
Abstract
The effect of pre-oxidation on the corrosion behavior of pure Ti covered with a solid NaCl deposit in the humid O2 flow at 600 °C is studied. The oxide scale, formed by pre-oxidation, protects the substrate from the NaCl induced corrosion during the initial stage. However, the corrosion of the pre-oxidized sample is severely accelerated by solid NaCl after an incubation period. The chlorine, generated from the decomposition of solid NaCl, diffuses into the oxide/substrate interface as ions during the incubation period, which was observed by ToF–SIMS. The chlorine at the oxide/substrate interface induces the fast corrosion after the incubation period although the pre-oxidation scale is complete and compact.
Similar content being viewed by others

Introduction
Compressor blades of airplanes and ships suffer severe corrosion in marine environment, which is mainly due to the fact that marine air contains abundant salts (especially NaCl) and water vapor. At the temperature of 300–600 °C where compressor blades operate, NaCl appears in solid state and gets deposited on the metal surface. The corrosion of several metals and alloys of compressor blades gets accelerated under a synergistic effect of the solid NaCl deposit and humid oxygen.
Extensive works have been done on the corrosion behavior of several metals and alloys of compressor blades. The authors in paper1,2,3,4,5,6,7,8,9,10,11 demonstrated that the corrosion behavior of pure Fe, pure Cr, Fe–Cr alloy, Ti alloy and Ni alloy at 600 ˚C under a deposit of solid NaCl deteriorated sharply. Similarly, the authors investigated the influence of KCl on the oxidation of the 304-type (Fe18Cr10Ni) austenitic stainless steel at 600 ˚C in 5% O2 and in 5% O2 + 40% H2O in paper12,13,14,15,16,17, and the results showed that small additions of potassium chloride strongly accelerated high temperature corrosion. The fast corrosion is mainly due to a chemical reaction of the solid NaCl/KCl deposit with oxides. This reaction destroys the protective oxide scale on the surface and forms a porous non-protective scale (i.e., the scale has numerous holes and voids1,6,8,9,15,17). It is also reported in these investigations that the key factor for the fast corrosion is the chlorine, HCl or Cl2, which cyclically reacts with the substrate at the oxide/substrate interface. The gaseous molecule of chlorine, which is formed during the chemical reaction of solid NaCl and oxide1,8,18, diffuses inward quickly through macro defects (like cracks and holes) in the scale of corrosion products. The chlorine then reacts cyclically with the substrate to generate volatile products (for example CrCl3) which break the protective scale 1,3,6,8.
In our previous work11, we observe that the scale of corrosion products of Ti60 alloys can be divided into an outer the corrosion layer and an inner corrosion layer after exposure at 600 ˚C in the environment of NaCl + H2O + O2. The chlorine exists in a Ti-Cl bond which is formed by replacing O with Cl within lattices of Ti oxides of the inner corrosion layer. We think the chlorine can diffuse inward as ion. In this case, Cl- comes from the decomposition of solid NaCl and the residual sodium reacts with oxides on the surface to form metallic acid salts (like Na2CrO41 and Na4Ti5O1210). In order to make clear the corrosion mechanism, we need to first figure out the state of the chlorine and study its diffusion in the scale of corrosion products, especially during the destruction process of the scale of protective oxide, like TiO2 and Cr2O3.
When pure Ti is oxidized in the pure O2 condition at 600 ˚C, a compact and even oxide scale forms on the surface10. This oxide scale can simulate the passive film to protect the substance from the corrosive environment. When the pre-oxidized pure Ti is exposed in the NaCl + H2O + O2 environment, this oxide scale can slow down the destruction process of the protective passive film such that the behavior of the solid NaCl can be investigated in detail. Thus, to clarify the micro-mechanism under the specific environment of NaCl + H2O + O2, we need to study the effects of pre-oxidation on the corrosion behavior of pure Ti.
In this paper, we investigate the effects of pre-oxidation on the corrosion behavior of pure Ti underneath a solid NaCl deposit in humid O2 flow at 600 ˚C, using scanning electron microscope (SEM) equipped with an energy dispersive spectrometer (EDS), X-ray diffraction (XRD) and time of flight-secondary ion mass spectrometry (ToF–SIMS). After examining the detailed properties of the corrosion products and the diffusion of chlorine in the oxide scale, we discuss possible micro acceleration mechanisms of the chlorine based on experiment results.
Result
Corrosion kinetics
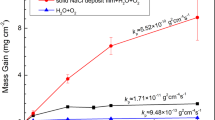
The mass gain curves of pure Ti under different conditions are shown in Fig. 1. For the pre-oxidized samples, the mass gain under the condition of H2O + O2 (about 0.17 mg/cm2) is slightly larger than that under the condition of O2 (about 0.09 mg/cm2). In fact, under both the above two conditions the corrosion is minor. In the initial stage (incubation period), the mass gain is slight when the pre-oxidized pure Ti samples under NaCl deposit. In contrast, the mass gain increases rapidly after this incubation period. After 20 h exposure in NaCl + H2O + O2 or NaCl + O2, the mass gains are about 1.8 mg/cm2 and 1.4 mg/cm2, respectively, which are about one order of magnitude larger than that in the absence of the solid NaCl deposit. Thus, we reach the conclusion that the solid NaCl accelerates the corrosion of the pre-oxidized pure Ti in O2 or humid O2.

The mass gain curves for the corrosion at 600 ˚C of the bare Ti under NaCl + H2O + O2 (red line) and the pre-oxidized Ti under O2 (cyan line), H2O + O2 (black line), NaCl + O2 (green line) and NaCl + H2O + O2 (purple line).
For the bare samples, the mass gain increases dramatically during the whole exposure in NaCl + H2O + O2. After exposed for 20 h, the mass gain is about 6.0 mg/cm2, which is increased by 3.5 times compared with the pre-oxidized samples. Therefore, we can conclude that the pre-oxidation improves the corrosion resistance of pure Ti coated with a solid NaCl deposit in humid O2 at 600 ˚C, especially in the incubation period.
Phase composition and microstructure of the pre-oxidized sample
For the pure Ti samples after pre-oxidation in O2 flow for 20 h, the surface and the cross-sectional morphologies are shown in Fig. 2. As can be observed in Fig. 2a,b, a continuous, compact and even oxide scale is formed on the surface. The thickness of the oxide scale is about 1 μm (Fig. 2b). We show the results of EDS analysis on the oxide scale in Fig. 2c. As can be seen, it consists of Ti and O. Through XRD analysis (which is not shown here), the phase composition of the pre-oxidized sample surface is only TiO2.

Surface morphologies (a), cross-sectional morphologies (b) and EDS patterns (c) of pure Ti oxidized in dry O2 at 600 ˚C for 20 h.
Phase composition and microstructure of the hot corrosion products forming on the bare samples
For the bare samples after 20 h exposure in NaCl + H2O + O2, the surface and the cross-sectional morphologies are shown in Fig. 3. As can be observed, the scale of corrosion products contains two parts: one with a compact structure, and the other with a grid structure part. The corrosion products with a compact structure (marked in Fig. 3a,b) are very complete, uniform and compact. The corrosion products with a grid structure (marked in Fig. 3a,b) contain lots of holes (filled with Ni plating as shown in Fig. 3b), which penetrate the corrosion scale. These through-holes are the fast diffusion path for the corrosion medium. Below the corrosion scale, a loose layer, containing some holes (close pores) and cracks, is formed within the substrate due to the outward diffusion of Ti. The thickness of the corrosion product scale and the loose layer is about 45 μm and15 μm, respectively.

Surface morphologies (a) and cross-sectional morphologies (b) of the bare samples after corrosion in NaCl + H2O + O2 at 600 ˚C for 20 h.
In Fig. 4, we present the XRD patterns of the bare samples exposed in NaCl + H2O + O2 up to 20 h. The results show that the corrosion products mainly consist of TiO2 and some Na4Ti5O12, with the residual NaCl.

X-ray diffraction patterns of the bare samples exposed in NaCl + H2O + O2 up to 20 h at 600 ˚C.
Phase composition and microstructure of the hot corrosion products forming on the pre-oxidized samples
For the pre-oxidized pure Ti samples after 100 h exposure in dry O2 flow, the surface and the cross-sectional morphologies are shown in Fig. 5. As can be seen in Fig. 5a,b, a very thin, compact and continuous scale is formed on the pre-oxidized samples after 100 h exposure in dry O2 flow. From Fig. 5c, we can see that the thickness of the oxide scale is about 2.5 μm.

Surface morphologies (a), the high magnification image (b) and cross-sectional morphologies (c) of the pre-oxidized samples after oxidized in dry O2 at 600 ˚C for 100 h.
For the pre-oxidized samples after 100 h exposure in humid O2 flow, the surface and the cross-sectional morphologies are presented in Fig. 6. As can be observed from Fig. 6a,b, a very thin, compact and continuous scale which contains both granulated corrosion products and compact products was formed on the pre-oxidized samples. From Fig. 6c, we can see that the oxide scale is divided into a loose outer layer and a compact inner layer, with a thickness of about 1.2 μm and 1 μm, respectively.

Surface morphologies (a), the high magnification image (b) and cross-sectional morphologies (c) of the pre-oxidized samples after oxidized in humid O2 at 600 ˚C for 100 h.
For the pre-oxidized samples after 20 h exposure in NaCl + O2, the surface and the cross-sectional morphologies are displayed in Fig. 7. As can be seen in Fig. 7a, the corrosion products on the surface are in clusters. As can be observed in Fig. 7b,c, the corrosion product scale can be divided into an outer layer and an inner layer. The outer layer is filled with clustered corrosion products, and the inner layer whose thickness is about 5 μm contains loose corrosion products.

Surface morphologies (a) and cross-sectional morphologies (b) and the high magnification image (c) of the pre-oxidized samples after corrosion in NaCl + O2 at 600 ˚C for 20 h.
For the pre-oxidized samples after 20 h exposure in NaCl + H2O + O2, the surface and the cross-sectional morphologies are depicted in Fig. 8. As can be observed in Fig. 8a,b, corrosion products have a very thin, compact and continuous scale, with some blind holes forming on the surface where we can also observe some defects (like pore). Furthermore, the scale can be divided into a loose inner corrosion layer and a compact outer corrosion layer. As shown in Fig. 8c, the thickness of the outer corrosion layer and the inner corrosion layer is 8 μm and 10 μm, respectively.

Surface morphologies (a) and cross-sectional morphologies (b) and the high magnification image (c) of the pre-oxidized samples after corrosion in NaCl + H2O + O2 at 600 ˚C for 20 h.
For the pre-oxidized samples after 1 h exposure in NaCl + H2O + O2, the surface and the cross-sectional morphologies are shown in Fig. 9. From Fig. 9a, we can observe a compact scale on which some loose corrosion products are scattered. In Fig. 9b, we can observe that under the loose product, the scale still remains complete and compact. This implies that the loose products are formed due to the reaction of the outward diffusion metal ions with the corrosive species. From Fig. 9d, we can observe that the thickness of this scale is about 1 μm.

Surface morphologies (a), the high magnification image (b, c) and cross-sectional morphologies (d) of the pre-oxidized samples under a solid NaCl deposit layer in humid O2 for 1 h at 600 ˚C.
For the pre-oxidized samples after 5 h exposure in NaCl + H2O + O2, the surface and the cross-sectional morphologies are depicted in Fig. 10. From Fig. 10a, we can observe that the porous corrosion products spread over the entire surface. This is consistent with the fact in Fig. 1 that the mass gain increased rapidly after the incubation period and the corrosion is severe. From Fig. 10b, we can observe that the scale of the corrosion products is compact and its thickness is about 1.2 μm. We can also observe some loose parts at the interface between the scale and metal.

Surface morphologies (a) and cross-sectional morphologies (b) of the pre-oxidized samples under a solid NaCl deposit layer in humid O2 for 5 h at 600 ˚C.
The corrosion products of the pure Ti samples are identified by XRD. The XRD patterns in Fig. 11 show that only TiO2 was formed on the pre-oxidized samples after 100 h exposure in dry O2 flow (Fig. 11a) or in H2O + O2 flow (Fig. 11b). In Fig. 12, we can observe that the corrosion products are TiO2, Ti2O and Na4Ti5O12 on the pre-oxidized samples after 20 h exposure in NaCl + O2 (Fig. 12b), whereas the Na4Ti5O12 cannot be identified by XRD after 5 h exposure (Fig. 12a). As shown in Fig. 13c, after the pre-oxidized samples are exposed in NaCl + H2O + O2 for 20 h, the corrosion products contain both TiO2 and Na4Ti5O12. There still remains residual NaCl as well as the substrate (pure Ti) on the surface. As shown in Fig. 13b, after 5 h exposure in NaCl + H2O + O2 (which is after the incubation period), the corrosion products also contain Na4Ti5O12. Since the diffracted intensity of Na4Ti5O12 in Fig. 13c are larger than that in Fig. 13b, we can infer that more Na4Ti5O12 is formed when the pre-oxidized samples are exposed in NaCl + H2O + O2 for a longer time period. As shown in Fig. 13a, after 1 h exposure in NaCl + H2O + O2 (which is during the incubation period), there are not Na4Ti5O12 among the corrosion products. We conclude the corrosion products under the above different conditions in Table 1.

X-ray diffraction patterns of the pre-oxidized samples after exposed in dry O2 flow (a) and in humid O2 flow (b) for 100 h.

X-ray diffraction patterns of the pre-oxidized samples after exposed in NaCl + O2 for 5 h (a) and for 20 h (b).

X-ray diffraction patterns of the pre-oxidized samples after exposed in NaCl + H2O + O2 for 1 h (a), 5 h (b) and 20 h (c).
Time of flight-secondary ion mass spectrometry, ToF–SIMS
Figure 14 shows the depth profiles for the negative ions from the pre-oxidized samples after 1 h and 5 h exposure in NaCl + H2O + O2. The \({\text{O}}_{2}^{ - }\) ion signal in Fig. 14 is applied here to indicate the distribution of oxygen in the scale and to identify the interface between the corrosion products scale and the substrate. In Fig. 14a, the \({\text{O}}_{2}^{ - }\) ion exhibits two levels of concentration, and the transition region is around the interface of the corrosion product scale and the substrate. Therefore, the average of logarithmic value is pinpointed as the interface as shown in Fig. 14a. Similarly, this interface is also identified in Fig. 14b.

ToF–SIMS sputter depth profiles in the corrosion products scales formed on the pre-oxidized samples under a NaCl deposit in an atmosphere of humid O2 at 600 ˚C for 1 h (a) and 5 (b).
In Fig. 14a, we can also observe that after 1 h exposure in NaCl + H2O + O2, Cl- ions first enrich on the surface of the pre-oxidized samples (which was caused by the residual NaCl) and then decrease rapidly in the corrosion product scale. Afterwards, Cl- is enriched at the oxide/metal interface, which indicates that the chlorine penetrates the oxide and accumulates at the oxide/metal interface. In Fig. 14b, after the pre-oxidized samples are exposed in NaCl + H2O + O2 for 5 h, a similar distribution of Cl- in the corrosion product scale was observed. Particularly, the Cl- ion is still enriched when the \({\text{O}}_{2}^{ - }\) ion decreases around the oxide/metal interface, which implies that some chlorine diffuses into the metal substrate. The intensity of Cl- around the oxide/metal interface is stronger in Fig. 14b than that in Fig. 14a. This implies that more chlorine penetrates into the oxide after a longer exposure in the presence of solid NaCl.
The OH- signal in Fig. 14 is applied here to indicate the distribution of H in the scale. From Fig. 14a, the distribution of the OH- is similar with Cl-. First, the OH- ion enriches on the surface and then decrease rapidly in the corrosion product scale. This is the information of water vapor. Afterwards, OH- is enriched at the oxide/metal interface where Cl- is accumulated, which indicates that the H penetrates the oxide along with the chlorine.
Discussion
The acceleration of NaCl to the corrosion of pure Ti
As shown in Fig. 2, after pure Ti samples were oxidized in the pure O2 flow at 600 ˚C for 20 h, a compact TiO2 scale with a thickness of about 1 μm was formed on the surface, which is caused by the following reaction between the metal substrate and O2:
In contrary, the mass gain of pure Ti under a solid NaCl deposit layer (NaCl + H2O + O2) increased quickly and the mass gain is about 6.0 mg/cm2 after 20 h exposure (Fig. 1), which is much larger than that in the absence of solid NaCl (about 0.3 mg/cm2 10). The resulting scale consists of plentiful and porous corrosion products (Fig. 3) and its thickness is about 45 μm. The above phenomena indicate that pure Ti suffered severe corrosion when exposed in NaCl + H2O + O2. This severe corrosion has been discussed in our previous work10 and the main reason is that the protective TiO2 scale cannot be formed due to the occurrence of a series of chemical reactions, and the corrosion becomes active. In the previous mechanism, NaCl first destroys the protective scale as follow:
The TiCl4 reacts with H2O to form HCl:
Then, HCl reacts with the substrate cyclically:
These reactions are thermodynamically spontaneous due to the negative ΔGº at 600 ˚C, as presented in Table 2.
As shown in Fig. 3b, in the corrosion product scale there are holes which provide rapid diffusion channels for corrosive species (for example, oxygen, chlorine and water vapor). Meanwhile, a corrosion scale is formed by the outward diffusion of Ti which results in a 15 μm loose layer that is observed under the corrosion products. Hence, the corrosion product scale is non-protective and the corrosion of pure Ti is greatly accelerated by solid NaCl in an O2 + H2O environment at 600 ˚C, which is similar with the observation for several Ti alloy11,18,19,20.
The protection of pre-oxidation scale to the pure Ti
After pre-oxidation in O2 flow for 20 h, a continuous, compact and even oxide scale is formed on the surface (Fig. 2). The pre-oxidation scale is only TiO2 (Fig. 2c) and its thickness is about 1 μm (Fig. 2b). Since this scale can protect the substrate, the mass gain of the pre-oxidized samples is small in the dry O2 or humid O2 (Fig. 1).
When pre-oxidized samples are exposed in the presence of solid NaCl (NaCl + O2 and NaCl + H2O + O2), the mass gain of the pre-oxidized samples is smaller than that of the bare samples (Fig. 1), especially in the incubation period (the mass gain increases slowly). The resulting scale on the pre-oxidized samples (which is compact as shown in Figs. 7 and 8) is thinner than that on the bare samples (which is porous as shown in Fig. 3). Thus, the pre-oxidation can protect the pure Ti samples from the corrosion under NaCl deposit. This is mainly attributed to the compact TiO2 scale formed on the surface in the pre-oxidation (Fig. 2). During the pre-oxidation, the thickness of the formed TiO2 scale is about 1 μm, and the pre-oxidation scale is larger thicker than the passive film forming on the bare sample. Thus, the TiO2 scale can be a barrier layer for the diffusion of corrosive species (for example, oxygen, water vapor and HCl) and reduce the corrosion to a certain extent, especially in the incubation period.
The destruction of pre-oxidation scale
When exposed in humid O2, the pre-oxidized samples suffer a slightly more serious oxidation than when exposed in dry O2 (Fig. 1). This is because H2O tends to dissociate at the defects on TiO2/(110) planes into free H atoms and OH- groups21,22,23 when the pre-oxidized samples is exposed in humid O2. The papers21,22 reported that along the crystal channels in c-axis direction, the diffusion rate of hydrogen is at least one order of magnitude larger than that in the perpendicular direction of c-axis. Thus, the generated hydrogen atoms are more prone to diffuse into TiO2 through the former one. This dissolution of hydrogen in TiO2 will form hydrogen defects, thus the hydrogen can increase the concentration of crystal defects and increase the outward diffusion of Ti ions23. This fast diffusion in the scale causes a larger corrosion rate and hence the hole forms in the oxide scale (Fig. 6), which make the corrosion product scale thicker and more porous (Fig. 6c). Thus, the water vapor slightly reduces the protection of the pre-oxidation scale by forming the H defects.
From Fig. 1, we can observe the following: (1) after the incubation period, the mass gain of the pre-oxidized samples increases more rapidly when exposed in the presence of the solid NaCl (NaCl + O2 or NaCl + H2O + O2) than when exposed in the absence of the solid NaCl (O2 or H2O + O2); (2) after 20 h exposure, the pre-oxidized samples has a larger mass gain under the condition of NaCl + O2 or NaCl + H2O + O2 than under the condition of O2 or H2O + O2. Additionally, we can observe that the corrosion product scale of the pre-oxidized samples is thicker when exposed in the presence of the solid NaCl (Figs. 7 and 8) than when exposed in the absence of the solid NaCl (Figs. 5 and 6). Thus, we can claim that the corrosion of the pre-oxidized samples is accelerated by solid NaCl. The fast corrosion and the formation of Na4Ti5O12 on the surface (Fig. 9c,d) indicate that the “protective oxide” formed in the pre-oxidation is not completely inert to solid NaCl.
The diffusion of the chlorine in the pre-oxidation scale
The evidence from XRD and SEM/EDX show that Na4Ti5O12 formed on the surface (see Figs. 4 and 9c,d). This indicated that the solid NaCl reacted with the TiO2 scale. Similar observations have been reported for Ti alloy using the same experimental conditions18. In that case, it was argued that the fast corrosion in an O2 + H2O environment is triggered by titanate formation. Thus, in an O2 + H2O + NaCl environment, the formation of titanate destroys the protective scale formed on the surface, and then the gathered chlorine circularly reacts with the substrate. Thus, for the corrosion of pure Ti/Ti alloy in the NaCl + H2O + O2 environment, the chlorine is the main factor.
For the pre-oxidized samples, a thick TiO2 scale is formed on the surface; thus, it takes more time to destroy the protective TiO2 scale by Na4Ti5O12 formation and the mass gain changes little during the incubation period (Fig. 1). Furthermore, after the sodium forms Na4Ti5O12, the gathered chlorine diffuses inward to the metal/oxide interface (Fig. 14a). Shu et al. 18 report that the chlorine permeates into the corrosion product scale as molecule, like HCl or Cl2. For the pre-oxidized samples, a compact TiO2 formed on the surface protects the substrate from the inward diffusion of chlorine and the fast corrosion. Nevertheless, during the incubation period, the chlorine also diffuses into the metal/oxide interface (Fig. 14a) while the TiO2 scale is complete (Fig. 9). This implies that the chlorine cannot be molecule. In our previous work, the chlorine forms Ti-Cl bond in the inner corrosion layer by the dopant Cl replacing O in the Ti oxides lattice when Ti60 alloy was exposed in the NaCl + H2O + O2. On the other hand, the diffusion of ion in the TiO2 is fast and some NaCl is split into ions (Na+ and Cl-) at 600 ˚C. Hence, we think the chlorine permeates into the corrosion product scale as ion (Cl-).
As shown in Fig. 14b, when the pre-oxidized samples suffer from severe corrosion in NaCl + H2O + O2, the chlorine is also observed around the metal/oxide interface by SIMS. Additionally, the intensity of chlorine is still large when the intensity of oxygen decreases to a relatively small value. As aforementioned, the substrate locates at the places where the intensity of oxygen is small. Hence, we can infer that the chlorine diffuses into the substrate and directly binds with the metal of the substrate during the fast corrosion. The gathered compound diffuses outward, resulting in the loss of metal in the substrate. After 20 h exposure, a mass of the metal reacts with the chlorine and diffuses outward, and the inner corrosion layer is formed under the outer corrosion layer (Fig. 8b).
Conclusion
During the pre-oxidation, a compact and thick TiO2 scale forms on the surface of pure Ti. This TiO2 scale is the barrier layer for the diffusion of corrosive species (for example, oxygen, chlorine, and water vapor), which can protect the substrate from fast corrosion. However, the “protective oxide” is not completely inert to solid NaCl and the corrosion rate of the pre-oxidized sample is also greatly accelerated by solid NaCl after an incubation period. During the incubation period, the chlorine, generated in the decomposition of solid NaCl at 600 ˚C, diffuses into the oxide/substrate interface as ions to enhance the corrosion rate, and then the residual sodium reacts with the oxide on the surface. After the incubation period, the chlorine diffuses into the substrate and directly reacts with the substrate, resulting in the fast corrosion.
Experiment
Materials preparation
The material used in this study was pure Ti and the samples were cut into the size of 10 mm × 15 mm × 2.5 mm. Prior to experiments, all samples were mechanically grinded with 800# SiC paper, ultrasonically degreased in alcohol for about 20 min, and dried in the air. The bare samples were then exposed in the pure O2 flow at 600 ˚C for 20 h for the pre-oxidization. The surfaces of preheated samples were covered with a layer of NaCl deposit by repeatedly brushing and drying with saturated NaCl solution1,6,8,9,18,24, until about 4 ± 0.2 mg/cm2 solid NaCl was deposited on.
Corrosion experiments
The corrosion tests were carried out in a thermo-balance 1,6,8,9,24. The continuous mass gain during the corrosion experiment was obtained with a thermo-gravimetric analysis (TGA).
The pure O2 was bubbled into the distilled water with a glass bubbler (when the inner diameter of the tube was about 3.2 cm, the flow rate of pure O2 was about 140 mL/min in this study) to produce the test atmosphere (humid O2). According to the relationship between the vapor pressure of water and its temperature, we precisely set up the temperature of the distilled water in the glass bubbler to control the amount of the water vapor. In this study, the temperature of the distilled water was about 70 ˚C, producing about 30.8 vol.% water vapor. In order to avoid the water vapor condensing inside the thermo-balance, a counter flow of pure N2 was passed through the thermo-balance, whose flow rate was about 400 mL/min. After the furnace reached 600 ˚C and the gas flows of humid O2 as well as pure N2 stabilized, the samples were quickly lowered into the constant temperature zone of the furnace tube.
In this study, the corrosion experiments were respectively carried out under the following four conditions: the first condition was with a solid NaCl deposit layer in a humid O2 flow at 600 ˚C (denoted as NaCl + H2O + O2); the second condition was carried out in a humid O2 flow at 600 ˚C (denoted as H2O + O2); the third condition was with a solid NaCl deposit layer in a dry O2 flow at 600 ˚C (denoted as NaCl + O2); and the fourth condition was carried out in a dry O2 flow at 600 ˚C (denoted as O2). We present the explicit environmental parameters in Table 3.
Morphologies and chemical composition analysis
The surface morphologies and the cross-sectional morphologies of corrosion products scale were collected by SEM–EDS. The chemical composition of corrosion products was identified by XRD.
To protect the oxide scale from fracture and spall during the metallographic preparation, the corroded samples were wrapped into a thin nickel foil by electroless plating and were embedded into the epoxy resin. Then, the corroded samples were grinded to 3000 grit with SiC paper. Finally, the corroded samples were polished with diamond paste. The samples were washed with distilled water to remove the residual solid NaCl, and then dried in air, before surface investigation by SEM. Under the condition of H2O + O2 and O2, the mass gains of the pre-oxidized sample were very small (Fig. 1) and the corrosion was minor. The surface and the cross-sectional morphologies after a longer corrosion time (100 h) were investigated.
Time of flight-secondary ion mass spectrometry, ToF–SIMS
The pre-oxidized samples were first exposed in either NaCl + H2O + O2 or NaCl + O2. Then, they are ultrasonically washed with distilled water to remove the residual solid NaCl. After the pre-oxidized samples were further dried in the air, they were analyzed using a ToF-SIMS5 instrument (ION-TOF GmbH), which allowed parallel mass registration with high sensitivity and high mass resolution. A cesium liquid–metal ion (LMI) gun at 20 keV beam energy was used for spatially resolved ToF–SIMS analysis.
References
Shu, Y., Wang, F. & Wu, W. Corrosion behavior of pure Cr with a solid NaCl deposit in O2 plus water vapor. Oxid. Met. 54, 457–471. https://doi.org/10.1023/a:1004690518225 (2000).
Liu, L., Li, Y., Zeng, C. & Wang, F. Electrochemical impedance spectroscopy (EIS) studies of the corrosion of pure Fe and Cr at 600 °C under solid NaCl deposit in water vapor. Electrochim. Acta 51, 4736–4743. https://doi.org/10.1016/j.electacta.2006.01.033 (2006).
Tang, Y., Liu, L., Li, Y. & Wang, F. The electrochemical corrosion mechanisms of pure Cr with NaCl deposit in water vapor at 600 °C. J. Electrochem. Soc. 158, C237. https://doi.org/10.1149/1.3596167 (2011).
Tang, Y., Liu, L., Li, Y. & Wang, F. Evidence for the occurrence of electrochemical reactions and their interaction with chemical reactions during the corrosion of pure Fe with solid NaCl deposit in water vapor at 600 °C. Electrochem. Commun. 12, 191–193. https://doi.org/10.1016/j.elecom.2009.11.021 (2010).
Tang, Y., Liu, L., Fan, L., Li, Y. & Wang, F. The corrosion behavior of pure iron under solid Na2SO4 deposit in wet oxygen flow at 500 °C. Materials 7, 6144–6157. https://doi.org/10.3390/ma7096144 (2014).
Wang, F. & Shu, Y. Influence of Cr content on the corrosion of Fe-Cr alloys: the synergistic effect of NaCl and water vapor. Oxid. Met. 59, 201–214. https://doi.org/10.1023/A:1023083309041 (2003).
7Pujilaksono, B. et al. Oxidation of binary FeCr alloys (Fe–2.25Cr, Fe–10Cr, Fe–18Cr and Fe–25Cr) in O2 and in O2 + H2O environment at 600 °C. Oxidation of Metals75, 183–207, https://doi.org/10.1007/s11085-010-9229-z (2011).
Shu, Y., Wang, F. & Wu, W. Synergistic effect of NaCl and water vapor on the corrosion of 1Cr-11Ni-2W-2Mo-V steel at 500–700 ˚C. Oxid. Met. 51, 97–110. https://doi.org/10.1023/a:1018854202982 (1999).
Wang, F., Geng, S. & Zhu, S. Corrosion behavior of a sputtered K38G nanocrystalline coating with a solid NaCl deposit in wet oxygen at 600 to 700 ˚C. Oxid. Met. 58, 185–195. https://doi.org/10.1023/a:1016072726338 (2002).
Fan, L. et al. Corrosion behavior of pure Ti under a solid NaCl deposit in a wet oxygen flow at 600 degrees C. Metals 6, 11. https://doi.org/10.3390/met6040072 (2016).
11Fan, L. et al. Corrosion behavior of Ti60 alloy under a solid NaCl deposit in wet oxygen flow at 600 degrees C. Scientific Reports6, https://doi.org/10.1038/srep29019 (2016).
Pettersson, J., Asteman, H., Svensson, J. E. & Johansson, L. G. KCl induced corrosion of a 304-type austenitic stainless steel at 600 °C; The role of potassium. Oxid. Met. 64, 23–41. https://doi.org/10.1007/s11085-005-5704-3 (2005).
Pettersson, C., Pettersson, J., Asteman, H., Svensson, J. E. & Johansson, L. G. KCl-induced high temperature corrosion of the austenitic Fe–Cr–Ni alloys 304L and Sanicro 28 at 600 °C. Corros. Sci. 48, 1368–1378. https://doi.org/10.1016/j.corsci.2005.05.018 (2006).
Pettersson, C., Johansson, L. G. & Svensson, J. E. The influence of small amounts of KCl(s) on the initial stages of the corrosion of alloy sanicro 28 at 600 °C. Oxid. Met. 70, 241–256. https://doi.org/10.1007/s11085-008-9118-x (2008).
Jonsson, T. et al. The influence of KCl on the corrosion of an austenitic stainless steel (304L) in oxidizing humid conditions at 600 °C: a microstructural study. Oxid. Met. 72, 213–239. https://doi.org/10.1007/s11085-009-9156-z (2009).
Pettersson, J., Svensson, J. E. & Johansson, L. G. KCl-Induced corrosion of a 304-type austenitic atainless ateel in O2 and in O2 + H2O environment: the influence of temperature. Oxid. Met. 72, 159–177. https://doi.org/10.1007/s11085-009-9153-2 (2009).
17Jonsson, T., Folkeson, N., Svensson, J. E., Johansson, L. G. & Halvarsson, M. An ESEM in situ investigation of initial stages of the KCl induced high temperature corrosion of a Fe-2.25Cr-1Mo steel at 400 degrees C. Corrosion Science53, 2233–2246, https://doi.org/10.1016/j.corsci.2011.03.007 (2011).
Shu, Y., Wang, F. & Wu, W. Corrosion behavior of Ti60 alloy coated with a solid NaCl deposit in O2 plus water vapor at 500–700 ˚C. Oxid. Met. 52, 463–473. https://doi.org/10.1023/a:1018864216554 (1999).
Dumas, P. & Stjohn, C. Nacl-induced accelerated oxidation of a titanium alloy. Oxid. Met. 10, 127–134. https://doi.org/10.1007/Bf00614242 (1976).
Yao, Z. & Marck, M. NaCl-induced hot corrosion of a titanium aluminide alloy. Mater. Sci. Eng., A 192–193, 994–1000. https://doi.org/10.1016/0921-5093(95)03345-9 (1995).
Johnson, O. W., Paek, S. H. & Deford, J. W. diffusion of h and d in TIO2 - suppression of internal fields by isotope-exchange. J. Appl. Phys. 46, 1026–1033. https://doi.org/10.1063/1.322206 (1975).
Fowler, J. D., Chandra, D., Elleman, T. S., Payne, A. W. & Verghese, K. T Diffusion in AL2O3 and BEO/D BEO. J. Am. Ceram. Soc. 60, 155–161. https://doi.org/10.1111/j.1151-2916.1977.tb15493.x (1977).
Douglass, D. L., Kofstad, P., Rahmel, A. & Wood, G. C. International workshop on high-temperature corrosion. Oxid. Met. 45, 529–620. https://doi.org/10.1007/bf01046850 (1996).
Wang, C., Jiang, F. & Wang, F. Corrosion inhibition of 304 stainless steel by nano-sized Ti/silicone coatings in an environment containing NaCl and Water Vapor at 400–600°C. Oxid. Met. 62, 1–13. https://doi.org/10.1023/B:OXID.0000038782.77162.5b (2004).
Acknowledgments
The investigation is supported by the National Key Research and Development Program of China (2017YFF0210406), the Shanxi National Science Foundation (2019JQ-213) and the Basic Research and Strategic Reserve Technology Research Fund (project of China National Petroleum Corporation) (2018Z-01).
Author information
Authors and Affiliations
Contributions
L.F. designed and performed experiment. L.F., L.L., C.X.Y, A.Q.F, J.T.Y, F.H.W. and Y.L. discussed and analyzed data. L.F. and L.L. wrote the manuscript. L.L., Y.H.L, H.W revised the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, L., Liu, L., Lv, Y. et al. Effects of pre-oxidation on the corrosion behavior of pure Ti under coexistence of solid NaCl deposit and humid oxygen at 600 °C: the diffusion of chlorine. Sci Rep 10, 16291 (2020). https://doi.org/10.1038/s41598-020-73034-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73034-y
This article is cited by
-
Corrosion behavior of pure Ti under continuous NaCl solution spraying at 600 °C
npj Materials Degradation (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
