
Abstract
Functionality of the accessory gene regulator (agr) quorum sensing system is an important factor promoting either acute or chronic infections by the notorious opportunistic human and veterinary pathogen Staphylococcus aureus. Spontaneous alterations of the agr system are known to frequently occur in human healthcare-associated S. aureus lineages. However, data on agr integrity and function are sparse regarding other major clonal lineages. Here we report on the agr system functionality and activity level in mecC-carrying methicillin resistant S. aureus (MRSA) of various animal origins (n = 33) obtained in Europe as well as in closely related human isolates (n = 12). Whole genome analysis assigned all isolates to four clonal complexes (CC) with distinct agr types (CC599 agr I, CC49 agr II, CC130 agr III and CC1943 agr IV). Agr functionality was assessed by a combination of phenotypic assays and proteome analysis. In each CC, isolates with varying agr activity levels were detected, including the presence of completely non-functional variants. Genomic comparison of the agr I–IV encoding regions associated these phenotypic differences with variations in the agrA and agrC genes. The genomic changes were detected independently in divergent lineages, suggesting that agr variation might foster viability and adaptation of emerging MRSA lineages to distinct ecological niches.
Similar content being viewed by others


Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) is among the leading causes of opportunistic infectious diseases in human and veterinary medicine worldwide1,2. Mobile genetic elements (MGEs) originally denominated as staphylococcal cassette chromosome mec (SCCmec) cause horizontal spread of methicillin resistance among staphylococci3, with either mecA or mecC4,5 as the resistance-mediating gene. So far, thirteen distinct SCCmec elements have been described, including the mecC-harbouring element SCCmec XI5,6. When these MRSA were first isolated from specimens of human and ruminant hosts back in 20117,8,9, detection and verification of mecC was challenging due to the limited sequence similarity to mecA and its common association with a moderate or even low minimal inhibitory concentration (MIC) for oxacillin5,9,10,11. Since then, mecC-MRSA were not only found in samples from humans and dairy cattle5,12, but also in domestic-, zoo-, and wild animals as well as in wastewater8,10,13,14,15. Human cases of infection associated with mecC-MRSA were reported from the United Kingdom, Germany, Austria, Spain and many other European countries7,8,9,16 suggesting that mecC-harbouring isolates have become ubiquitous10,14.
Mechanisms enabling opportunistic bacteria to cross species barriers, to infect a new host or to enhance their viability in the environment are still poorly understood. Changes expected to be involved in (niche) adaptation processes of S. aureus encompass (among others) alterations of metabolic pathways17, escape of host defence mechanisms1, biofilm formation18, iron acquisition abilities19, generation of pseudogenes20,21 or nucleotide changes altering promoter structures22. In addition, production of cytotoxins23,24 and other virulence factors as well as their regulation circuits might have a strong influence on host adaptation23,25,26. Acquisition of specific virulence factors from a host-specific gene pool1,27 by horizontal gene transfer as well as core genome diversification contributes to broadening the host range of S. aureus27, leading to the definition of extended host spectrum genotypes28.
In S. aureus, the accessory gene regulator (agr) locus represents a quorum sensing system that orchestrates the switch from expressing of surface associated factors (needed for initial attachment) to the expression of exotoxins at high cell density via the effector molecule RNAIII29. Notably, agr activity appears to be essential for skin and soft tissue infections30. Previous research demonstrated the important role of agr for virulence factor transcription in animal models of acute infections31,32,33, while agr defective mutants seemed to be frequently associated with chronic diseases34,35,36,37,38,39, persistent bacteremia40 and cystic fibrosis41, indicating adaptation of S. aureus to the infected host42.
The staphylococcal agr locus is an autoinducing control unit that combines bacterial quorum sensing with a classical bacterial two-component regulatory system (TCRS) and the employment of a regulatory RNA (i.e. RNAIII) as effector molecule29. The locus consists of two divergently arranged transcription units, which are transcribed from promoters P2 and P3, respectively. The P2-derived RNAII harbours the agrBDCA operon, while P3 drives transcription of RNAIII. In S. aureus, the cell–cell communication (i.e. quorum sensing) depends mainly on autoinducing, short peptides (AIPs) encoded by agrD, which are exported by the transmembrane endopeptidase (AgrB) and finally trimmed by a type I extracellular signal peptidase (SspB; reviewed in43). At sufficient extracellular AIP concentrations, the peptides binds to the sensor histidine kinases (AgrC) which then phosphorylates the response regulator AgrA (AgrA-P)43,44. While a weak baseline transcription from the P2 and P3 promoters is detectable even in the absence of AgrA-P43,44, the activated transcription factor strongly (auto)induces expression of both agrBDCA and RNAIII44. In addition, AgrA-P directly leads to transcription of phenol-soluble modulin (PSM) genes. The majority of agr-regulated genes, however, is controlled via RNAIII which influences their transcription and translation29,43,45. RNAIII represents a dual-function regulatory RNA that harbours, in addition to its base-pairing functions, a small open reading frame (hld) encoding staphylococcal delta-haemolysin (Hld). The hld Shine-Dalgarno site was shown to be accessible to ribosomes, indicating that hld represents a translatable unit on the RNAIII molecule46, which in turn makes Hld detection a suitable proxy to determine RNAIII expression and Agr activity in general.
S. aureus virulence often depends on the secretion of large amounts of toxins, including exotoxins with superantigenic functions, i.e., the staphylococcal enterotoxins (SEA, SEB, etc.) and toxic shock syndrome toxin (TSST-1)47. Expression of ordinary virulence determinants such as proteases, lipases or nucleases, are promoted by an activated agr system, whereby expression of surface binding proteins is downregulated. Nonetheless, this generalized summary still includes exceptions and lineage-specific differences48. In addition, primary transcription regulation of virulence factors not encoded by the core genome, e.g. genes associated with the enterotoxin gene cluster (egc) harbouring staphylococcal enterotoxins (SE), appears to be less depending on increased agr activation49.
Biofilm formation is another important feature in S. aureus pathogenesis which is influenced by agr. The effect of a cellular increase of the agr transcript RNAIII, however, is not entirely clear. While previous studies demonstrated enhanced biofilm formation in agr deficient strains42,50, recent reports, focusing on non-lab-adapted strains, found lineage-specific differences51, or a variable role of the agr system38,52.
Occurrence of independent genomic changes in divergent clonal lineages is generally considered to reflect the adaptation power of bacteria to changing environments53. Aim of the study presented here was to analyse the genome structures of emerging mecC-MRSA isolates of animal and human origin with the overarching goal to identify genomic patterns that might be associated with the adaptation of such strains to novel hosts and ecological niches. While differences in exotoxin and virulence factor endowments of the isolates were mainly associated with distinct clonal complexes (CC), we found independent genomic variations of the agr locus across different mecC-MRSA clonal lineages. These changes were associated with varying agr activity patterns and kaleidoscopic phenotypes regarding haemolysis and biofilm architecture, suggesting a role of the agr system in niche adaptation of the isolates.
Materials and methods
Sample collection and pre-screening
Staphylococcus aureus isolates obtained from samples of various animal species were collected from January 2014 to December 2016 by IDEXX Laboratories in Ludwigsburg, Germany. Inclusion criteria for isolates were (1) a methicillin resistant phenotype based on growth in the presence of 6 μg/ml cefoxitin according to the manufacturer’s VITEK 2 Advanced Expert System (Nürtingen, Germany) instruction together with (2) oxacillin MICs < 4 µg/ml, as reported for mecC-MRSA before14. Isolates from seven different European countries were included. Species identity and methicillin resistance mediated by mecC was confirmed by PCR as described elsewhere54. Antimicrobial susceptibility testing (AST) was carried out using the VITEK 2 system (BioMérieux, Germany) according to the standards given by CLSI VET01-A4 and M100-S2455,56. For comparative analysis, additional 12 mecC-positive isolates of human origin belonging to corresponding clonal backgrounds collected by the national reference laboratory for staphylococci at the Robert Koch Institute in Germany were included (Table 1).
Whole genome sequencing and genomic analysis of mecC-MRSA
MRSA isolates positive for mecC were whole-genome sequenced (WGS) using Illumina MiSeq 300 bp paired-end sequencing with an obtained coverage > 90X. Raw reads were used for de novo assembly into contiguous sequences (contigs) and subsequently into scaffolds using SPAdes v3.1257. Assembled draft genomes of the isolates were annotated using Prokka58. WGS data were used for genotypic characterization including the determination of the sequence type (ST) (MLST v2.0)59. Genomic sites of interest and genes encoding for major regulators including the accessory gene regulator (agr) system were investigated using Geneious 11.1.5 (Biomatters Ltd., Australia)60. Genomic data were further analysed with ResFinder-2.2 (threshold: 90% ID, 80% minimum length), VirulenceFinder-1.6 (threshold: 95% ID, 80% minimum length) and spaTyper59,61,62,63.
In order to compare the genomes at high resolution, we used the maximum common genome (MCG) that is defined by those orthologous genes present in all genomes64. The coding sequences were clustered based on the parameters of nucleotide sequence similarity (≥ 90%) and gene coverage (≥ 90%). The MCG was defined as those genes that were present in each genome and fulfilled the threshold parameters, yielding 2,094 genes. Allelic variants of these genes were subsequently extracted from all genomes by a blast-based approach, then aligned individually for each gene and concatenated, resulting in an alignment of 1.95 Mbp for these isolates. The alignment was used to generate a maximum likelihood phylogenetic tree using RAxML 8.2.965 which was visualized together with the distribution of the accessory gene content using phandango66.
Phenotypical assessment of accessory gene regulator (agr) activity
In this study, complementary phenotypical methods including colony spreading and haemolysin production were employed to investigate the activity of the agr system. Synergistic production of different haemolysins (SPDH) on columbia agar plates (Oxoid, Germany) supplemented with 5% sheep blood (SBA) was investigated by cross-streaking tested bacteria perpendicularly to laboratory strain S. aureus RN4220 as described before1. RN4220 is characterized by a strong β-haemolysin production while α-haemolysin secretion is missing and PSM α peptides are produced only at non-considerable amounts35, 67,68. Of note, RN4220 was reported to produce the α-haemolysin encoding mRNA (hla), indicating that hla transcription cannot necessarily be correlated with α-haemolysin production and/or secretion in S. aureus69. In addition, the capacity to cause synergistic haemolysis with β-haemolysin is to a large extend due to AgrA-induced expression of PSMs67, while β-haemolysin inhibits α-haemolysins’ effect on SBA35. After overnight incubation of the plates at 37 °C, the haemolysis zones were examined and pictures were taken.
A modified CAMP test for verification of β-haemolysin production was performed using Streptococcus agalactiae (ATCC12386) and the β-haemolysin producing S. aureus strain ATCC25923 (positive control) as described recently1. Briefly, staphylococci of interest were streaked perpendicularly to CAMP-factor producing ATCC12386. A positive interaction of CAMP-factor from Group B streptococci with β-haemolysin (phospholipase C) of S. aureus is characterized by completely lysed sheep blood erythrocytes forming a clear semilunar-shaped area70.
We also performed a colony spreading assay on soft agar described by Kaito et al.71, since an activated state of the agr system72 as well as AgrA-depending production PSM α3 are necessary for S. aureus to slide on wet surfaces73. Beyond that, a mutation in the membrane-bound transpeptidase sortase A (SrtA), or for instance the lack of its substrates fibronectin binding protein A and B (FnBPA, FnBPB), clumping factor A and B (ClfA, ClfB) enhances spreading74. Briefly, 20 ml tryptic soy soft agar plates (0.24%) were used to investigate spreading activity of all MRSA. After overnight incubation of the plates at 37 °C, the spreading zones were examined, and pictures were taken.
The standard laboratory strains USA300 (FPR3757), the USA300 agrA::Tn mutant (NE1532) and RN4220 were included in all phenotype assays as controls for wild type (wt) agr functionality, a loss-of-function- and a functionally impaired agr system, respectively69. In lab strain RN4220, a frameshift mutation in the agrA-coding region adds three amino acids to the C-terminus of AgrA, a variation known to cause a considerable delay in RNAIII transcription compared to the wild type70. To verify production of TSST, the TST-RPLA Kit TD940 (Thermo Fisher Scientific, USA) was used according to the manufacturer’s instructions.
All surveys were repeated thrice for each of the isolates including the reference strains.
Proteomic analysis of α-, β- and δ-haemolysin production in mecC-MRSA
Protein abundance values for Hla/Hlb/Hld from whole bacterial cell preparations were measured to assess RNAIII transcription capabilities by Hld production and Hla and Hlb (pre-) proteins of the bacteria and their general ability to produce them.
Sample preparation by easy extraction and digestion (SPEED)
Samples were prepared in triplicates using SPEED as previously described75. In brief, cells were re-suspended in trifluoroacetic acid (TFA) (Uvasol for spectroscopy, Merck, Darmstadt, Germany) (sample/TFA 1:4 (v/v)) and incubated at 70 °C for 3 min. Samples were neutralized with 2 M TrisBase using 10 × volume of TFA and further incubated at 95 °C for 5 min after adding Tris(2-carboxyethyl)phosphine (TCEP) to a final concentration of 10 mM and 2-Chloroacetamide (CAA) to a final concentration of 40 mM. Protein concentrations were determined by turbidity measurements at 360 nm, adjusted to 0.25 µg/µl using a 10:1 (v/v) mixture of 2 M TrisBase and TFA and then diluted 1:5 with water. Digestion was carried out for 20 h at 37 °C using Trypsin (Promega, Fitchburg, WI, USA) at a protein/enzyme ratio of 50:1. Resulting peptides were desalted using StageTips C1876.
Liquid chromatography and mass spectrometry
Peptides were analyzed on an EASY-nanoLC 1,200 (Thermo Fisher Scientific, Bremen, Germany) coupled online to a Q Exactive™ Plus mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). 1 µg peptides were loaded on an Acclaim PepMap trap column (20 mm × 75 μm i.d., 100 Å, C18, 3 μm; Thermo Fisher Scientific, Bremen, Germany) and were subsequently separated on a 200 cm μPAC column (PharmaFluidics, Ghent, Belgium). The flow rate was set to 800 nl/min and a stepped 40 min gradient was applied: 3–10% B in 4 min, 10–33% B in 17 min, 33–49% B in 4 min, 49–80% B in 7.5 min and 80% B for 7.5 min. Solvent A was 0.1% (v/v) formic acid (FA) in water, solvent B consisted of 80% (v/v) acetonitrile in 0.1% (v/v) FA.
The Q Exactive Plus was operated in data-independent (DIA) manner in the m/z range of 350–1,150. Full scan spectra were recorded with a resolution of 70,000 using an automatic gain control (AGC) target value of 3 × 106 with a maximum injection time of 100 ms. The Full scans were followed by 53 DIA scans of dynamic window widths using an overlap of 0.5 Th (Supplemental Table 1). DIA spectra were recorded at a resolution of 17,500@200 m/z using an AGC target value of 3 × 106 with a maximum injection time of 55 ms and a first fixed mass of 200 Th normalized collision energy (NCE) was set to 25% and default charge state was set to 3.
Mass spectrometric data analysis
The mass spectra were searched in DIA-NN (Version 1.7.6)77 using the deep-learning based spectra and RT prediction for sequences from the complete proteome of S. aureus strain NCTC 8,325 (UP000008816, 2,889 sequences, downloaded 4/10/18) and sequences of selected genes (hla, hlb and hld) obtained from whole genome sequencing. Spectra were searched with a tolerance of 10 ppm in MS1 and 20 ppm in MS2 mode, strict trypsin specificity (KR not P) and allowing up to one missed cleavage site. Cysteine carbamidomethylation and N-terminal methionine excision were set as modifications. Peptide length was restricted to 7–30 amino acids. The m/z ranges were 350–1,150 for full scans and 200–1,800 for DIA scans. A false discovery rate of 1% was applied for precursor and protein identifications.
Biofilm formation assay on polystyrene tissue culture plates
Biofilm formation to inert artificial surfaces was tested in 96‐well polystyrene tissue culture plates (Greiner Bio‐One, Cellstar, F‐form) as described previously78. Briefly, S. epidermidis RP62A and S. carnosus TM300 were used as positive and negative controls, respectively. Bacterial overnight cultures in triplicates were grown in Trypticase Soy Broth (TSB; Becton Dickinson) which contains, according to the standard composition, 2.5 g/l glucose. Cultures were diluted in fresh TSB to an OD600 of 0.05 and 200 µl filled in each well (four wells per biological replicate) and incubated under static condition at 30 °C for 18 h. Supernatant was discarded and adherent cells were washed twice with 1 × PBS buffer before the remaining cells were heat-fixed at 65 °C for 1 h. Plates were then stained with 10 mg/ml crystal violet for 2 min, washed twice with double-distilled water before proceeding with measuring the absorbance at OD492 by an ELISA plate reader (Multiskan Ascent).
Imaging of the biofilm architecture by confocal laser scanning microscopy (CLSM)
In order to study the putative effect of different agr non-wt variants on growth/biofilm characteristics on glass surfaces, overnight cultures of closely related isolates were subjected to comparative analysis: S. aureus isolates belonging to CC130 IMT38119 (agr III wt), RKI5966 (agr III non-wt AgrC variant) and IMT31819 (agr III non-wt AgrA variant) were diluted to 105 bacteria per ml. One milliliter diluted suspension was used to inoculate the wells of a 24-well plate with glass bottom (µ-Plate 24 Well Black, ibidi GmbH, Germany), which was then cultivated for 20 h at 37 °C with 150 rpm on an orbital shaker. Afterwards, samples were photographed (Lumix GM1, Panasonic, Japan) on a light table, stained with LIVE/DEAD (LIVE/DEAD Cell Viability Assay, ThermoFisher Scientific, Germany) according to the manufacturer’s instructions and imaged with a confocal laser scanning microscope (LSM780, Carl Zeiss AG, Germany) using the Plan-Apochromat 20 × /0.8 objective.
When necessary, images were cropped, adjusted for optimal brightness and contrast (applied to the whole image) using Adobe Photoshop CS6 (Adobe Systems, San Jose, CA, USA).
Database accession numbers
Genomic sequencing data used are available for download from the National Center for Biotechnology Information (NCBI) under BioProject accessions PRJNA588740. Accession numbers of whole genomes sequences are provided in Supplemental Table 2. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD016486.
Results
General features of the mecC-MRSA strain collection
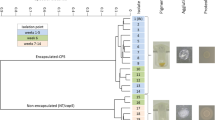
A total of 33 mecC-MRSA were obtained from seven different European countries and six different animal species for whole genome sequencing (WGS). Isolates from cats dominated the collection (24/33), followed by those obtained from dogs (4/33) and other companion-, wild- and livestock animals (Table 1). We also included 12 isolates from human patients belonging to matching genotype lineages (Table 1, Fig. 1) for further phenotype assays and subsequent comparative genome analysis. All 45 isolates displayed low or moderate oxacillin MICs < 4 µg/ml, while being able to grow in the presence of 6 μg/ml cefoxitin (Table 1), which is typical for mecC-MRSA14, 79. AST results revealed additional resistance to aminoglycosides (gentamicin, kanamycin) for isolate RKI5962 only. A positive latex agglutination test verified production of the TSST toxin for all isolates harbouring a tst-bov variant80. Interestingly, we noticed considerable phenotypic differences regarding haemolysis on SBA between the isolates, even among those sharing the same phylogenetic background. This prompted further investigations into putative genomic changes which might account for these variations.

The core genome phylogeny based on the Maximum Common Genome. The core genome phylogeny based on the Maximum Common Genome comprising 2,094 orthologous genes present in all isolates show four distinct clusters, whereby the genetic diversity within the clusters is rather low. Furthermore, the isolates metadata show no significant association with the core genome clusters. The 2,003 accessory genes show a distribution pattern that is highly correlated with the core genome clusters (right side), suggesting a lineage-specific gene content. Genes for aureolysin (aur), leucotoxins D and E (lukD, lukE), gamma-haemolysin component A–C (hlgA, hlgB, hlgC) and proteases SpIA or SpIB are present in all isolates. All isolates belonging to ST-1943 as well as some CC130 and CC599 were positive for different variants of the Staphylococcal pathogenicity island (SaPI) harbouring a toxic shock toxin encoding gene (tst), which were variants of tst-bov80. Moreover, 48.5% of the mecC-positive isolates harboured staphylococcal enterotoxins (SE). The protease SpIE can just be found in 23/33 isolates and is not associated with any sequence type. The epidermal cell differentiation inhibitor B (edinB) cannot be determined in the isolates of ST-1943, ST-2361, ST-49 and ST-599.
Phylogenetic relationship and distribution of virulence-associated factors among mecC-MSRA
WGS of the isolates revealed that the 45 mecC-positive MRSA belonged to four CCs: CC599 (12), CC130 (21), CC1943 (10) and CC49 (2) (Table 1, Fig. 1). Pairwise SNP-distances between the core genomes were calculated for all MRSA isolates (Supplemental Table 3), showing a very close phylogenetic relationship of those genomes belonging to the same CC. While the differences within one clonal complex ranged between 100 and 500 SNPs, the genomes belonging to different CCs differed by more than 10,000 SNPs, resulting in a clear clustering of MRSA from the same CC (Fig. 1). Metadata such as geographic origin, disease or animal species showed no significant association with the core genome clustering, but a lineage-specific association of the variable gene content was obvious (Fig. 1).
Nevertheless, a core set81 of S. aureus virulence factors was present in all isolates, including aureolysin (aur), bi-component leukotoxin (lukD/E), γ-haemolysin components A–C (hlgA, hlgB, hlgC) and genes encoding for the serine proteases SpIA and SpIB. Presence of the epidermal cell differentiation inhibitor B (edinB) and exfoliative-like toxin D282 was associated with CC130. When considering factors promoting biofilm production in S. aureus, all isolates harbour complete ica loci (icaR/icaADBC) but not the biofilm-associated surface protein bap. For iron acquisition, all isolates harboured amongst others the iron-regulated surface determinant gene cluster isdABCDEF.
While only one isolate lacked the gene encoding α-haemolysin (hla) completely, three isolates belonging to CC130 carried hla variations which resulted in aa sequence alterations and two further isolates harboured insertions (details are provided in Supplemental Table 4). Bacteriophages converting the β-haemolysin gene (hlb) were identified in four isolates (for details see Supplemental Table 4). Some of the mecC-MRSA, especially those belonging to CC599 and CC1943 harboured variants of previously described staphylococcal pathogenicity islands, including SaPIbov80 encoding SEC, a TSSTbov variant and SElL (Fig. 1). Isolates belonging to CC1943 were positive for an egc cluster variant (seg, sei, selm, seln, selo, selu), while all isolates were positive for selX (Fig. 1). Furthermore, all isolates were positive for intact and identical genes encoding phenol-soluble modulins (PSM α1 to PSM α4 and PSM β1 and PSM β2), and differences in the promoter regions of these operons seemed to particularly mirror the genomic lineage (data not shown).
mecC-MRSA belonging to CC599, CC49, CC130 and CC 1943 harbour agr variations
According to the typing scheme used by Shopsin et al.83, CC599 MRSA were assigned to the agr type I, CC49 to type II, CC130 to type III and CC1943 to type IV. Of the isolates harbouring the type I agr system, two isolates showed aa changes for AgrA and all AgrB aa sequences showed the variation A182T when compared to the S. aureus strain N315 sequences (Supplemental Table 4), while the respective aa sequences for AgrC and AgrD were identical (Fig. 2). All isolates belonging to CC599 and CC49 showed a nucleotide insertion (T) at bp 55 within the region encoding RNAIII, resulting in 514 + 1 bp length and an additional uracil (U) in the hairpin 2, according to the secondary structure model of RNAIII proposed by Benito and colleagues46. Since this insertion was found in all isolates associated with the agr types I and II in this study, this variation was considered as the wild type (wt) sequence here. Only two isolates belonged to the agr type II, and variation among them was detected in terms of a premature stop codon created by an insertion in the AgrA encoding gene in isolate IMT36945 (Fig. 2). Most variants were detected among the aa sequences for AgrA (one insertion, two changes) and AgrC (four changes) of the CC130 isolates harbouring the type III agr system (Fig. 2, and Supplemental Table 4). For the CC1943 isolates (type IV agr), only IMT36952 showed an aa sequence variation in AgrA (Fig. 2). The nucleotide sequence regions encoding for the 26-aa δ-haemolysin integrated in RNAIII and the promoter sequences for RNAII (P2) and RNAIII (P3) were conserved in all 45 genomes investigated here (Fig. 2a–d).

Sequence alignments of agr regions in mecC-MRSA. Sequence alignments for (a) CC599 (agr I), (b) CC49 (agr II), (c) CC130 (agr III) and (d) CC1943 (agr IV) isolates. First row shaded in yellow indicates the wild type (wt) exemplarily shown for all isolates sharing 100% coverage and 100% nucleotide and amino acid sequence identity. For each of the non-wt isolates on the display, changes within the upper gray line indicates a nucleotide sequence alteration while changes in the second line indicates amino acid sequence alteration. For all details and the reference sequences used for each CC see Supplemental Table 4. (a) First row, wt agr I in CC599 shared by IMT32509, IMT32513, IMT36943, IMT 36,947, IMT36948, IMT38116, IMT39825, RKI5962, RKI5963 and RKI5964. Second row, IMT31818 has a triplet nucleotid deletion resulting in ΔN177 in AgrA; third row, IMT39824 shows a non synonymous substitution (from A to G in position 289) leading to F196S in AgrA. (b) First row, wt agr II in CC49 represented by RKI5972 while the second row shows IMT36945 with an insertion creating a premature stop codon in AgrA. (c) First row, wt agr III in CC130 shared by IMT34480, IMT34488, IMT34491, IMT36946, IMT36950, IMT38115, IMT38119, IMT39816, IMT39819, IMT40506, IMT41554, RKI5965, RKI5967 and RKI5968. Rows 2, 4, 9 and 10 show amino acid variations in AgrC for IMT34479 (N5S), RKI5966 (Q16H), IMT32929 (E216K) and IMT32510 (G284D) generated by corresponding non-synonymous substitutions. RKI5967 (row 3), IMT38119 (row 5) and IMT38115 (row 6), harbour a nucleotide substitution (c to a) at position -2 upstream the RNAIII sequence start, respectively. Rows 7, 8 and 11 harbour variants of AgrA for IMT34489 (G68D), IMT34478 (S215P) caused by non-synonymous substitutions and IMT31819 shows an insertion at position 711 bp in agrA causing an alternate stop codon. (d) First row, WT agr IV in CC1943 shared by RKI5973, IMT34485, IMT40507, IMT40504, IMT38113. Second to fourth row (upper gray line) show a synonymous substitution (a to t at position 442) in agrA for RKI5971, RKI5970 and RKI5969 while the fifth row indicates a further variation of agrA (C199R) generated by a non-synonymous substitution.
Assessment of genes associated with virulence factor transcription in mecC-MRSA
Only in isolate IMT41554 (CC130), a C to T change within the − 35 promoter region previously identified as the AgrA binding region of the psm α operon84 was detected at position − 27. As the phenotype did not deviate from closely related isolates, this single nucleotide change is unlikely to affect the AgrA-binding abilities upstream of psm α1–4 in IMT41554. Yet, many two component regulatory systems (TCRS) and nucleic acid-binding proteins seem to modulate S. aureus virulence factor expression, especially of exotoxins with haemolytic activity85. Since TCRS variations might influence the particular phenotype appearance of S. aureus, the isolate collection was screened for obvious deletions, insertions or changes generating for instance amino acid (aa) substitutions or premature stop codons. Among the TCRS included in the analysis were those either positively or negatively influenced by agr such as the staphylococcal accessory regulator nucleic acid-binding protein (SarA) and the regulator of toxins (Rot). Overall, amino acid changes (referred to the respective reference sequence) in global regulators were rare and mostly lineage-specific (Supplemental Table 5), indicating a limited role of these variations for the phenotype differences observed for each CC.
Haemolysis and colony spreading of mecC-MRSA carrying agr variations
While analysing the mecC-MRSA belonging to four different phylogenetic lineages, we noticed a broad range of different phenotypes with respect to haemolysis on SBA plates.
Initially, haemolytic activities were assessed using a SPDH cross-streaking test utilizing strain RN4220 (Fig. 3). Here, only 30 of 45 isolates showed the typical haemolysis pattern for α-, β- and δ-haemolysin production (Fig. 4, Supplemental Figures 1 and 2), suggesting that some of the isolates might harbour genomic alterations affecting haemolytic activities (Table 2). We then used the CAMP test which indicates secretion of β-haemolysin by S. aureus through a characteristic arrow-shaped synergistic haemolysis zone on SBA. 10/45 isolates and two of the reference strains failed to produce the corresponding phenomenon (Table 2, Supplemental Figure 1), with four isolates (and two reference strains) harbouring a phage disrupting the hlb gene (Supplemental Table 4). The remaining six isolates lacked the hlb-converting phage but showed instead AgrA variations that clearly deviated from the wildtype (Fig. 2, Supplemental Table 4).

Haemolytic activities of S. aureus isolates on sheep blood agar (SBA) plates. (a) Scheme for assessment of haemolytic activity based on Geisinger et al.118. The isolates were tested by cross-streaking perpendicularly to S. aureus RN4220 on sheep blood agar (SBA) plates. The turbid zone induced by β-haemolysin production of RN4220 enhanced lysis by δ-haemolysin and PSMs (clear zone at the intersection) and inhibited α-haemolysin (V-shaped zone at the intersection). (b) Haemolytic activity of mecC-positive S. aureus belonging to different agr types on SBA plates. The lack of a corresponding phenotype in the isolate collection is indicated by a grey rectangle. One exemplarily image was used to illustrate the differences, respectively.

Illustration showing protein abundance values, relevant genomic variation and phenotype results of mecC-MRSA. (a) δ-haemolysin (Hld) protein abundance and the synergistic production of different haemolysins (SPDH test). (b) δ-haemolysin (Hld) protein abundance and the isolates’ capability for colony spreading.
Since functionality of the agr system is also reflected by the ability of S. aureus to slide over wet surfaces72, we performed a colony spreading assay on semisolid agar plates. As a result, 38/45 isolates showed the characteristics of “sliding bacteria”, which are exemplarily shown in Fig. 5. A comprehensive summary of all phenotype characteristics of the isolate collection and the reference strains included is presented in Table 2.

Colony spreading assay results for mecC-S. aureus with different agr functionalities on soft agar plates. A TSA soft agar plate (0.24%) was inoculated with 2 µl overnight culture of S. aureus. Examples shown here include isolate RKI5966 associated with a weak agr functionality, which was not able to spread on semisolid agar plates, while isolate IMT38119 (strong agr functionality) showed spreading. As controls we have employed the standard laboratory strains RN4220 (weak agr functionality) and USA300 (strong agr functionality).
Agr activity assessment by measuring δ-haemolysin production in mecC-MRSA using proteomics
The protein abundance of Hld (δ-haemolysin) was used to assess RNAIII transcription capabilities of S. aureus isolates, as the corresponding coding gene (hld) is integrated in the RNAIII transcript. Hld abundance was measured using whole cell proteomics to cover the actual expression level. Therefore, the assessment of direct whole-cell δ-haemolysin production appears as a suitable method to predict RNAIII transcription, as long as occurring sequence variations do not affect the hld encoding gene or the promoter regions in in the agr operon (Figs. 2 and 4a).
The protein abundance levels for δ-haemolysin measured by mass spectrometry are shown in Supplemental Table 6, and putative CC-specific differences were noticed (Fig. 4a). Isolates sharing the CC1943 background and lacking agr alterations, for example, showed higher overall δ-haemolysin abundance values than those belonging to CC130 or CC8 (Supplemental Table 6, Fig. 4a). Based on the δ-haemolysin detection level, we categorized the individual isolates’ agr activity as follows: 0, lack of detectable agr activity; +, weak agr activity and ++, strong agr activity (Fig. 4a). The reference strains included showed Hld abundances which clearly correlate with their previously reported agr functionality: For RN4220, a delay towards production of Hld due to an agrA mutation is known86. This alteration is also mirrored by a decreased protein abundance (Fig. 4a) and was consequently considered as “weak” agr activity. Moreover, the USA300 agrA::Tn strain (NE1532) completely lacked Hld detection, as expected. Thus, detection of Hld production together with a genomic inspection of the agr encoding region is indeed suitable to predict RNAIII transcription and agr functionality.
Comparison of phenotype assay results and haemolysin abundances
We then plotted the proteomic data against the results obtained from the phenotype assays described above.
The δ-haemolysin protein abundance and haemolysis phenotype associated with the isolates and reference strains are shown in Fig. 4a. Strikingly, all isolates (and reference strains) showing a strong reduction or lack of δ-haemolysin production were found to carry non-wt variants of either AgrA or AgrC (Fig. 4a). In addition, these AgrA or AgrC non-wt variants were associated with non-wt haemolysis patterns, too (Table 2, Supplemental Table 4, Fig. 4a).
The results for β-haemolysin (Hlb) protein abundances and the isolates’ capability to produce a synergistic haemolysis (CAMP phenomenon) are presented in Supplemental Figure 2 and Table 2, with isolates harbouring phage-disrupted hlb genes and agr alterations being indicated. Interestingly, all isolates with non-wt AgrA variants also failed to produce the CAMP phenomenon, even when whole-cell β-haemolysin abundance levels (e.g. for IMT36945) were obviously sufficient to induce the phenotype in matching agr wt isolates (e.g. RKI5972). Two further isolates harbouring non-wt variants of AgrC (IMT32510 and IMT32929) produced only a weak CAMP phenomenon (blue dots in Supplemental Figure 2). Thus, a functionally active agr system seems to be necessary for S. aureus to produce the exhibit CAMP phenomenon on SBA.
When comparing the results of whole-cell α-haemolysin detection with the haemolysis phenotypes obtained by the SPDH test we noted considerable discrepancies. Thus, we clearly found Hla protein production in some of the isolates harbouring AgrA and AgrC variations, but the strains did not exhibit an α-haemolysin phenotype on SBA (Fig. 3 and Supplemental Figure 1). Vice versa, in five isolates showing α-, β-, δ-haemolysin activity on SBA (blue dots in Supplemental Figure 1), no Hla protein levels were detectable. This could for instance be a result from repeated mis-judgement of the phenotype, from significant differences for Hla abundances between proteome- and secretome or the detection limit of the mass spectrometry. Of note, the α-haemolysis phenotype was absent in all isolates harbouring AgrA or AgrC variants (Supplemental Figure 1), again indicating that a functional agr system is required for inducing an α-haemolysis zone on SBA.
Finally, colony spreading on wet surfaces is known to be directly induced by AgrA-P. As shown in Table 2, all isolates lacking this feature displayed non-wt variants of AgrA or AgrC, while some of the isolates with AgrA or AgrC variants were still capable to spread (Figs. 4b,5).
Agr variation is not associated with differences in biofilm formation on polystyrene tissue culture plates
All 45 mecC-MRSA isolates were tested for their ability to form stable biofilms on the inert surfaces of polystyrene tissue culture plates in a standard crystal violet-staining assay. The test allows for the detection and quantification of sturdy biofilms that are firmly attached to artificial surfaces and which are not removable by washing. With respect to the 12 isolates belonging to CC599, only two (IMT36947 and IMT36948) formed a visible biofilm (value-1-biofilm) in the crystal violet assay (Table 2, detailed information in Supplemental Table 7). Among the 21 CC130 isolates, 12 were biofilm negative (value-0-biofilm), eight displayed moderate biofilm levels (value-1-biofilm) and one isolate produced a strong biofilm (value-2-biofilm). In addition, two out of ten CC1943 mecC-MRSA were strong biofilm producers (value-2-biofilms), while the remaining isolates lacked this ability completely. Summarizing the results of Table 2 and Supplemental Table 3, neither the agr type nor the non-wt variants of AgrA or AgrC were attributable to the ability to form a stable, non-removable biofilm on inert polystyrene surfaces.
AgrA and AgrC variations influence biofilm architecture
While the crystal violet-staining assay exclusively detects robust biofilms that remain permanently attached to surfaces upon washing, CLSM imaging allows for monitoring the biofilm development in situ. By this approach, even delicate interactions of bacterial communities on surfaces can be visualized. In order to elucidate the potential impact of agr functionality on biofilm architecture, we selected three representative isolates of the CC130 carrying different genomic variants of agr (marked by * in Table 2 and Supplemental Table 4) and analyzed them by CLSM imaging. Although the selected isolates were biofilm-negative in the crystal violet-staining assay on polystyrene, they clearly displayed an in situ biofilm on glass slides during CLSM monitoring, with remarkable differences between the isolates. Thus, the biofilm of the Hld-producing isolate IMT38119 harbouring the CC130-wt agr system, formed a flat, dense biofilm that covered almost the entire area of the well (Fig. 6a,b). In contrast, isolate RKI5966 (a non-wt AgrC-variant lacking Hld production) built a dense, local aggregate on the slide (Fig. 6c), with the biofilm mass growing much higher than that of agr-WT isolate IMT38119 (i.e. 307 µm vs. 174 µm) (Fig. 6b,d). Similarly, isolate IMT31819, a non-wt AgrA variant lacking Hld production, also formed a local aggregate (Fig. 6e) and displayed a tall biofilm mass whose architecture, however, appeared less dense than those of the other two isolates tested (Fig. 6f). The combined data suggest that the agr system may rather influence the biofilm architecture than the overall biofilm-forming capacity of S. aureus.

Agr activity and biofilm formation differences among mecC-MRSA belonging to clonal complex 130. (a) Macroscopic camera image of an S. aureus isolate (mecC-MRSA) harbouring the agr III wild type (wt) variant (IMT38119) grown in a 24-well plate and (b) the confocal laser scanning picture showing the biofilm profile at the indicated spot (red square). (c) Isolate harbouring a agr III variant (non-wt agrC variant) lacking agr activity (RKI5966) and (d) its corresponding biofilm. (e) Isolate lacking agr activity (IMT31819) and (f) its corresponding biofilm.
Discussion
Agr variants fine-tune virulence levels in mecC-MRSA belonging to CC130, CC1943, CC599 and CC49
In the past decade, MRSA have established both as commensals and as infectious agents in animals at alarming rates87,88. In addition to classical mecA-carrying MRSA clonal lineages, emerging mecC-MRSA further add to this problem by affecting various animal species as well as humans, suggesting a broad host range of such strains8,14,16. The mecC-MRSA isolates analysed in our study were found to belong to clonal complexes CC130, CC1943, CC599 and CC49, confirming the widespread occurrence of these lineages, at least in Europe8, 14,16. Each of the four CCs harboured distinct isolates with altered agr loci, resulting in reduced or even total loss of agr activity. By employing mass spectrometry, we determined Hld production which served as a proxy for RNAIII transcription in our experiments. Agr functionality was further assessed by testing haemolysis on SBA and a colony spreading assay on semisolid agar whose accuracy for predicting Agr activity was shown before35,71. Based on the δ-haemolysin amounts detected, the 45 isolates (and three reference strains) were assigned to distinct agr activity groups (i.e. 0, +, ++) which matched well with synergistic haemolysis on SBA (Fig. 4a) and the results of the colony spreading assay (Fig. 4b).
Isolates showing amino acid changes in the C-terminal DNA-binding domain of the AgrA response regulator89 such as IMT31818 (ΔN177), IMT39824 (F196S), IMT34478 (S215P) and IMT36952 (C199R) did not produce δ-haemolysin, indicating that these aberrations silenced the agr system. However, IMT34478 was still capable of colony spreading, suggesting residual or agr-independent PSM production by a mechanism that still needs to be established.
With respect to non-wt AgrC variants, RKI5966 (Q16H) showed a variation in the transmembrane 1 domain of the protein, while the aa exchange E216K in IMT32929 is located in the C-terminal dimerization/histidine phosphotransfer subdomain of the protein90. These changes are prone to impair AgrC-mediated AIP sensing and signal transduction90, and are therefore likely to cause the agr-negative phenotype observed in the isolates.
Further, comparative analysis of Hld production values with the CAMP test results confirmed that an active agr system is necessary to induce the lunar-shaped synergistic haemolysis on SBA, with Hld and/or PSMs known to contribute to the phenotype67. With respect to β-haemolysin production, agr was shown to have a major impact on hlb transcription91. However, this association is obviously not straightforward and applicable for all strains. Thus, high levels of Hlb were also detected in S. aureus isolates displaying low agr activity, such as RN422086, which is in good agreement with the Hlb detection results obtained in our isolates (Supplemental Figure 2). Moreover, our data indicate that at least a weak agr activity is necessary to induce a Hlb-mediated β-haemolysis phenotype on SBA, even when the intracellular Hlb levels were apparently sufficient to induce haemolysis in corresponding agr wt-isolates (Supplemental Figure 2).
Production of many secreted enzymes involved in lipid and protein degradation and haemolysins are influenced or even directly controlled by agr. The main effector of this locus, RNAIII, is known to promote α-haemolysin expression on the transcriptional and post-transcriptional level92. As expected, Hla production was not detected in isolates lacking the gene (IMT34489) and in those carrying hla frameshift mutations (IMT34479 and IMT34480). Surprisingly, Hla abundance values measured in whole cells of isolates with intact hla genes did not correlate with the agr activity levels (Supplemental Figure 1). Also, for some of the isolates with AgrA/AgrC aberrations and negative α-haemolysis on SBA (IMT31818, IMT32510, IMT32929, IMT36945, IMT39824, RKI5966, NE1532, and RN4220) we noticed intracellular Hla protein amounts that were comparable to that of agr-wt strains showing a haemolytic phenotype (Supplemental Figure 1). A study by Montgomery et al. confirmed notable agr-independent transcription of Hla in an agr-deficient strain (USA300 lineage), while the protein was not detected in the corresponding culture supernants69, which is commonly explained by the promoting role of RNAIII for hla transcript translation92. Since our Hlb and Hla protein abundances were measured from overnight-grown cells after washing with PBS, our results indicate that some agr-activity is needed for toxin release. Interestingly, extracellular vesicles released from cells of the USA300 lineage were found to contain Hla93. In that particular study, PSMα was identified to promote biogenesis of extracellular vesicles filled with proteins by destabilisation of the cytoplasmic membrane93. Thus, is tempting to speculate that baseline agr-independent hla transcription and translation might occur in the variants, leading to intracellular accumulation of α-haemolysin. Lacking PSM activity (due to AgrA/C variations) might prevent release of Hla in the supernatant, resulting in the haemolysis-negative phenotypes observed. More experimental work, however, is needed to substantiate this hypothesis in the future.
Agr variants influence biofilm structure and density
Biofilm formation is a key factor in pathogenesis of persistent staphylococcal infections94, and downregulation of agr is supposed to facilitate biofilm development in staphylococci50,95,96,97 with strain-specific differences occurring particularly among clinical isolates98. When testing the mecC-MRSA isolates in a standard crystal violet-staining biofilm assay, we did not find an association between agr functionality and stable biofilm formation in polystyrene tissue culture plates (Table 2), which is in good agreement with previous findings in clinical MRSA clonal lineages38. Generally, we found only a few biofilm-forming isolates by this assay, although biofilm-associated genes were present and intact in all strains. In comparison to S. epidermidis, S. aureus is known to show a much weaker biofilm detection performance in the standard biofilm assay. Rather than suggesting a lower overall biofilm-forming capacity of S. aureus, the phenomenon may reflect a general mechanically instable contact of biofilm-associated S. aureus cells to inert surfaces, leading to removal of loosely attached biofilm structures upon washing of the plates. Indeed, when using CLSM imaging, which does not involve washing of the cover slips, we detected visible biofilms, at least in the three CC130 isolates analysed (Fig. 6). Interestingly, strains possessing different agr activity levels displayed differences regarding biofilm thickness and architecture. Thus, while the WT-agr isolate formed a well-organized biofilm (Fig. 6b), the two agr-deficient variants displayed a much higher biofilm mass which, however, appeared less-structured (Fig. 6d,f). This might be due to the lack of agr-dependent production of PSMs which were previously shown to play an eminent role in functional biofilm architecture99,100. S. aureus biofilm formation is a complex and highly dynamic process which comprises, according to a recently newly defined five-stage model, attachment, multiplication, exodus, maturation and dispersal of the biofilm101. Apart from its function in maturation and dispersal (involving agr-controlled proteases and PSMs), agr also plays a significant role during initial attachment by facilitating (in the early growth stage) expression of cell wall-anchored proteins that mediate host matrix protein binding as well as contact to abiotic surfaces102. We currently speculate that the loose biofilm structure in the CC130 agr-variants might be associated with a diminished initial attachment of the bacteria to the surface and/or to each other. However, it is conceivable that other processes known to shape the biofilm architecture such as programmed autolysis and eDNA release103 might be (indirectly) affected by the Agr variations as well. But, clearly more experimental work is needed to substantiate this hypothesis.
Adaptation strategies of mecC-MRSA might involve agr defectiveness and carriage of agr-independent virulence factors
Evolution and changes of bacterial virulence is highly dynamic and difficult to predict104,105. Attenuated virulence favouring host colonization and/or persistence of infection (e.g. small colony variants) is a common concept in bacterial evolution106,107,108. The mecC-MRSA lineages CC130, CC49, CC1943 and CC599 were obviously not among those CC’s frequently reported for nasal colonization in humans and animals such as CC22 and CC398109,110,111,112. Consequently, other strategies might promote their viability or even spread among mammalian hosts and the environment. On one hand, the disability to produce the agrD-encoded auto-inducer peptide (AIP) may prevent costly competition of invading strains with an “incompatible” agr system harboured by other S. aureus lineages or even other staphylococcal species in a particular host113. One the other hand, presence of a compatible AIP system allows the cells to take advantage of the agr-induced factors produced by co-habiting staphylococci29. A highly instructive study, using a wax moth larva virulence model, revealed that a functional agr system is necessary for a cooperative and beneficial “behaviour” of the local population, while agr defective mutants exploit (“cheat”) on the cells with a functional agr system, allowing them to prevail when grown in mixed populations with cooperators113. Based on earlier published results of growth competition experiments113, 114, an increased viability of agr defective S. aureus mutants among resident staphylococci of different host species seems to be likely. This view is further supported by a recent study, showing that distinct external stress conditions drive the selection of Agr quorum-sensing mutants that may confer a fitness advantage to the S. aureus population115.
Unspecific and unregulated T-cell stimulators (superantigens) such as enterotoxins, enterotoxin-like proteins, and the toxic shock syndrome toxin contribute to host cell damages inducible by S. aureus. Combinations of genes encoding these superantigens were identified in varying frequencies for all four lineages reported on here (Fig. 1). With respect to enterotoxins116, Enterotoxin C (SEC), which is commonly located on a SaPI (reviewed in49), was identified in isolates belonging to CC130, CC599 and CC1943. However, SEC production requires agr-depending rot degradation117, which is defective in some of the isolates reported on here. While classical enterotoxins such as SEA-C are regulated by the agr system, the “novel” enterotoxins and variants of the toxic shock syndrome toxin seem to be agr independent (reviewed in49). In line with this, we have shown expression of the protein encoded by tst-bov in isolates showing lacking detectable agr activity (Fig. 1, Table 2), exemplified by isolates IMT31818 and IMT39824. The variants of tst-bov were harboured by S. aureus pathogenicity islands showing mosaic structures of known and novel SaPIs, an observation which has been reported (e.g. for human clinical isolates) before118.
Conclusion
Comparative genomics of the agr encoding region allow identification of variants deviating from the wildtype in mecC-MRSA belonging to CC130, CC599, CC49 and CC1943, and subsequent proteomics revealed the capability of each altered agr system to transcribe RNAIII, which was directly mirrored by the corresponding Hld protein values. Our research indicates that mecC-MRSA with agr variations are defective for agr-depending quorum sensing, harbour additional agr-independent virulence factors and exhibit varying biofilm properties as a likely part of their survival strategy. In bacteria, adaptation to a changing environment is often associated with the selection for mutations in (virulence factor) genes that become dispensable or disadvantageous in the novel niche (reviewed in104). This concept can obviously be extended to global regulators such as the agr quorum-sensing system as well, resulting in pleiotropic effects and the generation of phenotypic heterogeneity that might further support the establishment of emerging clonal lineages in new hosts and niches.
Data availability
Genomic sequencing data used are available for download from the National Center for Biotechnology Information (NCBI) under BioProject accessions PRJNA588740. Accession numbers of whole genomes sequences are provided in Supplemental Table 2. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD016486.
References
Walther, B. et al. Equine methicillin-resistant sequence type 398 Staphylococcus aureus (MRSA) harbor mobile genetic elements promoting host adaptation. Front. Microbiol. 9, 2516. https://doi.org/10.3389/fmicb.2018.02516 (2018).
Lowy, F. D. Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532. https://doi.org/10.1056/nejm199808203390806 (1998).
Kriegeskorte, A. & Peters, G. Horizontal gene transfer boosts MRSA spreading. Nat. Med. 18, 662–663. https://doi.org/10.1038/nm.2765 (2012).
Peacock, S. J. & Paterson, G. K. Mechanisms of methicillin resistance in Staphylococcus aureus. Annu. Rev. Biochem. 84, 577–601. https://doi.org/10.1146/annurev-biochem-060614-034516 (2015).
Garcia-Alvarez, L. et al. Meticillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect. Dis. 11, 595–603. https://doi.org/10.1016/s1473-3099(11)70126-8 (2011).
Baig, S. et al. Novel SCCmec type XIII (9A) identified in an ST152 methicillin-resistant Staphylococcus aureus. Infect. Genet. Evol. 61, 74–76. https://doi.org/10.1016/j.meegid.2018.03.013 (2018).
Kerschner, H., Harrison, E. M., Hartl, R., Holmes, M. A. & Apfalter, P. First report of mecC MRSA in human samples from Austria: molecular characteristics and clinical data. New Microb. New Infect. 3, 4–9. https://doi.org/10.1016/j.nmni.2014.11.001 (2015).
Paterson, G. K., Harrison, E. M. & Holmes, M. A. The emergence of mecC methicillin-resistant Staphylococcus aureus. Trends Microbiol. 22, 42–47. https://doi.org/10.1016/j.tim.2013.11.003 (2014).
Cuny, C., Layer, F., Strommenger, B. & Witte, W. Rare occurrence of methicillin-resistant Staphylococcus aureus CC130 with a novel mecA homologue in humans in Germany. PLoS ONE 6, e24360. https://doi.org/10.1371/journal.pone.0024360 (2011).
Ford, B. A. mecC-harboring methicillin-resistant Staphylococcus aureus: hiding in plain sight. J. Clin. Microbiol. 5, 6. https://doi.org/10.1128/jcm.01549-17 (2018).
Kil, E. H., Heymann, W. R. & Weinberg, J. M. Methicillin-resistant Staphylococcus aureus: an update for the dermatologist, part 4: additional therapeutic considerations. Cutis 81, 343–347 (2008).
Shore, A. C. et al. Detection of staphylococcal cassette chromosome mec type XI carrying highly divergent mecA, mecI, mecR1, blaZ, and ccr genes in human clinical isolates of clonal complex 130 methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 55, 3765–3773. https://doi.org/10.1128/aac.00187-11 (2011).
Worthing, K. A. et al. Isolation of mecC MRSA in Australia. J. Antimicrob. Chemother. 71, 2348–2349. https://doi.org/10.1093/jac/dkw138 (2016).
Walther, B. et al. MRSA variant in companion animals. Emerg. Infect. Dis. 18, 2017–2020. https://doi.org/10.3201/eid1812.120238 (2012).
Loncaric, I. et al. Characterization of methicillin-resistant Staphylococcus spp. carrying the mecC gene, isolated from wildlife. J. Antimicrob. Chemother. 68, 2222–2225. https://doi.org/10.1093/jac/dkt186 (2013).
Garcia-Garrote, F. et al. Methicillin-resistant Staphylococcus aureus carrying the mecC gene: emergence in Spain and report of a fatal case of bacteraemia. J. Antimicrob. Chemother. 69, 45–50. https://doi.org/10.1093/jac/dkt327 (2014).
Mekonnen, S. A. et al. Metabolic niche adaptation of community- and hospital-associated methicillin-resistant Staphylococcus aureus. J. Proteom. 193, 154–161. https://doi.org/10.1016/j.jprot.2018.10.005 (2019).
Paharik, A. E. & Horswill, A. R. The Staphylococcal biofilm: adhesins, regulation, and host response. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.VMBF-0022-2015 (2016).
Hammer, N. D. & Skaar, E. P. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu. Rev. Microbiol. 65, 129–147. https://doi.org/10.1146/annurev-micro-090110-102851 (2011).
Lerat, E. & Ochman, H. Recognizing the pseudogenes in bacterial genomes. Nucl. Acids Res. 33, 3125–3132. https://doi.org/10.1093/nar/gki631 (2005).
Mcgavin, M., Arsic, B. & Nickerson, N. Evolutionary blueprint for host- and niche-adaptation in Staphylococcus aureus clonal complex CC30. Front. Cell. Infect. Microbiol. https://doi.org/10.3389/fcimb.2012.00048 (2012).
Tavares, A. et al. Insights into alpha-hemolysin (Hla) evolution and expression among Staphylococcus aureus clones with hospital and community origin. PLoS ONE 9, e98634. https://doi.org/10.1371/journal.pone.0098634 (2014).
Yoong, P. & Torres, V. J. Counter inhibition between leukotoxins attenuates Staphylococcus aureus virulence. Nat. Commun. 6, 8125. https://doi.org/10.1038/ncomms9125 (2015).
Chadha, A. D. et al. Host response to Staphylococcus aureus cytotoxins in children with cystic fibrosis. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 15, 597–604. https://doi.org/10.1016/j.jcf.2015.12.023 (2016).
Benson, M. A. et al. Evolution of hypervirulence by a MRSA clone through acquisition of a transposable element. Mol. Microbiol. 93, 664–681. https://doi.org/10.1111/mmi.12682 (2014).
Busche, T. et al. Comparative secretome analyses of human and zoonotic Staphylococcus aureus Isolates CC8, CC22, and CC398. Mol. Cell. Proteom. MCP 17, 2412–2433. https://doi.org/10.1074/mcp.RA118.001036 (2018).
Richardson, E. J. et al. Gene exchange drives the ecological success of a multi-host bacterial pathogen. Nat. Ecol. Evol. 2, 1468–1478. https://doi.org/10.1038/s41559-018-0617-0 (2018).
Walther, B. et al. Comparative molecular analysis substantiates zoonotic potential of equine methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 47, 704–710. https://doi.org/10.1128/jcm.01626-08 (2009).
Wang, B. & Muir, T. W. Regulation of virulence in Staphylococcus aureus: molecular mechanisms and remaining puzzles. Cell Chem. Biol. 23, 214–224. https://doi.org/10.1016/j.chembiol.2016.01.004 (2016).
Jenul, C. & Horswill, A. R. Regulation of Staphylococcus aureus virulence. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.GPP3-0031-2018 (2018).
Cheung, A. L. et al. Diminished virulence of a sar-/agr-mutant of Staphylococcus aureus in the rabbit model of endocarditis. J. Clin. Investig. 94, 1815–1822. https://doi.org/10.1172/jci117530 (1994).
Kobayashi, S. D. et al. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J. Infect. Dis. 204, 937–941. https://doi.org/10.1093/infdis/jir441 (2011).
Abdelnour, A., Arvidson, S., Bremell, T., Rydén, C. & Tarkowski, A. The accessory gene regulator (agr) controls Staphylococcus aureus virulence in a murine arthritis model. Infect. Immun. 61, 3879–3885 (1993).
Fowler, V. G. Jr. et al. Persistent bacteremia due to methicillin-resistant Staphylococcus aureus infection is associated with agr dysfunction and low-level in vitro resistance to thrombin-induced platelet microbicidal protein. J. Infect. Dis. 190, 1140–1149. https://doi.org/10.1086/423145 (2004).
Traber, K. E. et al. agr function in clinical Staphylococcus aureus isolates. Microbiology (Reading, England) 154, 2265–2274. https://doi.org/10.1099/mic.0.2007/011874-0 (2008).
Schweizer, M. L. et al. Increased mortality with accessory gene regulator (agr) dysfunction in Staphylococcus aureus among bacteremic patients. Antimicrob. Agents Chemother. 55, 1082–1087. https://doi.org/10.1128/aac.00918-10 (2011).
Painter, K. L., Krishna, A., Wigneshweraraj, S. & Edwards, A. M. What role does the quorum-sensing accessory gene regulator system play during Staphylococcus aureus bacteremia?. Trends Microbiol. 22, 676–685. https://doi.org/10.1016/j.tim.2014.09.002 (2014).
Yang, X. et al. Accessory gene regulator (agr) dysfunction was unusual in Staphylococcus aureus isolated from Chinese children. BMC Microbiol. 19, 95. https://doi.org/10.1186/s12866-019-1465-z (2019).
He, L. et al. Resistance to leukocytes ties benefits of quorum sensing dysfunctionality to biofilm infection. Nat. Microbiol. 4, 1114–1119. https://doi.org/10.1038/s41564-019-0413-x (2019).
Altman, D. R. et al. Genome plasticity of agr-defective Staphylococcus aureus during clinical infection. Infect. Immun. https://doi.org/10.1128/iai.00331-18 (2018).
Goerke, C. et al. Direct quantitative transcript analysis of the agr regulon of Staphylococcus aureus during human infection in comparison to the expression profile in vitro. Infect. Immun. 68, 1304–1311. https://doi.org/10.1128/iai.68.3.1304-1311.2000 (2000).
Suligoy, C. M. et al. Mutation of Agr is associated with the adaptation of Staphylococcus aureus to the host during chronic osteomyelitis. Front. Cell. Infect. Microbiol. 8, 18. https://doi.org/10.3389/fcimb.2018.00018 (2018).
Le, K. Y. & Otto, M. Quorum-sensing regulation in staphylococci—an overview. Front. Microbiol. 6, 1174. https://doi.org/10.3389/fmicb.2015.01174 (2015).
Reynolds, J. & Wigneshweraraj, S. Molecular insights into the control of transcription initiation at the Staphylococcus aureus agr operon. J. Mol. Biol. 412, 862–881. https://doi.org/10.1016/j.jmb.2011.06.018 (2011).
Abisado, R. G., Benomar, S., Klaus, J. R., Dandekar, A. A. & Chandler, J. R. Bacterial quorum sensing and microbial community interactions. mBio 9, e02331-02317. https://doi.org/10.1128/mBio.02331-17 (2018).
Benito, Y. et al. Probing the structure of RNAIII, the Staphylococcus aureus agr regulatory RNA, and identification of the RNA domain involved in repression of protein A expression. RNA (New York, N.Y.) 6, 668–679. https://doi.org/10.1017/s1355838200992550 (2000).
Joo, H. S. et al. Mechanism of gene regulation by a Staphylococcus aureus toxin. mBio https://doi.org/10.1128/mBio.01579-16 (2016).
Cheung, G. Y., Wang, R., Khan, B. A., Sturdevant, D. E. & Otto, M. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect. Immun. 79, 1927–1935. https://doi.org/10.1128/iai.00046-11 (2011).
Fisher, E. L., Otto, M. & Cheung, G. Y. C. Basis of Virulence in enterotoxin-mediated staphylococcal food poisoning. Front. Microbiol. https://doi.org/10.3389/fmicb.2018.00436 (2018).
Ferreira, F. A. et al. Impact of agr dysfunction on virulence profiles and infections associated with a novel methicillin-resistant Staphylococcus aureus (MRSA) variant of the lineage ST1-SCCmec IV. BMC Microbiol. 13, 93. https://doi.org/10.1186/1471-2180-13-93 (2013).
Tasse, J. et al. Association between biofilm formation phenotype and clonal lineage in Staphylococcus aureus strains from bone and joint infections. PLoS ONE 13, e0200064. https://doi.org/10.1371/journal.pone.0200064 (2018).
Jang, H. C. et al. Difference in agr dysfunction and reduced vancomycin susceptibility between MRSA bacteremia involving SCCmec types IV/IVa and I-III. PLoS ONE 7, e49136. https://doi.org/10.1371/journal.pone.0049136 (2012).
Sheppard, S. K., Guttman, D. S. & Fitzgerald, J. R. Population genomics of bacterial host adaptation. Nat. Rev. Genet. 19, 549–565. https://doi.org/10.1038/s41576-018-0032-z (2018).
Merlino, J. et al. Detection and expression of methicillin/oxacillin resistance in multidrug-resistant and non-multidrug-resistant Staphylococcus aureus in Central Sydney Australia. J. Antimicrob. Chemother. 49, 793–801 (2002).
Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Fourth Informational Supplement M100-S24 (2014).
Clinical and Laboratory Standards Institute (Wayne, PA, 2013).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. https://doi.org/10.1093/bioinformatics/btu153 (2014).
Larsen, M. V. et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. https://doi.org/10.1128/jcm.06094-11 (2012).
Kearse, M. et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199 (2012).
Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. https://doi.org/10.1093/jac/dks261 (2012).
Joensen, K. G. et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. https://doi.org/10.1128/jcm.03617-13 (2014).
Bartels, M. D. et al. Comparing whole-genome sequencing with Sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 52, 4305–4308. https://doi.org/10.1128/jcm.01979-14 (2014).
von Mentzer, A. et al. Identification of enterotoxigenic Escherichia coli (ETEC) clades with long-term global distribution. Nat. Genet. 46, 1321. https://doi.org/10.1038/ng.3145 (2015).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics (Oxford, England) 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 (2014).
Hadfield, J. et al. Phandango: an interactive viewer for bacterial population genomics. Bioinformatics https://doi.org/10.1093/bioinformatics/btx610 (2017).
Cheung, G. Y. C., Duong, A. C. & Otto, M. Direct and synergistic hemolysis caused by Staphylococcus phenol-soluble modulins: implications for diagnosis and pathogenesis. Microbes Infect. 14, 380–386. https://doi.org/10.1016/j.micinf.2011.11.013 (2012).
Nair, D. et al. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J. Bacteriol. 193, 2332–2335. https://doi.org/10.1128/jb.00027-11 (2011).
Montgomery, C. P., Boyle-Vavra, S. & Daum, R. S. Importance of the global regulators Agr and SaeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS ONE 5, e15177. https://doi.org/10.1371/journal.pone.0015177 (2010).
Gase, K., Ferretti, J. J., Primeaux, C. & McShan, W. M. Identification, cloning, and expression of the CAMP factor gene (cfa) of group A streptococci. Infect. Immun. 67, 4725–4731 (1999).
Kaito, C. & Sekimizu, K. Colony spreading in Staphylococcus aureus. J. Bacteriol. 189, 2553–2557. https://doi.org/10.1128/JB.01635-06 (2007).
Tsompanidou, E. et al. Requirement of the agr locus for colony spreading of Staphylococcus aureus. J. Bacteriol. 193, 1267–1272. https://doi.org/10.1128/jb.01276-10 (2011).
Lin, M.-H., Ke, W.-J., Liu, C.-C. & Yang, M.-W. Modulation of Staphylococcus aureus spreading by water. Sci. Rep. 6, 25233. https://doi.org/10.1038/srep25233 (2016).
Tsompanidou, E. et al. The sortase A substrates FnbpA, FnbpB, ClfA and ClfB antagonize colony spreading of Staphylococcus aureus. PLoS ONE 7, e44646. https://doi.org/10.1371/journal.pone.0044646 (2012).
Doellinger, J., Schneider, A., Hoeller, M. & Lasch, P. Sample preparation by easy extraction and digestion (SPEED)—a universal, rapid, and detergent-free protocol for proteomics based on acid extraction. Mol. Cell. Proteom. MCP https://doi.org/10.1074/mcp.TIR119.001616 (2019).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906. https://doi.org/10.1038/nprot.2007.261 (2007).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods https://doi.org/10.1038/s41592-019-0638-x (2019).
Christensen, G. D. et al. Adherence of coagulase-negative staphylococci to plastic tissue culture plates: a quantitative model for the adherence of staphylococci to medical devices. J. Clin. Microbiol. 22, 996–1006 (1985).
Becker, K., Ballhausen, B., Köck, R. & Kriegeskorte, A. Methicillin resistance in Staphylococcus isolates: the “mec alphabet” with specific consideration of mecC, a mec homolog associated with zoonotic S. aureus lineages. Int. J. Med. Microbiol. 304, 794–804. https://doi.org/10.1016/j.ijmm.2014.06.007 (2014).
Fitzgerald, J. R. et al. Characterization of a putative pathogenicity island from bovine Staphylococcus aureus encoding multiple superantigens. J. Bacteriol. 183, 63–70. https://doi.org/10.1128/jb.183.1.63-70.2001 (2001).
Bosi, E. et al. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. USA 113, E3801-3809. https://doi.org/10.1073/pnas.1523199113 (2016).
Monecke, S. et al. Detection of mecC-positive Staphylococcus aureus (CC130-MRSA-XI) in diseased European hedgehogs (Erinaceus europaeus) in Sweden. PLoS ONE 8, e66166. https://doi.org/10.1371/journal.pone.0066166 (2013).
Shopsin, B. et al. Prevalence of agr specificity groups among Staphylococcus aureus strains colonizing children and their guardians. J. Clin. Microbiol. 41, 456–459. https://doi.org/10.1128/jcm.41.1.456-459.2003 (2003).
Queck, S. Y. et al. RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol. Cell 32, 150–158. https://doi.org/10.1016/j.molcel.2008.08.005 (2008).
Morrison, J. M., Anderson, K. L., Beenken, K. E., Smeltzer, M. S. & Dunman, P. M. The staphylococcal accessory regulator, SarA, is an RNA-binding protein that modulates the mRNA turnover properties of late-exponential and stationary phase Staphylococcus aureus cells. Front. Cell. Infect. Microbiol. 2, 26. https://doi.org/10.3389/fcimb.2012.00026 (2012).
Traber, K. & Novick, R. A slipped-mispairing mutation in AgrA of laboratory strains and clinical isolates results in delayed activation of agr and failure to translate δ- and α-haemolysins. Mol. Microbiol. 59, 1519–1530. https://doi.org/10.1111/j.1365-2958.2006.04986.x (2006).
Vincze, S. et al. Alarming proportions of methicillin-resistant Staphylococcus aureus (MRSA) in wound samples from companion animals, Germany 2010–2012. PLoS ONE 9, e85656. https://doi.org/10.1371/journal.pone.0085656 (2014).
Kaspar, U. et al. Zoonotic multidrug-resistant microorganisms among small companion animals in Germany. PLoS ONE 13, e0208364. https://doi.org/10.1371/journal.pone.0208364 (2018).
Sidote, D. J., Barbieri, C. M., Wu, T. & Stock, A. M. Structure of the Staphylococcus aureus AgrA LytTR domain bound to DNA reveals a beta fold with an unusual mode of binding. Structure 16, 727–735. https://doi.org/10.1016/j.str.2008.02.011 (2008).
George Cisar, E. A., Geisinger, E., Muir, T. W. & Novick, R. P. Symmetric signalling within asymmetric dimers of the Staphylococcus aureus receptor histidine kinase AgrC. Mol. Microbiol. 74, 44–57. https://doi.org/10.1111/j.1365-2958.2009.06849.x (2009).
Recsei, P. et al. Regulation of exoprotein gene expression in Staphylococcus aureus by agar. Mol. Gen. Genet. MGG 202, 58–61. https://doi.org/10.1007/bf00330517 (1986).
Morfeldt, E., Taylor, D., von Gabain, A. & Arvidson, S. Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA RNAIII. EMBO J. 14, 4569–4577 (1995).
Wang, X., Thompson, C. D., Weidenmaier, C. & Lee, J. C. Release of Staphylococcus aureus extracellular vesicles and their application as a vaccine platform. Nat. Commun. 9, 1379. https://doi.org/10.1038/s41467-018-03847-z (2018).
Dastgheyb, S. S. & Otto, M. Staphylococcal adaptation to diverse physiologic niches: an overview of transcriptomic and phenotypic changes in different biological environments. Future Microbiol. 10, 1981–1995. https://doi.org/10.2217/fmb.15.116 (2015).
Dai, L. et al. Staphylococcus epidermidis recovered from indwelling catheters exhibit enhanced biofilm dispersal and “self-renewal” through downregulation of agr. BMC Microbiol. 12, 102. https://doi.org/10.1186/1471-2180-12-102 (2012).
Cafiso, V. et al. agr-Genotyping and transcriptional analysis of biofilm-producing Staphylococcus aureus. FEMS Immunol. Med. Microbiol. 51, 220–227. https://doi.org/10.1111/j.1574-695X.2007.00298.x (2007).
Kong, K. F., Vuong, C. & Otto, M. Staphylococcus quorum sensing in biofilm formation and infection. Int. J. Med. Microbiol. IJMM 296, 133–139. https://doi.org/10.1016/j.ijmm.2006.01.042 (2006).
Beenken, K. E. et al. Epistatic relationships between sarA and agr in Staphylococcus aureus biofilm formation. PLoS ONE 5, e10790. https://doi.org/10.1371/journal.pone.0010790 (2010).
Wang, R. et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 13, 1510–1514. https://doi.org/10.1038/nm1656 (2007).
Periasamy, S. et al. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. USA 109, 1281–1286. https://doi.org/10.1073/pnas.1115006109 (2012).
Moormeier, D. E. & Bayles, K. W. Staphylococcus aureus biofilm: a complex developmental organism. Mol. Microbiol. 104, 365–376. https://doi.org/10.1111/mmi.13634 (2017).
Foster, T. J., Geoghegan, J. A., Ganesh, V. K. & Hook, M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 12, 49–62. https://doi.org/10.1038/nrmicro3161 (2014).
Sadykov, M. R. & Bayles, K. W. The control of death and lysis in staphylococcal biofilms: a coordination of physiological signals. Curr. Opin. Microbiol. 15, 211–215. https://doi.org/10.1016/j.mib.2011.12.010 (2012).
Diard, M. & Hardt, W. D. Evolution of bacterial virulence. FEMS Microbiol. Rev. 41, 679–697. https://doi.org/10.1093/femsre/fux023 (2017).
Priest, N. K. et al. From genotype to phenotype: can systems biology be used to predict Staphylococcus aureus virulence?. Nat. Rev. Microbiol. 10, 791–797. https://doi.org/10.1038/nrmicro2880 (2012).
Kahl, B. C., Becker, K. & Loffler, B. Clinical significance and pathogenesis of staphylococcal small colony variants in persistent infections. Clin. Microbiol. Rev. 29, 401–427. https://doi.org/10.1128/cmr.00069-15 (2016).
Tuchscherr, L. et al. Clinical S. aureus isolates vary in their virulence to promote adaptation to the host. Toxins (Basel) https://doi.org/10.3390/toxins11030135 (2019).
Fraunholz, M. & Sinha, B. Intracellular Staphylococcus aureus: live-in and let die. Front. Cell. Infect. Microbiol. 2, 43. https://doi.org/10.3389/fcimb.2012.00043 (2012).
Cuny, C. et al. Methicillin-resistant Staphylococcus aureus from infections in horses in Germany are frequent colonizers of veterinarians but rare among MRSA from infections in humans. One Health (Amsterdam, Netherlands) 2, 11–17. https://doi.org/10.1016/j.onehlt.2015.11.004 (2016).
Golubchik, T. et al. Within-host evolution of Staphylococcus aureus during asymptomatic carriage. PLoS ONE 8, e61319. https://doi.org/10.1371/journal.pone.0061319 (2013).
Skallerup, P., Espinosa-Gongora, C., Jorgensen, C. B., Guardabassi, L. & Fredholm, M. Genome-wide association study reveals a locus for nasal carriage of Staphylococcus aureus in Danish crossbred pigs. BMC Vet. Res. 11, 290. https://doi.org/10.1186/s12917-015-0599-y (2015).
Holtfreter, S. et al. Molecular epidemiology of Staphylococcus aureus in the general population in Northeast Germany: results of the study of health in Pomerania (SHIP-TREND-0). J. Clin. Microbiol. 54, 2774–2785. https://doi.org/10.1128/jcm.00312-16 (2016).
Pollitt, E. J., West, S. A., Crusz, S. A., Burton-Chellew, M. N. & Diggle, S. P. Cooperation, quorum sensing, and evolution of virulence in Staphylococcus aureus. Infect. Immun. 82, 1045–1051. https://doi.org/10.1128/iai.01216-13 (2014).
Paulander, W. et al. Antibiotic-mediated selection of quorum-sensing-negative Staphylococcus aureus. mBio 3, e00459-00412. https://doi.org/10.1128/mBio.00459-12 (2013).
George, S. E. et al. Oxidative stress drives the selection of quorum sensing mutants in the Staphylococcus aureus population. Proc. Natl. Acad. Sci. USA 116, 19145–19154. https://doi.org/10.1073/pnas.1902752116 (2019).
Schlievert, P. M. et al. Pyrogenic toxin superantigen site specificity in toxic shock syndrome and food poisoning in animals. Infect. Immun. 68, 3630–3634. https://doi.org/10.1128/iai.68.6.3630-3634.2000 (2000).
Regassa, L. B. & Betley, M. J. High sodium chloride concentrations inhibit staphylococcal enterotoxin C gene (sec) expression at the level of sec mRNA. Infect. Immun. 61, 1581–1585 (1993).
Amissah, N. A. et al. Virulence potential of Staphylococcus aureus isolates from Buruli ulcer patients. Int. J. Med. Microbiol. 307, 223–232. https://doi.org/10.1016/j.ijmm.2017.04.002 (2017).
Acknowledgements
We thank Aline Poppe, Andy Schneider, ZBS4 photo lab and the team of MF2 of the Robert Koch Institute for their individual contribution to this project. This research was funded by the Federal Ministry of Education and Research (BMBF) under Project Numbers 01KI1727D, 01KI1727F, 01KI1727E and 01KI1725F as part of the Research Network Zoonotic Infectious Diseases as well as by the Deutsche Forschungsgemeinschaft (DFG) through Project ZI665/3-1. CH was supported by a Grant from Akademie für Tiergesundheit e.V. (scholarship). MB acknowledges support from the German Ministry for Education and Research (01KI1301B).
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
C.H. and B.W. designed the project. C.H., B.W., C.S., W.Z. and J.D. conceived and designed the experiments. I.S., B.S., C.C. and W.W. collected and screened the isolates. A.T. sequenced the isolates and T.S. and L.E. analysed the phylogenetic relationship based on WGS data. C.H., G.J., T.M., C.S., A.L-B. and J.D. performed the laboratory experiments. C.H., B.W., W.Z., G.M., M.B., J.D., C.S., L.E., T.S., A. L-B. and L.H.W. analysed the data. C.H., W.Z., M.B., J.D., C.S., T.S., L.H.W. and B.W. wrote the manuscript. All authors have read and approved the final draft of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huber, C., Stamm, I., Ziebuhr, W. et al. Silence as a way of niche adaptation: mecC-MRSA with variations in the accessory gene regulator (agr) functionality express kaleidoscopic phenotypes. Sci Rep 10, 14787 (2020). https://doi.org/10.1038/s41598-020-71640-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-71640-4
This article is cited by
-
The role of Staphylococcus aureus quorum sensing in cutaneous and systemic infections
Inflammation and Regeneration (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
