal DNA (rDNA) to differentiate among cryptic Anopheles species. Insect Mol. Biol. 5, 1–9. https://doi.org/10.1111/j.1365-2583.1996.tb00034.x (1996).
Conn, J. E. News from Africa: Novel anopheline species transmit Plasmodium in western Kenya. Am. J. Trop. Med. Hyg. 94, 251–252. https://doi.org/10.4269/ajtmh.16-0020 (2016).
Ogola, E. O., Chepkorir, E., Sang, R. & Tchouassi, D. P. A previously unreported potential malaria vector in a dry ecology of Kenya. Parasit. Vectors 12, 80. https://doi.org/10.1186/s13071-019-3332-z (2019).
St Laurent, B. et al. Molecular characterization reveals diverse and unknown malaria vectors in the western Kenyan highlands. Am. J. Trop. Med. Hyg. 94, 327–335. https://doi.org/10.4269/ajtmh.15-0562 (2016).
Coetzee, M. Anopheles crypticus, new species from South Africa is distinguished from Anopheles coustani (Diptera: Culicidae). Mosq. Syst. 26, 125 (1994).
Hackett, B. J. et al. Ribosomal DNA internal transcribed spacer (ITS2) sequences differentiate Anopheles funestus and An. rivulorum, and uncover a cryptic taxon. Insect Mol. Biol. 9, 369–374. https://doi.org/10.1046/j.1365-2583.2000.00198.x (2000).
Spillings, B. L. et al. A new species concealed by Anopheles funestus Giles, a major malaria vector in Africa. Am. J. Trop. Med. Hyg. 81, 510–515 (2009).
Crawford, J. E. et al. Evolution of GOUNDRY, a cryptic subgroup of Anopheles gambiae s.l., and its impact on susceptibility to Plasmodium infection. Mol. Ecol. 25, 1494–1510. https://doi.org/10.1111/mec.13572 (2016).
Riehle, M. M. et al. A cryptic subgroup of Anopheles gambiae is highly susceptible to human malaria parasites. Science (New York, N.Y.) 331, 596–598. https://doi.org/10.1126/science.1196759 (2011).
Barrón, M. et al. A new species in the major malaria vector complex sheds light on reticulated species evolution. Sci. Rep. https://doi.org/10.1038/s41598-019-49065-5 (2019).
Stevenson, J. et al. Novel vectors of malaria parasites in the western highlands of Kenya. Emerg. Infect. Dis. 18, 1547–1549. https://doi.org/10.3201/eid1809.120283 (2012).
Scott, J. A., Brogdon, W. G. & Collins, F. H. Identification of single specimens of the Anopheles gambiae complex by the polymerase chain reaction. Am. J. Trop. Med. Hyg. 49, 520–529. https://doi.org/10.4269/ajtmh.1993.49.520 (1993).
Dusfour, I. et al. Polymerase chain reaction identification of three members of the Anopheles sundaicus (Diptera: Culicidae) complex, malaria vectors in Southeast Asia. J. Med. Entomol. 44, 723–731. https://doi.org/10.1603/0022-2585(2007)44[723:pcriot]2.0.co;2 (2007).
Norris, L. C. & Norris, D. E. Phylogeny of anopheline (Diptera: Culicidae) species in southern Africa, based on nuclear and mitochondrial genes. J. Vector Ecol. 40, 16–27. https://doi.org/10.1111/jvec.12128 (2015).
Beebe, N. W. & Saul, A. Discrimination of all members of the Anopheles punctulatus complex by polymerase chain reaction–restriction fragment length polymorphism analysis. Am. J. Trop. Med. Hyg. 53, 478–481. https://doi.org/10.4269/ajtmh.1995.53.478 (1995).
Paskewitz, S. M., Ng, K., Coetzee, M. & Hunt, R. H. Evaluation of the polymerase chain reaction method for identifying members of the Anopheles gambiae (Diptera: Culicidae) complex in Southern Africa. J. Med. Entomol. 30, 953–957. https://doi.org/10.1093/jmedent/30.5.953 (1993).
Bourke, B. P., Oliveira, T. P., Suesdek, L., Bergo, E. S. & Sallum, M. A. A multi-locus approach to barcoding in the Anopheles strodei subgroup (Diptera: Culicidae). Parasit. Vectors 6, 111. https://doi.org/10.1186/1756-3305-6-111 (2013).
Kumar, N. P., Rajavel, A. R., Natarajan, R. & Jambulingam, P. DNA barcodes can distinguish species of Indian mosquitoes (Diptera: Culicidae). J. Med. Entomol. 44, 1–7. https://doi.org/10.1603/0022-2585(2007)44[1:dbcdso]2.0.co;2 (2007).
Ratnasingham, S. & Hebert, P. D. N. BOLD: The barcode of life data system. Mol. Ecol. Notes 7, 355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x (2007).
Beebe, N. W. DNA barcoding mosquitoes: Advice for potential prospectors. Parasitology 145, 622–633. https://doi.org/10.1017/s0031182018000343 (2018).
Carter, T. E., Yared, S., Hansel, S., Lopez, K. & Janies, D. Sequence-based identification of Anopheles species in eastern Ethiopia. Malar. J. 18, 135–135. https://doi.org/10.1186/s12936-019-2768-0 (2019).
WHO. Global Technical Strategy for Malaria 2016–2030 (World Health Organization, Geneva, 2015).
Githeko, A. K., Serddvice, M. W., Mbogo, C. M., Atieli, F. K. & Juma, F. O. Plasmodium falciparum sporozoite and entomological inoculation rates at the Ahero rice irrigation scheme and the Miwani sugar-belt in western Kenya. Ann. Trop. Med. Parasitol. 87, 379–391. https://doi.org/10.1080/00034983.1993.11812782 (1993).
Bass, C. et al. PCR-based detection of Plasmodium in Anopheles mosquitoes: A comparison of a new high-throughput assay with existing methods. Malar. J. 7, 177. https://doi.org/10.1186/1475-2875-7-177 (2008).
Russell, T. L. et al. Increased proportions of outdoor feeding among residual malaria vector populations following increased use of insecticide-treated nets in rural Tanzania. Malar. J. 10, 80. https://doi.org/10.1186/1475-2875-10-80 (2011).
Russell, T. L., Beebe, N. W., Cooper, R. D., Lobo, N. F. & Burkot, T. R. Successful malaria elimination strategies require interventions that target changing vector behaviours. Malar. J. 12, 56. https://doi.org/10.1186/1475-2875-12-56 (2013).
Pombi, M. et al. Unexpectedly high Plasmodium sporozoite rate associated with low human blood index in Anopheles coluzzii from a LLIN-protected village in Burkina Faso. Sci. Rep. 8, 12806. https://doi.org/10.1038/s41598-018-31117-x (2018).
Kibret, S. & Wilson, G. G. Increased outdoor biting tendency of Anopheles arabiensis and its challenge for malaria control in Central Ethiopia. Public Health 141, 143–145. https://doi.org/10.1016/j.puhe.2016.09.012 (2016).
Machani, M. G. et al. Resting behaviour of malaria vectors in highland and lowland sites of western Kenya: Implication on malaria vector control measures. PLoS ONE 15, e0224718. https://doi.org/10.1371/journal.pone.0224718 (2020).
Massebo, F., Balkew, M., Gebre-Michael, T. & Lindtjørn, B. Zoophagic behaviour of anopheline mosquitoes in southwest Ethiopia: Opportunity for malaria vector control. Parasit. Vectors 8, 645. https://doi.org/10.1186/s13071-015-1264-9 (2015).
Afrane, Y. A., Githeko, A. K. & Yan, G. The ecology of Anopheles mosquitoes under climate change: Case studies from the effects of deforestation in East African highlands. Ann. N. Y. Acad. Sci. 1249, 204–210. https://doi.org/10.1111/j.1749-6632.2011.06432.x (2012).
Gillies, M. T. & Coetzee, M. A supplement to the Anophelinae of Africa south of the Sahara (Afrotropical Region). Publ. S. Afr. Inst. Med. Res. 55, 1–143 (1987).
Shanks, G. D., Hay, S. I., Omumbo, J. A. & Snow, R. W. Malaria in Kenya’s western highlands. Emerg. Infect. Dis. 11, 1425–1432. https://doi.org/10.3201/eid1109.041131 (2005).
Wanjala, C. L., Waitumbi, J., Zhou, G. & Githeko, A. K. Identification of malaria transmission and epidemic hotspots in the western Kenya highlands: Its application to malaria epidemic prediction. Parasit. Vectors 4, 81. https://doi.org/10.1186/1756-3305-4-81 (2011).
Ototo, E. N. et al. Surveillance of malaria vector population density and biting behaviour in western Kenya. Malar. J. 14, 244. https://doi.org/10.1186/s12936-015-0763-7 (2015).
Degefa, T. et al. Indoor and outdoor malaria vector surveillance in western Kenya: Implications for better understanding of residual transmission. Malar. J. 16, 443–443. https://doi.org/10.1186/s12936-017-2098-z (2017).
Degefa, T. et al. Evaluation of the performance of new sticky pots for outdoor resting malaria vector surveillance in western Kenya. Parasit. Vectors 12, 278. https://doi.org/10.1186/s13071-019-3535-3 (2019).
Gillies, M. T. & Meillon, B. D. The Anophelinae of Africa south of the Sahara (Ethiopian zoogeographical region). Publ. S. Afr. Inst. Med. Res. 54, 1–343 (1968).
Koekemoer, L. L., Kamau, L., Hunt, R. H. & Coetzee, M. A cocktail polymerase chain reaction assay to identify members of the Anopheles funestus (Diptera: Culicidae) group. Am. J. Trop. Med. Hyg. 66, 804–811. https://doi.org/10.4269/ajtmh.2002.66.804 (2002).
Erlank, E., Koekemoer, L. L. & Coetzee, M. The importance of morphological identification of African anopheline mosquitoes (Diptera: Culicidae) for malaria control programmes. Malar. J. 17, 43. https://doi.org/10.1186/s12936-018-2189-5 (2018).
Dahan-Moss, Y. et al. Member species of the Anopheles gambiae complex can be misidentified as Anopheles leesoni. Malar. J. 19, 89. https://doi.org/10.1186/s12936-020-03168-x (2020).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Veron, V., Simon, S. & Carme, B. Multiplex real-time PCR detection of P. falciparum, P. vivax and P. malariae in human blood samples. Exp. Parasitol. 121, 346–351. https://doi.org/10.1016/j.exppara.2008.12.012 (2009).
Shokoples, S. E., Ndao, M., Kowalewska-Grochowska, K. & Yanow, S. K. Multiplexed real-time PCR assay for discrimination of Plasmodium species with improved sensitivity for mixed infections. J. Clin. Microbiol. 47, 975–980. https://doi.org/10.1128/jcm.01858-08 (2009).
Hall, T. A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 41, 95–98 (1999).
Brown, N. P., Leroy, C. & Sander, C. MView: A web-compatible database search or multiple alignment viewer. Bioinformatics 14, 380–381. https://doi.org/10.1093/bioinformatics/14.4.380 (1998).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Hammer, Ø, Harper, D. A. T. & Ryan, P. D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 4, 9 (2001).
Hutcheson, K. A test for comparing diversities based on the Shannon formula. J. Theor. Biol. 29, 151–154. https://doi.org/10.1016/0022-5193(70)90124-4 (1970).
Garrett-Jones, C. The human blood index of malaria vectors in relation to epidemiological assessment. Bull. World Health Organ. 30, 241–261 (1964).
Newcombe, R. G. Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat. Med. 17, 857–872. https://doi.org/10.1002/(sici)1097-0258(19980430)17:8%3c857::aid-sim777%3e3.0.co;2-e (1998).
Acknowledgements
We thank our field team of the ICEMR project in Kenya for collecting mosquito samples. We thank Sijia Yang, Jiazheng Chen, Andrew Shin, Zhewei Liu, Hannah Li, Stacia Octaviani, Ethan Teng, Viola Lwin for helping with mosquito DNA extraction and PCR. We are grateful to Dr. John Githure for his critical comments and helpful suggestions on the manuscript. We thank the editor and three anonymous reviewers for their constructive comments that help to improve this manuscript. This work is supported by the National Institutes of Health (NIH) Grants: U19AI129326, R01AI050243, R01AI123074, and D43TW001505.
Author information
Authors and Affiliations
Contributions
D.Z., E.H.S. and G.Y. contributed to the conception of the study. H.A., Y.A.A. and A.K.G. participated in the design and implementation of entomological studies. D.Z. carried out the PCR and sequence analysis, interpretation of data and drafted the manuscript. X.W., S.K. and G.Z. contributed to the data interpretation. D.Z., E.H.S., S.K., X.W. and G.Y. provided substantial contributions to conception and design, interpretation of data and drafting the article. All authors contributed to the critical revision of the manuscript for important intellectual content.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhong, D., Hemming-Schroeder, E., Wang, X. et al. Extensive new Anopheles cryptic species involved in human malaria transmission in western Kenya. Sci Rep 10, 16139 (2020). https://doi.org/10.1038/s41598-020-73073-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73073-5
This article is cited by
-
Whole transcriptomic analysis reveals overexpression of salivary gland and cuticular proteins genes in insecticide-resistant Anopheles arabiensis from Western Kenya
BMC Genomics (2024)
-
Molecular surveillance of Kelch 13 polymorphisms in Plasmodium falciparum isolates from Kenya and Ethiopia
Malaria Journal (2024)
-
Vectorial drivers of malaria transmission in Jabi Tehnan district, Amhara Regional State, Ethiopia
Scientific Reports (2024)
-
Anopheles rufipes implicated in malaria transmission both indoors and outdoors alongside Anopheles funestus and Anopheles arabiensis in rural south-east Zambia
Malaria Journal (2023)
-
Changes in contributions of different Anopheles vector species to malaria transmission in east and southern Africa from 2000 to 2022
Parasites & Vectors (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
