
Abstract
Ensete ventricosum (Musaceae, enset) is an Ethiopian food security crop. To realize the potential of enset for rural livelihoods, further knowledge of enset diversity, genetics and genomics is required to support breeding programs and conservation. This study was conducted to explore the enset genome to develop molecular markers, genomics resources, and characterize enset landraces while giving insight into the organization of the genome. We identified 233 microsatellites (simple sequence repeats, SSRs) per Mbp in the enset genome, representing 0.28% of the genome. Mono- and di-nucleotide repeats motifs were found in a higher proportion than other classes of SSR-motifs. In total, 154,586 non-redundant enset microsatellite markers (EMM) were identified and 40 selected for primer development. Marker validation by PCR and low-cost agarose gel electrophoresis revealed that 92.5% were polymorphic, showing a high PIC (Polymorphism Information Content; 0.87) and expected heterozygosity (He = 0.79–0.82). In silico analysis of genomes of closely related species showed 46.86% of the markers were transferable among enset species and 1.90% were transferable to Musa. The SSRs are robust (with basic PCR methods and agarose gel electrophoresis), informative, and applicable in measuring enset diversity, genotyping, selection and potentially breeding. Enset SSRs are available in a web-based database at https://enset-project.org/EnMom@base.html (or https://enset.aau.edu.et/index.html, downloadable from Figshare).
Similar content being viewed by others


Introduction
Ensete ventricosum (Musaceae) is a giant monocarpic perennial herbaceous plant, wide spread in tropical East and Southern Africa, and domesticated in Ethiopia, where it provides the main starch staple for 20 million rural people1. It is known as Ethiopian, Abyssinian or false banana, ensete, or (as used here) enset. The genus extends from Africa to tropical eastern Asia, and is wild as well as cultivated in Ethiopia, a Vavilovian centre of plant diversity2,3. While banana and plantain (Musa spp.) are cultivated for their fruits, enset starch is extracted from the pseudostem, leaf sheaths and underground corm. Enset is harvested year-round, and is reportedly drought tolerant4 so is known as a food-security crop5. With good management and relatively low inputs, production per unit area is higher than most cereals6, so it can feed a large population7. Enset plants store starch, reaching a maximum shortly before flowering, and 40 kg starch can be harvested from 3 to 4 year old plants. Enset is also good sources for fibre, medicines and animal fodder and the leaves may be used for packaging, fibre, and roofing8.
Several thousand landraces with high genetic diversity have been reported in banana9 worldwide. For enset, multiple morphologically distinct landraces are grown in each small holding, and DNA markers10 show they are diverse. The diversity of the few hundred enset landraces, with local names, is now being surveyed from different agro-ecological regions in Ethiopia10,11. There are several enset germplasm collections maintained in Ethiopia, including Hawassa University, Wolkite University, Southern Agricultural Research Institute (SARI)12,13 and Ethiopian Biodiversity Institute (EBI)14.Unlike triploid banana, the diploid enset is able to produce plants from the seeds of its non-edible fruits, although most enset grown on farms is not permitted to flower and is propagated clonally. Enset genetic resource conservation and management rely on cultivation with regular renewal. Vegetative propagation, vernacular naming systems, and the long juvenility period makes enset improvement or breeding difficult and expensive15.
With the knowledge gap about enset genetics, distribution and diversity, several studies have been conducted to estimate genetic diversity and define relationships among the limited enset germplasm stocks. Most of these studies use DNA markers including Random Amplified Polymorphic DNA (RAPD)16, Inter-Simple Sequence Repeats (ISSR)11, and Amplified Fragment Length Polymorphism (AFLP)17, although some of these marker techniques are high-cost, show limited reproducibility, or identify only dominant alleles (from a heterozygous crop without systematic inbreeding). Simple sequence repeats (SSR) were identified by using a CT and GT repeat-enriched pyrosequencing (454) library by Temesgen et al.10, who tested 217 pairs of microsatellite primers, of which 67 showed amplification; 59 were polymorphic and 34 were published and used for their analysis10.
There is minimal breeding of enset as a crop, although genetic diversity analysis shows that cultivated accessions are genetically different to most wild accessions5,18,19,20; there is the opportunity for future application of marker-assisted breeding10 and marker based parental choice in crossing programmes in the crop. Genetic diversity and population structure studies are required for enset in Ethiopia for germplasm management, identifying landraces or cultivars, collection-management, and determining phylogenetic relationships. Genotyping-by-sequencing and transcriptome sequencing (RAD-seq or RNA-seq), and including SNP (Single Nucleotide Polymorphism) analysis, provide the deepest and highest coverage of genomic diversity, but both experimental and analytical costs are very substantial.
Microsatellites (SSRs, along with single-locus PCR markers such as cleaved amplified polymorphic sequence) are robust, have adequate genome-wide coverage for most targeted purposes, are relatively low cost, and can be used informatively on a small or large number of accessions. Development of large numbers of microsatellite markers—thousands to tens of thousands—from genomic DNA sequences is possible. Their applications include genetic diversity surveys21,22,23,24,25, population structure analysis26,27, genotyping28,29,30, association mapping31,32,33,34,35, linkage mapping36,37 and ultimately plant breeding. Microsatellite markers are usually robust with less dependence on DNA quality and laboratory environment, require only basic molecular-biology equipment (here, aiming to find polymorphisms detectable by agarose gel electrophoresis), are co-dominant, and often are transferable to related species. Draft whole genome sequences of Ensete ventricosum are available in the public domain38,39, although so far newly isolated sequences or transferable banana microsatellite markers have been used for enset genetic diversity study40. Large microsatellite databases have been developed for many crop plants41,42.
The present study aimed to exploit the draft whole-genome sequence of enset to (1) Identify microsatellite sequences and characterize their genome-wide landscape, including the nature of motifs, frequency, genomic distribution and, where appropriate, functional annotation; (2) Identify candidate primers for all microsatellites in a large-scale microsatellite database, and develop web-based open tools for access; (3) Validate a subset of candidate microsatellite primers both in silico and by PCR amplification of isolated DNA and fragment analysis; (4) Compare the genome-wide microsatellite landscape in enset with Musa species, assessing cross-taxa transferability both in silico and by PCR; (5) Recommend a sub-set of markers use for genetic diversity analysis.
Results
Microsatellite content in enset and cross-species comparisons
The pipeline for identification of microsatellites in whole genome sequences of four published enset landraces (Ensete ventricosum, ‘Bedadeti’ ‘Derea’ ‘Onjamo’ and ‘Jungle Seeds’), is shown in Fig. S1 and the complete data are given in the enset database at https://enset-project.org/EnMom@base.html. The database can be downloaded from https://figshare.com/s/20dd8c0d0a2994dbce8d with CC-BY-4.0 licence). Between 93,000 and 115,000 microsatellites were detected in the genome assemblies (Fig. 1, Tables 1 and S1), with an average microsatellite density of 233 per Mb (Table S1). Mono-, di-and tri- nucleotide repeats were frequent, with fewer tetra-, penta- and hexa- nucleotide repeats (Fig. 1a). Microsatellites were classified43 with the longer class I (> 20 nt) slightly more frequent that class II (≤ 20 nt) (Fig. 1b). AT-rich microsatellites were seven-fold more frequent than GC-rich microsatellites or those with equal AT/GC content (Fig. 1c).

Comparative microsatellite frequency distribution in four Ensete ventricosum genomes. (a) Motif type distribution, (b) motif class (Class I > 20 bp and Class II ≤ 20 bp) distributions, (c) motif distribution by nucleotide base composition (balance motif rich = motif compose equal ration of AT and GC content), (d) motif distribution among different genomic regions, (e) distribution of mono-hexa nucleotide repeat motifs.
Flanking regions (about 200 bp each side) of microsatellites were extracted and classified into intergenic regions (non-coding), coding regions and 3′ or 5′ UTRs. Microsatellites were over-represented near predicted coding sequences (24% of sequences compared to 16% in the entire genome) (Fig. 1d). The most common mono- to hexa-nucleotide microsatellite types in the E. ventricosum genome, are shown in Fig. 1e and Table S2.
Microsatellite content of E. ventricosum was compared with fourteen monocot and one gymnosperm species where sequence data are available using with similar microsatellite discovery methods (Tables S1 and Fig. S2). The microsatellite density (233 SSRs/Mbp and 0.28% of the genome) in E. ventricosum genome was similar to the four Musa species (average 207 SSRs/Mbp), and higher than other studied monocots (28–198 SSRs/Mbp, except Spirode lapolyrhiza (385 SSRs/Mbp; Table S1). Like the Musa species, most microsatellites in enset were AT-rich (67% vs. 69%; Table S1).
Marker development, functional annotation, cross-taxa transferability and comparative mapping
An automated strategy successfully designed primers in about half of the SSR flanking sequences. Primer redundancy or non-specificity often arises from duplicated regions within genomes, and we used a Perl-based script to eliminate redundant primers (6.39–10.12%), leaving a total of 210,404 unique primer pairs (Table 1). As shown above, there were no notable differences in microsatellite frequency between the four landraces, and clustering showed that 27% of primers were identified in two or more landraces, giving 154,586 unique primers (Table 1) deposited in the Enset Microsatellite Marker (EMM) database (at https://enset-project.org/EnMom@base.html). We found that 20 of the new unique microsatellite markers coincided with the 217 identified by targeted sequencing of a CT- and GT-enriched library10 (Table S3).
In silico comparative mapping of the enset marker amplicons to genomes of four Musa species revealed that 19,579 (12.67%; range 12.44–12.92%) were found in each Musa species (Table S4). The high-quality, chromosome-level assemblies of three Musa genomes showed wide spread distribution of the enset markers with 307–573 (average 435 ± 74) allocated to each Musa chromosome, and some clustering around the putative centromere (Table S4 and Fig. S3).
In silico PCR44 was used to explore transferability and polymorphisms of the microsatellite markers across the four enset landraces and three Musa species (Table 1; Tables S5). Between 3 and 7% of markers were landrace-specific (‘Bedadeti’, ‘Derea’, ‘JungleSeeds’ and ‘Onjamo’) and about half were common across the four genomes (Fig. 2a). Only 4353 (1.90%) were transferable to the four Musa genomes (Fig. 2b), mostly di- and tri-nucleotide (> 20 bp, class I) repeats. Analysis of in silico PCR polymorphisms showed that 83% of the markers were polymorphic, with slightly higher polymorphism frequencies among longer (class I), di-nucleotide, and AT or GC-rich microsatellites (Fig. 2c–f).

Venn diagram showing the number of common and specific EMM markers in (a) enset accessions and (b) Musa species. In silico characterization summary of the EMM markers (c–f).
Marker validation and genetic diversity
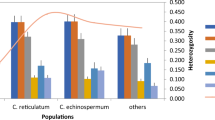
The 154,586 unique markers were filtered in silico to select monomorphic primers, those with functional annotation, and transferability to enset relatives (Tables S5, S6). From a sample of 40 primer pairs used for PCR amplification using DNA from four enset and one Musa samples (Fig. S4), 34 (85.0%) gave the expected primer size, of which 33 were polymorphic (Table S7). In total, 126 polymorphic alleles were scored with a range of 1–6 per locus and an average PIC value of 0.87. The expected heterozygosity (He) ranged from 0.79 to 0.82, while the observed heterozygosity (Ho) ranged from 0.41 to 0.63 (Table S7). From these primers, 15 markers were chosen for genotyping 45 E. ventricosum wild, cultivated and landrace germplasm collections as well as three related species E. superbum, E. glaucum and E. lecongkietii (Fig. S5 and Table S8). The He for all accessions was 0.48. The mean F (Fixation Index) value indicated moderate to high genetic differentiation between species (0.36) (Table S9). The AMOVA analysis for distinguished Ensete species are presented in Table S10, and result reveals that AMOVA analysis enabled some clustering of enset landraces by their genetic variation. Neighbor-joining phylogenetic analysis strongly supported the 45 E. ventricosum accessions as a sister group to the three other Ensete species, well-resolved with bootstrap values > 91%. Within the E. ventricosum accessions, there was weaker bootstrap support for a few phylogenetic groupings of accessions (Fig. 3).

Phylogenetic relation analysis of 45 Enseteventricosum landraces and three Ensete species. “Ensete-ventricosum” and “E-venticosum Maurelii” are commercially available in the UK. “Unknown Red” was a feral plant.
Enset microsatellite database architecture, features and utility
The enset microsatellite marker (EMM) information has been placed in the EMM-database (https://enset-project.org/EnMom@base.html) including search fields for microsatellite type, length, motif type, transferability, polymorphisms, and name (Fig. 4). The search returns a list of markers with Marker ID, microsatellite type and motif, forward- and reverse-primer sequences, and the name of source landrace, with links to additional information including genome position, transferability, predicted polymorphism, PCR product size, flanking sequences, any functional annotations, three sets of primer sequences and annealing temperatures. Query results can be downloaded in XLS and CSV file format for subsequent use.

The online Enset Microsatellite Marker (EMM) database. (a) Enset microsatellite marker search page, (b) search result, (c) linked page for detail information of each EMM.
Discussion
Analysis of microsatellites in enset defined microsatellite numbers, motifs and polymorphisms, and allowed development and testing of a genomic resource of microsatellite markers for landrace identification and analysis of diversity in the species and its relatives. The microsatellite analysis pipeline found an average of 233 SSRs per Mb, within the range known for both monocotyledonous and dicotyledonous species43,44,45,46. As expected, we did not find significant differences in microsatellite occurrence between the four enset landraces across our analyses. As in a range of monocotyledons, dicotyledons and a gymnosperm, mono-, di- and tri-nucleotide microsatellite motifs in enset represented the majority of all motifs between 1- and 6-bases long. Despite reanalysis of the abundance of microsatellite motifs in genome assemblies of four Musa, eight grasses, two other monocots and a gymnosperm, there were few other notable patterns in the abundance of different motif lengths. Tri-nucleotide repeats are twice as frequent as 1- and 2-bp repeats in Poaceae species (except wheat and barley), while di-nucleotide repeats are twice as frequent as 1- and 3-bp repeats in Musaceae. Enset also has a low frequency of 3-bp repeats, with an equal frequency of 1- and 2-bp repeats.
Various approaches to microsatellite identification have been used, including sequencing microsatellite-targeted libraries (e.g.10 in enset), analysis of EST (expressed sequence tag) or RNA-sequence results, analysis of BAC or BAC-end-sequence (GSS, genomic survey sequences), and analysis of whole-genome assemblies. Whole genome sequences use various approaches (technology, depth, length, gap-filling) with widely different N50 values and proportions of reads left unassembled. There are technical limitations in assembly only based on short parid-end reads (Illumina), so repetitive regions (with satellite repeats or transposable elements) will not be assembled, and both microsatellites and primers will normally be included in single read pairs, so longer SSR stretches will be omitted or wrongly assembled. Thus assemblies around the microsatellites analysed here will lie mostly in low-copy-number genomic regions. Previous comparative analyses with diverse plant species including not only E. guineensis47, bamboo48, Triticeae cereal species49, and foxtail millet50, but also sweet orange51, Nicotiana52, cucumbers53 and others also have reported different relative abundances of mono-, di- and tri-nucleotide repeats. Victoria et al.54 reported that di-nucleotide motifs were more frequent in green algae and mosses, with tri-nucleotide repeats being more abundant in monocots and dicots; we note that di-nucleotide motifs also seem more abundant in enset and Musa species, as well as Spirodela and Picea (Table S1), so there is no clear correlation with evolutionary position nor phylogenetic branch, important in building a picture of SSR evolution across all plants.
Grouped motifs A/T, AT/AT, AAT/ATT, AAAT/ATTT and AATAT/ATATT were the major microsatellites in enset. AT-rich microsatellite were also abundant in other monocot species such as Musa spp., Sorghum bicolor, Elaeis guineensis, Phyllostachys edulis, and Piceaabies (Fig. S2). In contrast, some studies have found that AT-rich repeats were dominant in dicots and CG-rich repeats in monocot plant species53,55, so we suggest that base compositions of the most abundant microsatellites are not related to taxon nor genome sizes. A study of correlations of AT/GC-richness of microsatellite motif with genome-wide content51 demonstrated that AT/GC-richness was correlated with the genomic AT/GC-content, supported by our results. We report that more than 60% of microsatellite loci were located in the intergenic regions of enset genome supporting comparative data56.
For development of usable markers from the microsatellite survey, we found 56% of the loci were appropriate for primer modeling, similar to other genome-scale marker development studies45,46,50,51,57. We identified 27% of primer redundancy, some what higher than the 5–20% in other genome scale analyses in plants25,51, most likely reflecting an artifact of assembly where one primer-SSR read is mismatched with more than one SSR-primerread58,59. Cross-taxon transferability features eg Refs.49,54 facilitate comparison of multiple taxa, gene mapping, and identification of orthologous loci including discovery of gene polymorphisms. In silico analysis showed about half the E. ventricosum markers were transferable to three other Ensete species, but only 1.90% were transferable to all the four Musa genomes.
Marker validation by PCR showed that the in silico testing strategies were able to select markers with successful amplification and high levels of polymorphism. The 93% of those tested were polymorphic, similar to the percentage reported using a DNA microsatellite library in enset10 although our genome-wide selection had a higher average PIC value showing they are informative markers for population genetic studies. The mean expected heterozygosity (He = 0.80) was also higher than previously reported in enset microsatellites (He = 0.59)10 and eleven cross-transferred Musa SSRs (He = 0.55)40. The in silico selection strategy confirmed by the validation shows that the markers are robust and require little laboratory-based optimization for scoring polymorphisms by agarose gel electrophoresis, contrasting with RAPD, AFLP, ISSR10,16,17, which also showed lower levels of polymorphisms.
The AMOVA analysis distinguished Ensete species, and enabled some clustering of enset landraces by their genetic variation. This result is consistent with several studies10,17,40: for example AFLP marker based genetic diversity of 146 enset landraces from five geographical regions showed a small variation among geographical regions (4.8%), but a high variation (95.2%) within regions17. The PIC values in enset are similar to those reported in other species such as Aegilops60 although notably the enset heterozygosity was high (average Ho = 0.87), suggesting outbreeding among the parents, and contrasting with the low level seen in the largely inbreeding Aegilops. The phylogenetic relations of Ensete landraces use in this study in general agree with patterns in another group of landraces analysed in the SSR-based phylogeny of Olango et al.10. In this study we found that the clustering pattern of landraces of E. ventricosum does not reflect any division based on cultivation region. These findings further confirm the extensive practice of germplasm (young plant or sucker) exchange between enset growers communities.
The markers derived from the sequence analysis are freely available in the EMM-database (https://enset-project.org/EnMom@base.html) and the whole database can be downloaded from https://figshare.com/s/20dd8c0d0a2994dbce8d. Microsatellite marker databases have been developed for various crop plant species including foxtail millet41, eggplant61, tomato42, oil palm62. The Enset Marker database and contains flexible search and download features, enabling large numbers of markers to be extracted and providing a resource for enset crop improvement. Many hundreds of additional markers can be extracted for targeted genotyping, GWAS (genome wide association studies) or marker assisted selection (MAS) studies as well as genetic diversity analysis, understanding population structures, and landrace identification.
Materials and methods
Genome-wide microsatellite exploration, characterization and marker development
We explored the microsatellite landscape using the whole nuclear genome sequence assemblies of four enset landraces (E. ventricosum ‘Bedadeti’ GenBank assembly: GCA_000818735.2; E. ventricosum ‘JungleSeeds’ GenBank assembly: GCA_000331365.2; E. ventricosum ‘Onjamo’ GenBank assembly: GCA_001884845.1; E. ventricosum ‘Derea’ GenBank assembly: GCA_001884805.1). The microsatellite mining pipeline was built with a combination of open bioinformatics tools including MISA (https://pgrc.ipk-gatersleben.de/misa/), Primer363, e-PCR44 (in silico PCR) and Perl-scripts (Fig. S1). The microsatellite search was performed using the search parameters with the minimum repeat unit 12 for mono, 8 for di-; 5 for tri- and tetra-; 4 for penta- and hexa-nucleotides. Microsatellites were classified based on microsatellites locus length (ClassI > 20 and ClassII ≤ 20 nt)43 and nucleotide base composition of the microsatellites motif (AT rich, equal AT/GC content and GC rich). Microsatellites primers were designed with Primer3 with default parameters. Redundant primer sets were filtered using a Perl script (Table S12). SSR-containing flanking sequences were analysed with the ORF (open reading frame) finder Perl script using default parameters to predict the longest ORF within the SSR-containing flanking sequences. Then Augustus364 was use to predict CDS (coding sequence), UTR (Untranslated regions) and introns regions in the flanking sequences. After that both ORF-finder output and Augustus output compile with the SSR position and assign the SSR locations. For, further verification the available genome annotations data of cv. 'Bedadeti' was retrieved from NCBI and compiled with SSR-location.
Cross-taxa transferability, functional annotation and comparative mapping
In silico cross-taxa transferability of the enset to Musa was estimated using an e-PCR approach (permitting3 mismatches and 3 gaps). Predicted lengths of e-PCR amplicons were compared with the expected amplicon of each marker, if the length variation differs at least 6 bp, the markers were denoted as polymorphic. All the transferable markers were then mapped on the Musa acuminata65, Musa balbisiana66 and Musa schizocarpa genomes. Comparative mapping result was visualized by CIRCOS software67.
Enset microsatellite marker database
To maximize the utility and availability of the enset microsatellite markers, we set up a searchable database using CSS, HTML and JavaScript under MySQL; a PHP based script was used to bridge the search interface and database, with results visualization, and download in XLS or CSV format.
Tissue sampling, DNA extraction and PCR
Leaf samples of enset landraces were collected from across the distribution of enset in Ethiopia. Tissue samples were harvested from young cigar leaves and stored on silica gel. Genomic DNA was extracted using CTAB methods.
PCR amplification was performedfor microsatellite primer validation under the following conditions: 94 °C for 5 min, 35 cycles at 94 °C for 30 s, 56–60 °C (according to primer annealing temperature) for 30 s, and 72 °C for 45 s, followed by a final elongation at 72 °C for 5 min. PCR products were run on 1.5% agarose gels in 1 × Tris–Borate-EDTA (TBE) buffer and a 100-bp molecular ladder was used to estimate the amplicon size.
Phylogeny and genetic diversity
A phylogenetic tree constructed based on SSR marker assay data, from the most highly polymorphic 15 EMM markers among 48 enset landraces and Ensete species. These fragment size variations were used for phylogenetic tree construction and subsequent genetic parameters analysis. Parameters including Polymorphism Information Content (PIC) of each marker; observed (Ho) and expected (He) heterozygosity; pair-wise comparisons of species genetic distance68 and FST (genetic differentiation) were calculated by PowerMarker version 3.2569. A Principal Coordinate Analysis (PCoA) was performed using the dissimilarity matrix data using GenAlEx software version6.570. A dissimilarity matrix was estimated then transfered into Mega6 software71 and a Neighbor-joining (NJ) approach used to construct boot strap NJ-phylogenetic tree.
Data availability
Data generated in this study are included in the main table, figures, additional file and also deposited in the online portal with free accessibility (https://enset-project.org/EnMom@base.html or https://enset.aau.edu.et/index.html) and the database can be downloaded from https://figshare.com/s/20dd8c0d0a2994dbce8d.
References
Borrell, J. S. et al. Enset in Ethiopia: a poorly characterized but resilient starch staple. Ann. Bot. 123, 20. https://doi.org/10.1093/aob/mcy214 (2019).
Vavilov, N. I. The Origin, Variation, Immunity and Breeding of Cultivated Plants: Selected Writings of NI Vavilov, vol. 72 (Chronica Botanica, Leyden, 1951).
Haile, M. Cluster analysis for evaluation of genetic diversity in Enset (Ensete ventricosum (Welw.): Cheesman) clones at Areka Condition. J. Plant Sci. 2, 55–69 (2014).
Quinlan, R. J. et al. Vulnerability and resilience of Sidamaenset and maize farms in Southwestern Ethiopia. J. Ethnobiol. 35, 314–337 (2015).
Yemata, G. Ensete ventricosum: a multipurpose crop against hunger in Ethiopia. Sci. World J. 2020, 1–10 (2020).
Tsegaye, A. & Struik, P. Enset (Ensete ventricosum (Welw.) Cheesman) kocho yield under different crop establishment methods as compared to yields of other carbohydrate-rich food crops. NJAS Wagening. J. Life Sci. 49, 81–94 (2001).
Bayu, T. Y. Land use dynamics and challenges of enset (Ensete ventricosum) agriculture in the upper reaches of Baso–Deme watershed, Gamo Highland, SW Ethiopia. Glob. J. Interdiscip. Soc. Sci. 5, 8 (2016).
Spring, A. et al. Tree against hunger: enset-based agricultural systems in Ethiopia. Am. Assoc. Adv. Sci. 5, 1073–1084 (1997).
Heslop-Harrison, J. S. & Schwarzacher, T. Domestication, genomics and the future for banana. Ann. Bot. 100, 1073–1084 (2007).
Olango, T. M., Tesfaye, B., Pagnotta, M. A., Pè, M. E. & Catellani, M. Development of SSR markers and genetic diversity analysis in enset (Ensete ventricosum (Welw.) Cheesman), an orphan food security crop from Southern Ethiopia. BMC Genet. 16, 98 (2015).
Tobiaw, D. C. & Bekele, E. Analysis of genetic diversity among cultivated enset (Ensete ventricosum) populations from Essera and Kefficho, southwestern part of Ethiopia using inter simple sequence repeats (ISSRs) marker. Afr. J. Biotechnol. 10, 15697–15709 (2011).
Yeshitla, M. &Yemataw, Z. In Enset Research and Development Experiences in Ethiopia, Proceedings of the Enset National Workshop, Wolkite, Ethiopia. (2010) 19–20.
Yemataw, Z. et al. Morphological variation and inter-relationships of quantitative traits in enset (Ensete ventricosum (Welw.) Cheesman) germplasm from South and South-Western Ethiopia. Plants 6, 56 (2017).
Frankel, O. & Brown, A. Genetics: New Frontiers: Proceedings of the XV International Congress of Genetics/Editors, VL Chopra...[et al.] (Oxford & IBH Publishing Co., New Delhi, 1984).
Bezuneh, T. in Proceedings of Enset Research and Development Experiences in Ethiopia: National Workshop: 19–20 August 2010; Wolkite, Ethiopia. 1–21.
Birmeta, G., Nybom, H. & Bekele, E. RAPD analysis of genetic diversity among clones of the Ethiopian crop plant Ensete ventricosum. Euphytica 124, 315–325 (2002).
Negash, A., Tsegaye, A., van Treuren, R. & Visser, B. AFLP analysis of enset clonal diversity in south and southwestern Ethiopia for conservation. Crop Sci. 42, 1105–1111 (2002).
Nuraga, G. W., Feyissa, T., Tesfaye, K., Demissew, S. & Tadele, Z. Phenotypic diversity of enset (Ensete ventricosum (Welw.) Cheesman) landraces used in traditional medicine. Genet. Resour. Crop Evol. 66, 1761–1772 (2019).
Birmeta, G., Nybom, H. & Bekele, E. Distinction between wild and cultivated enset (Ensete ventricosum) gene pools in Ethiopia using RAPD markers. Hereditas 140, 139–148 (2004).
Gerura, F. N. et al. Genetic diversity and population structure of enset (Ensete ventricosum Welw Cheesman) landraces of Gurage zone, Ethiopia. Genet. Resour. Crop Evol. 66, 1813–1824 (2019).
Liang, T. et al. Genetic diversity of Ziziphus mauritiana germplasm based on SSR markers and ploidy level estimation. Planta 249, 1875–1887 (2019).
Kumar, M. et al. Genetic diversity and population structure analysis of Indian garlic (Allium sativum L.) collection using SSR markers. Physiol. Mol. Biol. Plants 25, 377–386 (2019).
Ronoh, R. et al. Development of next-generation sequencing (NGS)-based SSRs in African nightshades: tools for analyzing genetic diversity for conservation and breeding. Sci. Hortic. 235, 152–159 (2018).
Lassois, L. et al. Genetic diversity, population structure, parentage analysis, and construction of core collections in the French apple germplasm based on SSR markers. Plant Mol. Biol. Rep. 34, 827–844 (2016).
Biswas, M. K. et al. Genome-wide computational analysis of Musa microsatellites: classification, cross-taxon transferability, functional annotation, association with transposons & miRNAs, and genetic marker potential. PLoS ONE 10, e0131312 (2015).
Biswas, M. K. et al. Exploration and exploitation of novel SSR markers for candidate transcription factor genes in Lilium species. Genes 9, 97 (2018).
Chen, H. et al. Assessment of genetic diversity and population structure of mung bean (Vigna radiata) germplasm using EST-based and genomic SSR markers. Gene 566, 175–183 (2015).
Miyatake, K., Shinmura, Y., Matsunaga, H., Fukuoka, H. & Saito, T. Construction of a core collection of eggplant (Solanum melongena L.) based on genome-wide SNP and SSR genotypes. Breed. Sci. 69, 498–502 (2019).
Hayano-Kanashiro, C. et al. An SSR-based approach incorporating a novel algorithm for identification of rare maize genotypes facilitates criteria for landrace conservation in Mexico. Ecol. Evol. 7, 1680–1690 (2017).
Ashraf, H. et al. SSR based genetic diversity of pigmented and aromatic rice (Oryza sativa L.) genotypes of the western Himalayan region of India. Physiol. Mol. Biol. Plants 22, 547–555 (2016).
Patil, P. G. et al. Association mapping to discover significant marker-trait associations for resistance against fusarium wilt variant 2 in pigeonpea [Cajanus cajan (L.) Millspaugh] using SSR markers. J. Appl. Genet. 58, 307–319 (2017).
Bohra, A. et al. New hypervariable SSR markers for diversity analysis, hybrid purity testing and trait mapping in Pigeonpea [Cajanus cajan (L.) Millspaugh]. Front. Plant Sci. 8, 377 (2017).
Nie, X. et al. Genome-wide SSR-based association mapping for fiber quality in nation-wide upland cotton inbreed cultivars in China. BMC Genom. 17, 352 (2016).
Zhao, Y. et al. SSR-based association mapping of salt tolerance in cotton (Gossypium hirsutum L.). Genet. Mol. Res. 15, 15027370 (2016).
Abou-Elwafa, S. F. Association mapping for yield and yield-contributing traits in barley under drought conditions with genome-based SSR markers. C.R. Biol. 339, 153–162 (2016).
Yagi, M. et al. Construction of an SSR and RAD marker-based genetic linkage map for carnation (Dianthus caryophyllus L.). Plant Mol. Biol. Rep. 35, 110–117 (2017).
Ambawat, S. et al. QTL mapping of pearl millet rust resistance using an integrated DArT-and SSR-based linkage map. Euphytica 209, 461–476 (2016).
Harrison, J. et al. A draft genome sequence for Ensete ventricosum, the drought-tolerant “tree against hunger”. Agronomy 4, 13–33 (2014).
Yemataw, Z. et al. Genome sequence data from 17 accessions of Ensete ventricosum, a staple food crop for millions in Ethiopia. Data Brief 18, 285–293 (2018).
Getachew, S. et al. A look into genetic diversity of enset (Ensete ventricosum (Welw.) cheesman) using transferable microsatellite sequences of banana in Ethiopia. J. Crop Improv. 28, 159–183 (2014).
Muthamilarasan, M., Misra, G. & Prasad, M. FmMDb: a versatile database of foxtail millet markers for millets and bioenergy grasses research. PLoS ONE 8, e71418 (2013).
Iquebal, M. A., Arora, V., Verma, N., Rai, A. & Kumar, D. First whole genome based microsatellite DNA marker database of tomato for mapping and variety identification. BMC Plant Biol. 13, 197 (2013).
Temnykh, S. et al. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 11, 1441–1452 (2001).
Schuler, G. D. Sequence mapping by electronic PCR. Genome Res. 7, 541–550 (1997).
Sonah, H. et al. Genome-wide distribution and organization of microsatellites in plants: an insight into marker development in Brachypodium. PLoS ONE 6, e21298 (2011).
Ellegren, H. Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 5, 435 (2004).
Tranbarger, T. J. et al. SSR markers in transcripts of genes linked to post-transcriptional and transcriptional regulatory functions during vegetative and reproductive development of Elaeisguineensis. BMC Plant Biol. 12, 1 (2012).
Zhao, H. et al. Developing genome-wide microsatellite markers of bamboo and their applications on molecular marker assisted taxonomy for accessions in the genus Phyllostachys. Sci. Rep. 5, 8018 (2015).
Varshney, R. K., Graner, A. & Sorrells, M. E. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 23, 48–55 (2005).
Pandey, G. et al. Genome-wide development and use of microsatellite markers for large-scale genotyping applications in foxtail millet [Setariaitalica (L.)]. DNA Res. 20, 197–207 (2013).
Biswas, M. K., Xu, Q., Mayer, C. & Deng, X. Genome wide characterization of short tandem repeat markers in sweet orange (Citrus sinensis). PLoS ONE 9, e104182. https://doi.org/10.1371/journal.pone.0104182PONE-D-14-13890 (2014).
Wang, X. et al. Comparative genome-wide characterization leading to simple sequence repeat marker development for Nicotiana. BMC Genom. 19, 500 (2018).
Cavagnaro, P. F. et al. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom. 11, 569 (2010).
Victoria, F. C., da Maia, L. C. & de Oliveira, A. C. In silico comparative analysis of SSR markers in plants. BMC Plant Biol. 11, 15 (2011).
Morgante, M., Hanafey, M. & Powell, W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 30, 194 (2002).
Tóth, G., Gáspári, Z. & Jurka, J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 10, 967–981 (2000).
Biswas, M. K. et al. Exploiting BAC-end sequences for the mining, characterization and utility of new short sequences repeat (SSR) markers in Citrus. Mol. Biol. Rep. https://doi.org/10.1007/s11033-011-1338-5 (2012).
Crow, K. D. & Wagner, G. P. What is the role of genome duplication in the evolution of complexity and diversity?. Mol. Biol. Evol. 23, 887–892 (2005).
Alix, K., Gérard, P. R., Schwarzacher, T. & Heslop-Harrison, J. Polyploidy and interspecific hybridization: partners for adaptation, speciation and evolution in plants. Ann. Bot. 120, 183–194 (2017).
Saeidi, H., Rahiminejad, M. R. & Heslop-Harrison, J. Retroelement insertional polymorphisms, diversity and phylogeography within diploid, D-genome Aegilops tauschii (Triticeae, Poaceae) sub-taxa in Iran. Ann. Bot. 101, 855–861 (2008).
Portis, E. et al. Comprehensive characterization of simple sequence repeats in eggplant (Solanum melongena L.) genome and construction of a web resource. Front. Plant Sci. 9, 401 (2018).
Babu, K. et al. Development and validation of whole genome-wide and genic microsatellite markers in oil palm (Elaeis guineensis Jacq.): First microsatellite database (OpSatdb). Sci. Rep. 9, 1899 (2019).
Koressaar, T. & Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291 (2007).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467 (2005).
D’Hont, A. et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213 (2012).
Davey, M. W. et al. A draft Musa balbisiana genome sequence for molecular genetics in polyploid, inter-and intra-specific Musa hybrids. BMC Genom. 14, 683 (2013).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Nei, M. &Takezaki, N., Estimation of genetic distances and phylogenetic trees from DNAanalysis. In: Proceedings of the 5th World Congress on Genetics Applied to Livestock Production. Guelph, 21 (1994), 405–412.
Liu, K. & Muse, S. V. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129 (2005).
Smouse, R. P. P. & Peakall, R. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539 (2012).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Acknowledgements
Authors are thankful to Ramesh Patel, lab technician, for preparing some reagents, maintaining the plant material in the greenhouse. Tissue samples were exported under appropriate Access and Benefit Sharing Agreements and Material Transfer Agreements between the Ethiopian Biodiversity Institute, Royal Botanic Gardens, Kew and the University of Leicester.
Funding
This work was supported by the GCRF Foundation Awards for Global Agricultural and Food Systems Research, entitled, ‘Modelling and genomics resources to enhance exploitation of the sustainable and diverse Ethiopian starch crop enset and support livelihoods’ [Grant No. BB/P02307X/1].
Author information
Authors and Affiliations
Contributions
Conceptualization: M.K.B., T.S. and P.H.H.; Data curation: M.K.B., J.N.D., M.B., D.B.; Experiment: M.K.B., J.N.D., J.B., M.B.; Plant material collection: J.B., G.Z.; E-resource development: M.K.B., M.B. and D.B.; Funding acquisition: T.S., P.W., P.H.H.; Project administration: T.S., P.W., P.H.H., S.D.; Supervision: T.S. and P.H.H.; Writing -original draft: M.K.B.; Writing-reviewing and editing: M.K.B., J.B., T.S. and P.H.H.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Biswas, M.K., Darbar, J.N., Borrell, J.S. et al. The landscape of microsatellites in the enset (Ensete ventricosum) genome and web-based marker resource development. Sci Rep 10, 15312 (2020). https://doi.org/10.1038/s41598-020-71984-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-71984-x
This article is cited by
-
Genetic diversity, population structure and selection signatures in Enset (Ensete ventricosum, (Welw.) Cheesman), an underutilized and key food security crop in Ethiopia
Genetic Resources and Crop Evolution (2024)
-
Transcriptome wide SSR discovery cross-taxa transferability and development of marker database for studying genetic diversity population structure of Lilium species
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
