
Abstract
Ancient genomic studies have identified Yersinia pestis (Y. pestis) as the causative agent of the second plague pandemic (fourteenth–eighteenth century) that started with the Black Death (1,347–1,353). Most of the Y. pestis strains investigated from this pandemic have been isolated from western Europe, and not much is known about the diversity and microevolution of this bacterium in eastern European countries. In this study, we investigated human remains excavated from two cemeteries in Riga (Latvia). Historical evidence suggests that the burials were a consequence of plague outbreaks during the seventeenth century. DNA was extracted from teeth of 16 individuals and subjected to shotgun sequencing. Analysis of the metagenomic data revealed the presence of Y. pestis sequences in four remains, confirming that the buried individuals were victims of plague. In two samples, Y. pestis DNA coverage was sufficient for genome reconstruction. Subsequent phylogenetic analysis showed that the Riga strains fell within the diversity of the already known post-Black Death genomes. Interestingly, the two Latvian isolates did not cluster together. Moreover, we detected a drop in coverage of the pPCP1 plasmid region containing the pla gene. Further analysis indicated the presence of two pPCP1 plasmids, one with and one without the pla gene region, and only one bacterial chromosome, indicating that the same bacterium carried two distinct pPCP1 plasmids. In addition, we found the same pattern in the majority of previously published post-Black Death strains, but not in the Black Death strains. The pla gene is an important virulence factor for the infection of and transmission in humans. Thus, the spread of pla-depleted strains may, among other causes, have contributed to the disappearance of the second plague pandemic in eighteenth century Europe.
Similar content being viewed by others

Introduction
Yersinia pestis (Y. pestis) is the causative agent of plague that evolved from a relatively benign pathogen—Yersinia pseudotuberculosis—thousands of years ago1,2,3,4. Whilst plague is a zoonotic disease with rodents being the primary hosts of the pathogen5,6,7, it has also afflicted humans for at least 5,000 years1. In the last two millennia, Y. pestis was responsible for three major epidemics, of which the second was the most infamous. This pandemic began with the Black Death in the fourteenth century (1,346–1,353). It is thought to have originated in Asia from where it quickly spread to and across the European continent killing approximately 30–50% of the population within a few years8,9. The second pandemic persisted for over 400 years10 causing, among others, the Great Plague of London (1,665–1,666) and Marseille (1,720–1,722)11. In contrast to western Europe, the first major outbreaks of plague in the north-eastern part of the continent took place mainly in the post-Black Death period (after 1,353), for instance in Riga (1,601–3, 1,621–23 and 1,657) or during the Russian Plague (1,770–1,772)12,13,14.
Ancient DNA (aDNA) studies have shown that all second-plague pandemic strains cluster together in branch 1 of the Y. pestis phylogeny, close to present-day representatives15,16,17. Interestingly, pathogens involved in the post-Black Death outbreaks derived from two Black Death strains from London and Barcelona15,18. This post-Black Death subset includes strains from Germany (Starnberg 1,433–1,523, Ellwangen 1,486–1,627), England (Cambridge 1,433–1,523, London 1,560–1,635), Sweden (Stans 1,485–1,635) and France (Marseille 1,720–1,722)19. Since no modern descendants have yet been identified, this lineage probably went extinct10.
There are ongoing debates about the reasons as to why the disease disappeared from Europe in the eighteenth century. Improvements in living conditions, hygiene, and quarantine facilities as well as a replacement of the rodent host from black to brown rat have been suggested20. Other explanations revolved around the idea that evolutionary processes may have favored less virulent pathogen strains21 or may have resulted in increased immunity of the human population20.
Most of the second-pandemic strains analyzed to date have been isolated from human remains excavated in western Europe, with exception of two strains found in Russia (Bolgar 1,362–1,40018, Laishevo 1,300–1,40019). Therefore, not much is known about the diversity and microevolution of the strains responsible for disease outbreaks in eastern Europe, which could have served as a gateway for the spread of the pathogen towards the west.
At least three plague outbreaks in Riga (Latvia) dating to the seventeenth century have been previously reported12,13. To confirm the presence of Y. pestis in Riga, we analysed samples from 16 human skeletons excavated from two cemeteries (fifteenth–seventeenth century13) (Fig. 1 and Table 1). The individuals had been interred in multiple or mass burials and single graves (Table 1). Four of the samples yielded positive screening results for the presence of Y. pestis, leading to the successful reconstruction of two complete bacterial genomes. This new data allowed us to expand the geographical range of known Y. pestis strains to north-eastern Europe.

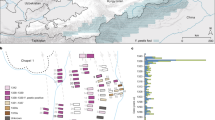
Cemeteries in Riga from which the samples in this study were obtained (left picture). Map of Latvia and Europe (right picture, upper right corner) with the location of Riga. (Figure was created using CorelDRAW Home & Student X8, URL: https://www.coreldraw.com/en/).
Results
Sixteen individuals excavated from two cemeteries located in Riga, Latvia were submitted for aDNA analysis. For 14 samples, authentic ancient shotgun sequences were generated. Of these, we identified four Y. pestis-positive individuals using MALT in BlastN mode (Table 1 and S1). Three of them (G103, G645, G488) were buried in two mass graves and one (G701) in the burial pit of the St. Gertrude’s Church cemetery. The presence of Y. pestis-specific reads was confirmed by mapping against a multi fasta reference containing several representatives of the genus Yersinia (Table S2). Furthermore, we calculated an endogenous based score (i.e. the number of Y. pestis-specific reads with respect to the number of total reads in the sample)2 (Table S3). Two of the samples (G488, G701) were identified as strong positives (endogenous based score > 0.005, Table S3). For these candidates, we subsequently generated 330 and 383 million reads, respectively, to reconstruct the complete bacterial genomes. The obtained reads were mapped against the Y. pestis reference (CO92, NC_003143.1) containing the chromosome and all three plasmids (pCD1, NC_003131.1; pMT1, NC_003134.1; pPCP1, NC_003132.1). Both samples exhibited a genome coverage above 92% for the chromosome as well as for the three plasmids (Table 2 and Fig. S1). Terminal deamination patterns displayed damage profiles typical of authentic aDNA (Fig. S2).
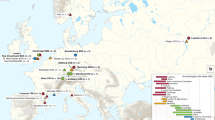
We computed maximum likelihood and Bayesian trees (Figs. 2, S3 and S4) with the two genomes from Riga (G488, G701) and previously published Y. pestis strains (228 modern (Table S4), 36 ancient (Table S5)) and a Y. pseudotuberculosis genome (NZ_CP008943.1). The strains from Riga clustered with other second-plague pandemic strains from Starnberg (STA001) and Cambridge (NMS002)19 with a strong bootstrap support of > 97% and a posterior probability of 1 (Figs. 2, S3 and S4).

Maximum-likelihood tree. The tree is based on the SNP alignment (15,976 positions) of 228 modern Y. pestis genomes, 36 published ancient Y. pestis strains, one Y. pseudotuberculosis genome, and the two genomes G488 and G701 from St. Gertrude’s Church cemetery (left panel, blue). Country abbreviation is given in brackets (DE = Germany, ES = Spain, FR = France, GB = Great Britain, US = United States, RU = Russia, LV = Latvia, CN = China, CG = Congo, FSU = Former Soviet Union, IN = India, IR = Iran, MG = Madagascar, MM = Myanmar, MN = Mongolia, NP = Nepal, UG = Uganda, KG = Kyrgyzstan, CH = Switzerland). Bootstrap values are shown on the nodes for 500 replicates. An asterisk (*) indicates a bootstrap support above 94. Strains in red exhibit no signs of the pla-negative plasmid. Strains in blue exhibit clear signs of the pla-negative plasmid. For the strains in black (within the Back Death and post-Black Death subclades), the pla status was inconclusive or could not be determined due to a general lack of coverage.
Both Riga isolates had SNP alleles (p1 and p214) diagnostic for branch 1 (Table S6). Furthermore, they showed differences in the SNPs p3 and p4 relative to the CO92 reference sequence. These substitutions also characterize other previously reported second-plague pandemic strains from London (England), Barcelona (Spain), Ellwangen (Germany), and l’Observance (France)10,15,18.
The two reconstructed genomes from Riga exhibited a drastic drop in coverage of sequences mapping to the pla gene region (NC_003132.1:6,428–8,530), which consists of the pla gene, a putative transcriptional regulator and a hypothetical protein on the pPCP1 plasmid (Fig. 3). We confirmed the presence of two variants of the plasmid, i.e. one with (pla+) and one without (pla−) the pla region, by identifying gap-spanning reads in each of the strains (Figs. 3C,D, S5). In contrast, we noted only one bacterial chromosome per strain, indicating that the two distinct pPCP1 plasmids existed in the same bacterium. To rule out that the pla gene originated from contamination, a competitive mapping against the pla gene from environmental bacteria (e.g. E. coli and Citrobacter koseri) and Y. pestis showed that the reads in this region were indeed specific for Y. pestis and exhibited the expected damage patterns (Table S7). This observation was verified by a competitive mapping against a pla+ and a pla− reference (Fig. 3 and Table S8). The observed ratio between the coverage of the region in pla+ and pla− plasmids was about 1:10 (Table S9). To further explore this finding, we re-analyzed 28 previously published Y. pestis genomes from the second pandemic10,15,18,19. Interestingly, in 14 strains (marked in blue, Fig. 2) we detected gap-spanning reads and a depletion of the pla region, also indicating the presence of both pla+ and pla− plasmids (Fig. S6 and Table S10). Four more strains showed no signs of pla− plasmids (marked in red, Figs. 2 and S6), whereas ten others, for which the overall pPCP1 plasmid coverage was poor, were excluded as their pla region could not be examined reliably (marked in black, Fig. 2). The possibility that the different plasmids could be the result of a co-infection with two different strains is highly unlikely as this pattern was found in several Y. pestis genomes from various sites and times, and there was no evidence for such an infection on the chromosomal level.

Coverage plots of pPCP1. (A, B) Coverage plot of the pla region in G701 (A) and G488 (B) depicting the pla gene in red, the putative transcriptional regulator in yellow and the hypothetical protein in white. G488 average coverage (ac) before the pla region 107x, ac of the pla region 7x; G701 ac before the pla region 47x, ac of the pla region 3x. Each grey line represents an obtained aDNA sequence. (C) Coverage plot showing reads of G701 (sequences in dark grey) specifically mapping to the gap-spanning region. A white color indicates sequences that could be mapped to both the pla+ and pla− references as they are not specific to the newly created region. (D) Coverage plot showing reads of G488 (sequences in dark grey) specifically mapping to the gap-spanning region.
With regard to (potentially) functional SNPs, we identified two variants of interest. First, both Riga genomes carried the T259 allele in the pla gene that is important for massive bacterial dissemination in the bubonic form of plague22. Second, in sample G488, SnpEff23 detected a variant in the ssrA-smpB region.
For five of the samples (G92, G143b, G161c, G488 and G701), the human endogenous DNA content in the metagenomic data was sufficient to investigate the population genetic ancestry using PCA, f3 and admixture analyses. The results indicate a northern European origin of the individuals who were genetically fairly homogeneous (Figs. S7, S8, S9 and S10).
Discussion
Here we analyzed the remains of 16 individuals who had been interred in two cemeteries (Dome Church cemetery, St. Gertrude’s Church cemetery) in Riga, Latvia (fifteenth–seventeenth century) (Fig. 1 and Table 1). Four of the individuals buried in two mass graves and a burial pit of St. Gertrude’s Church cemetery were infected with the Y. pestis bacterium. Previous analyses led to the hypothesis that people inhumed in the mass graves were rural immigrants. Our human population genetic data confirmed their northern European descent. Most likely they came to Riga during the famine of 1601–2 and stayed in shelters in the vicinity of St. Gertrude’s Church13. Inadequate nutrition and crowded, unhygienic conditions may have increased their susceptibility to infectious diseases, including plague24,25. Our finding showed that the individuals buried in the mass graves as well as the small burial pit were indeed victims of plague26,27. Thus, we corroborate historical reports of local plague outbreaks in Riga at the beginning of the seventeenth century (1,601–3, 1,621–23 and 1,657)12.
From two (G488 and G701) of the four infected individuals, high-quality whole genomes of Y. pestis were generated. They formed a subclade in branch 1 with other strains dating to the post-Black Death pandemic (Fig. 2). They all originated from Black Death genomes from London15 and Barcelona18. Although the two Riga strains were found in the same cemetery (in one mass grave and the burial pit), they showed a surprisingly large genetic distance. Based on this observation and the close proximity of G488 to NMS002 (Cambridge19) (Fig. 2), we assume that G488 had not evolved from G701 during a single outbreak. This may indicate that the two different strains were responsible for two subsequent plague events12,13.
As no modern strains fall into the post-Black Death subclade, which includes the two genomes from Riga, it could represent a Y. pestis lineage that went extinct10. Although the strains differ widely in terms of time and geography, their positions within the subclade—like pearls on a string—suggest continuous in situ evolution. This observation agrees with the hypothesis that the second plague pandemic reached Europe from Asia rather in one than in multiple waves. If the pandemic had multiple sources, one would expect a higher diversity. Thus, the post-Black Death outbreaks were likely due to established plague reservoirs within Europe. Little is known about natural plague foci and hosts during the second pandemic. Several rodent species, e.g. marmots and squirrels, from the Alps and the steppe have been mentioned as possible reservoirs20.
The majority (ratio 10:1) of the pPCP1 plasmid copies carried by the two Riga strains G488 and G701 did not possess the pla region. Based on the chromosome data we did not see any signs of coinfection with different strains that could explain the presence of two different pPCP1 plasmids. In a re-evaluation of already published second-plague pandemic genomes, we identified 14 strains carrying both plasmid types (pla− and pla+) (Table S10, Fig. S6). All 14 strains were dated to the post-Black Death period whereas four strains which only had copies of the pla+ plasmid were present during the Black Death (Fig. 2). The pla region consists of the pla gene, a putative transcriptional regulator and a hypothetical protein. Up to now, the role of the putative transcriptional regulator and the hypothetical protein with respect to pla expression and function has remained unclear. Several studies have shown that pla is an important factor for the infection of and transmission in humans. The pla protease activates host fibrinolysis and allows the spread of the bacteria to the lymphatic nodes28 as well as their extensive growth in the lower airways29,30. Pla− mutants of Y. pestis have been associated with reduced dissemination and virulence22,30,31. In mice, pla− strains transmitted by fleas were reported to cause primary septicemic plague while no bubonic forms were detected31. Therefore, pla seems to influence the infection routes leading to bubonic and pneumonic plague, while the septicemic route remains unaffected32. Therefore, the extremely high mortality of the Black Death could possibly be explained by the functional features of pla. The pandemic was mainly associated with a rapid spread of the severe bubonic form via the flea vector. In contrast, subsequent outbreaks could have been influenced by a slower transmission rate and a different disease course (primarily septicemic rather than bubonic) due to the pla-depleted strains (Fig. 2).
Horizontal gene transfer, selective gene loss, and changes in regulatory pathways have been described in the evolution and adaptation of Y. pestis to new host environments33,34. These mechanisms together with the genomic structure (e.g. IS-like elements or transposases) of the plasmids could have led to a loss of pla or a pla-depletion. Pla− Y. pestis strains have been isolated worldwide35,36 and pla− plasmids are known from Pestoides strains37—enzoonotic pathogens that infect rodents38, but rarely humans39. The pla-depleted post-Black Death strains could therefore be an adaptation to a specific host environment, perhaps in rodents.
The strain from individual G488 possessed a mutation in the ssrA gene, which has not yet been described in other strains. Together with the smpB protein, ssrA creates a complex responsible for maintaining cellular homeostasis40,41,42. The important role of this system in bacterial pathogenicity was shown in previous studies on several bacteria such as Salmonella, Y. pseudotuberculosis and Y. pestis43,44,45. Interestingly, in a mouse model, Y. pestis strains with a deletion of ssrA and smpB had limited ability to disseminate and colonize target organs. Moreover, mice which were intranasally immunized with this mutated strain were protected against pulmonary infection with a fully virulent strain46. In view of these findings, a SNP in the ssrA gene is intriguing. However, the functional implications of this single point mutation remain unclear.
Y. pestis strains responsible for the Post-Black Death plague showed a pla-depletion in the pPCP1 plasmid. This change in the pathogen genome can probably be attributed to an adaptation of the bacterium to a new host environment (e.g. rodents). In humans, the depletion may have favorably influenced the disease course, the probability of disease manifestation after a flea bite and/or the transmission rate. These positive effects fit with the evolutionary hypothesis that less virulent strains may, at least in part, explain the easing and ultimate disappearance of plague from Europe21. Nevertheless, it is imperative to obtain more Y. pestis genomes and conduct additional functional studies to create a more comprehensive picture of the bacteria responsible for the second plague pandemic.
Material and methods
Historical background and sample selection
Individuals excavated from the cemeteries Riga Dome Church and St. Gertrude’s Church cemetery, Riga, were selected for a bacterial pathogen screening (Fig. 1). At Riga Dome Church cemetery, three mass burial pits were found at the northern wall of the church. A total of 87 individuals were identified and archaeological finds suggest that they date to the seventeenth century47. Unlike people buried elsewhere in the cemetery, the individuals in the mass burial pits had been buried in coffins, and none of them showed evidence of peri-mortem trauma48. Therefore, it is likely that they had succumbed to a mass mortality event that was not related to violence. Indeed, they could be victims of one of the seventeenth century plague epidemics. Historical sources suggest that during the plague epidemic of 1,657, the churches of Riga ran out of intramural burial space, which was normally reserved for people of a higher social status48. It is possible that during the epidemic, the dead were buried in larger pits outside the church, albeit in a dedicated area and in coffins to signify their higher social status. In addition, smaller collective burials close to the aforementioned mass burial pits were found. In these, four to nine people were buried in several layers, without coffins. For DNA analysis, three samples were selected from the mass burial pits with coffins, and one from the smaller collective burial pits (Table 1).
The other site selected for aDNA analysis was St. Gertrude’s Church cemetery. The graveyard was in use between the fifteenth and seventeenth century, and back then located outside the city wall (Fig. 1). The cemetery was divided into four distinct burial areas: a general cemetery, two mass graves, and a small mass burial pit. In total, 709 individuals were excavated from this cemetery, and 283 of these were buried in the two mass graves (south-eastern mass grave, or mass grave 1, and north-western mass grave, or mass grave 2)13. The bodies in the mass graves were placed on top of each other, reaching seven to eight layers in total. Both archaeological (based on artefacts) and radiocarbon analysis dated the two mass graves to the end of the sixteenth or the beginning of the seventeenth century, although they are not necessarily contemporaneous49. The chronology of the small mass burial pit with 15 buried individuals is not entirely clear. The demographic profile of the mass graves with comparatively few children < 7 years of age and, again, the lack of evidence of peri-mortem trauma, suggested that these people had died in a non-violent mass mortality event13,48. Moreover, the date of both mass graves coincides with two historically documented plague epidemics (1,601–3 and 1,621–23) and a famine (1,601–2)12. For DNA analysis, four samples were selected from mass grave 1, three from mass grave 2, two from the grave pit and three from the general cemetery (Table 1).
DNA isolation, sequencing and postprocessing
All lab work was carried out in a dedicated ancient DNA facility. DNA extraction of 16 tooth samples from Riga, sequencing (Illumina HiSeq4000 (2 × 75 bp)) and subsequent clip and merge was performed as described in Krause-Kyora et al.50. Reads that were shorter than 25 bp were discarded after merging. Two samples (G83 and G157) did not yield sufficient DNA and were not processed further (Table 1).
Pathogen screening
Screening of the 14 samples was carried out with the software MALT using a custom database consisting of complete bacterial genomes downloaded from NCBI (24.01.2019)51. A sequence identity threshold of 90% was set and the alignment mode was changed to SemiGlobal. The analysis was done using the following command:
malt-run -mode BlastN -e 0.001 -id 85 -alignmentType SemiGlobal -index $index -inFile $FASTQCM -output $OUT
where $index is the index file, $FASTQCM is the clipped and merged file and $OUT is the output file.
The resulting alignments were visually inspected using MEGAN 652.
Y. pestis alignment
All four positive samples from the screening (G103, G488, G645, G701) were mapped against the Y. pestis reference genome including the three plasmids pCD1, pMT1 and pPCP1 (NC_0031431.1; NC_003131.1; NC_003134.1; NC_003132.1). The mapping was conducted using BWA with the following command line53:
bwa aln -n 0.01 -l 300 $INDEX $FASTQCM $OUT
where $INDEX is the reference, $FASTQCM is the input file and $OUT is the output file. Minimum mapping quality was set to 0. Subsequent duplicate removal was performed using DeDup version 0.11.3, part of the EAGER pipeline54, with default parameters. Subsequently, the Binary Alignment Map (BAM) files were filtered for a mapping quality of 30 using the following command in samtools55:
samtools view -bq $INT $IN > $OUT
where $INT is the mapping quality, $IN is the input BAM file and $OUT is the output BAM file.
Sample authentication
To further authenticate the positive samples, an endogenous score was used to calculate the number of Y. pestis-specific reads with respect to the number of total reads in the sample2. Positive samples were mapped against a multi fasta reference containing several representatives of the genus Yersinia (Table S2) and the three Y. pestis-specific plasmids (pCD1, pMT1, pPCP1). The mapping was done as described above followed by a duplicate removal step. Mapped reads were filtered for a mapping quality greater than 30 using samtools as described above.
Applying samtools idxstats, an endogenous based score was used to evaluate the potential of the sample being positive for Y. pestis. The score was calculated with the following formula:
where YPS is the number of reads specifically mapping to Y. pestis; YS is the maximum number of reads mapping specifically to any Yersinia species with the exception of Y. pestis and M is the total number of merged reads in the sample. By using the maximum number of reads mapping to another species of the genus Yersinia, the score takes into account different sources of contamination other than Y. pseudotuberculosis. Two of the four positive samples exhibited a score higher than 0.005, a value which indicates a strong positive candidate (Table S3).
VCF-generation and phylogenetic analysis
VFC files were generated for the two positive samples using GATK and the Y. pestis reference with the following command line56:
GenomeAnalysisTK.jar-T UnifiedGenotyper -R $REF -I $IN -out_mode EMIT_ALL_SITES -o $OUT
where $REF is the reference, $IN is the input BAM file and $OUT is the VCF output. These two VCFs together with a VCF dataset of 264 strains (Table S4 and S5, mapped (see Y. pestis alignment) and generated as described above) and one Y. pseudotuberculosis (NZ_CP008943.1) genome were used as an input for the MultiVCFAnalyzer to generate a SNP-based multiple alignment57. The analysis was carried out with the following command:
Java -jar MultiVCFAnalyzer_0-85-1.jar NA $REF NA $OUT F 30 3 0.9 0.9 NA
where $REF is the Y. pestis reference and $OUT is the output folder. Y. pseudotuberculosis-specific variants were removed from the SNP table to improve visual resolution of the tree. The resulting SNP table was also filtered for SNPs in repetitive regions and homoplasies16. The SNP table was reformatted to multi fasta and nexus format that were used as input for RAxML and MrBayes58,59. RAxML was executed with the following command line:
RaxmlHPC -f a -x 12345 -p 12345 -#500 -m GTRGAMMA -s $IN -o $OG -n $NAME
where $IN is the input file, $OG is the name of the outgroup and $NAME is the outfile name.
MrBayes was executed applying the GTR model with the default number of generations.
Resulting trees were visualized using FigTree60.
SNP effect analysis
VCFs from the Y. pestis-positive samples were filtered for SNPs with a coverage of at least 3x, a calling quality of 30 and a 90% majority call. This set of filtered SNPS was provided to SNPEff to analyze the effects of SNPs in the ancient genomes23. SNPEff was executed with the following command line:
java -jar snpEff.jar $REF $IN > $OUT
where $REF is the SNPEff reference, $IN is the input VCF file and $OUT is the annotated SNPEff output file. The annotated VCF file was filtered for high-impact SNPs and evaluated.
pPCP1 and pla analysis
To ascertain that the coverage in the low-coverage region was not introduced artificially by environmental contamination, we carried out a competitive mapping as described above. The fasta reference that was used contained the Y. pestis reference sequence of the pPCP1 plasmid and also the pla genes of Citrobacter and E. coli (Table S7). Gap-bridging reads were analyzed by removing the low-coverage region from the pPCP1 plasmid. Seqkit was used with the following command lines61:
seqkit subseq -r 1:6428 $IN > $OUT1
seqkit subseq -r 8530:9612 $IN > $OUT2
where $IN is the pPCP1 reference (NC_003132.1) and $OUT1/$OUT2 are parts of the new plasmid without the pla region. Both output files were concatenated to form a complete plasmid. An additional mapping with the pPCP1 plasmid from which the pla region was removed (pla-) and the regular pPCP1 plasmid (pla+) as references was performed (Table S8). These references were used to identify reads which were truly unique for the new region in the pla− plasmid. All the non-specific reads received a mapping quality of zero as the exact same region was present in both references. The reads mapping exclusively to the new region were extracted and compared with the reads from the initial mapping against Y. pestis and gap-bridging reads were identified. Based on these exclusive mapping reads, the coverage for the pla− plasmid was calculated. The coverage of the pla region (NC_003132.1, 6,428–8,529) in the regular mapping was used to estimate the number of pla+ plasmids. Based on these two values a ratio was calculated to estimate the proportions.
Human genetic analyses
Mapping and aDNA damage patterns
Shotgun sequence data was mapped to the human genome build hg19 using BWA with a reduced mapping stringency parameter “-n 0.01” to account for mismatches in aDNA54,62. Duplicates were removed. Authenticity of the aDNA fragments was assessed using mapDamage 2.063,64. After validation of the terminal damage patterns, the first two positions from the 5′ end of the fastq-reads were trimmed.
Genotyping
Alleles were drawn at random from a pool of 1,233,013 SNPs (commonly used for population genetic analyses) in a pseudo-haploid manner using a custom script as described previously65,66,67,68. Datasets had to have at least 10,000 SNPs to be considered for further analysis68. Five individuals from Riga (G92, G143b, G161c, G488 and G701) passed this filtering step.
Population genetic analyses
The five genotype datasets from Riga were merged with a dataset of previously published 59 West Eurasian populations genotyped on the aforementioned 1,233,013 SNPs using the program mergeit from the EIGENSOFT package69. Principal component analysis (PCA) was performed using the software smartpca and the ‘lsqproject’ option69. Admixture modelling was done with the software ADMIXTURE on the same populations as used in the PCA. The number of ancestral components k ranged from 4 to 1270. Cross-validation was performed for every admixture model and the model with the highest accuracy was determined by the lowest cross-validation error.
F3 outgroup statistics
f3 outgroup statistics were run as a part of the Admixtools package in the form of f3 (Riga; test; Mbuti) using for test the same populations as used in the PCA and ADMIXTURE analysis70.
Material transfer agreement
The authors confirm that the sampling and the ancient DNA analysis of human bones from the fifteenth–seventeenth century archaeological features unearthed in Riga, Latvia were performed with permission from the institution which headed the excavations and the anthropological analysis (Institute of Latvian History, University of Latvia).
Data availability
The aligned sequences are available through the European Nucleotide Archive under Accession Number PRJEB36413.
References
Rasmussen, S. et al. Early divergent strains of Yersinia pestis in Eurasia 5,000 years ago. Cell 163, 571–582 (2015).
Valtuena, A. A. et al. The Stone Age plague and its persistence in Europe. Curr. Biol. 27, 3683–3691 (2017).
Spyrou, M. A. et al. Analysis of 3800-year old Yersinia pestis genomes suggests Bronze Age origin for bubonic plague. Nat. Commun. 9, 2234. https://doi.org/10.1038/s41467-018-04550-9 (2018).
Rascovan, N. et al. Emergence and spread of basal lineages of Yersinia pestis during the Neolithic Decline. Cell 176, 295–305 (2019).
Anisimov, A. P., Lindler, L. E. & Pier, G. B. Intraspecific diversity of Yersinia pestis. Clin. Microbiol. Rev. 17, 434–464 (2004).
Gage, K. L. & Kosoy, M. Y. Natural history of plague: perspectives from more than a century of research. Annu. Rev. Entomol. 50, 505–528 (2005).
Roelle, J. E., Miller, B. J., Godbey, J. L. & Biggins, D. E. Recovery of the black-footed ferret progress and continuing challenges. US Geol. Surv. Sci. Invest. Rep. 5293, 213–231 (2006).
Morelli, G. et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Genet. 42, 1140–1143 (2010).
Ziegler, P. The Black Death (Harper Collins, New York, 1969).
Bos, K. I. et al. Eighteenth century Yersinia pestis genomes reveal the long-term persistence of an historical plague focus. Elife 5, e12994. https://doi.org/10.7554/eLife.12994 (2016).
Byrne, J. P. Encyclopedia of the Black Death (ABC-CLIO, Santa Barbara, 2012).
Napiersky, J. G. L. Bodeckers Chronik livländischer und rigascher Ereignisse [Bodecker’s chronicle of Livonian and Riga events] (in German) (Kymmel’s Buchhandlung, Riga, 1890).
Petersone-Gordina, E., Roberts, C., Millard, A. R., Montgomery, J. & Gerhards, G. Dental disease and dietary isotopes of individuals from St Gertrude Church cemetery, Riga, Lativia. PLoS ONE 13, e0191757. https://doi.org/10.1371/journal.pone.0191757 (2018).
John, T. A. Bubonic Plague in Early Modern Russia: Public Health and Urban Disaster (Oxford University Press, Oxford, 2003).
Bos, K. I. et al. A draft genome of Yersinia pestis from victims of the Black Death. Nature 478, 506–510 (2011).
Cui, Y. et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc. Natl. Acad. Sci. USA 110, 577–582 (2013).
Namouchi, A. et al. Integrative approach using Yersinia pestis genomes to revisit the historical landscape of plague during the Medieval Period. Proc. Natl. Acad. Sci. USA 115, E11790–E11797. https://doi.org/10.1073/pnas.1812865115 (2018).
Spyrou, M. A. et al. Historical Y pestis genomes reveal the European Black Death as the source of ancient and modern plague pandemics. Cell Host Microbe 19, 874–881 (2016).
Spyrou, M. A. et al. Phylogeography of the second plague pandemic revealed through analysis of historical Yersinia pestis genomes. Nat. Commun. 10, 4470. https://doi.org/10.1038/s41467-019-12154-0 (2019).
Appleby, A. B. The disappearance of plague: a continuing puzzle. Econ. Hist. Rev. 33, 161–173 (1980).
Lenski, R. E. Evolution of plague virulence. Nature 334, 473–474 (1988).
Zimbler, D. L., Schroeder, J. A., Eddy, J. L. & Lathem, W. W. Early emergence of Yersinia pestis as a severe respiratory pathogen. Nat. Commun. 6, 7487. https://doi.org/10.1038/ncomms8487 (2015).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92 (2012).
Dirks, R. et al. Social responses during severe food shortages and famine. Cur. Anthropol. 21, 21–44 (1980).
Wisner, B. Adams, J. Control of communicable diseases and prevention of epidemics. Rīgas pilsētas valde WHO. www.who.int/entity/water_sanitation_health/hygiene/emergencies/em2002chap11.pdf (2019)
Pīrangs, H. Rīgas kapsētas [Graveyards of Riga] (in Latvian) (Rīgas pilsētas valde, Riga, 1932).
Rusovs, B. Livonijas kronika [Chronicle of Livonia] (in Latvian) (Valters un Rapa, Riga, 1926).
Degen, J. L., Bugge, T. H. & Goguen, J. D. Fibrin and fibrinolysis in infection and host defense. J. Thromb. Haemost. 5, 24–31 (2007).
Lathem, W. W. et al. Post-transcriptional regulation of the Yersinia pestis cAMP receptor protein Crp and impact on virulence. MBio 5, e01038-13. https://doi.org/10.1128/mBio.01038-13 (2014).
Perry, R. D. & Fetherston, J. D. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 10, 35–66 (1997).
Sebbane, F., Jarrett, C. O., Gardner, D., Long, D. & Hinnebusch, B. J. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc. Natl. Acad. Sci. USA 103, 5526–5530 (2006).
Sebbane, F. et al. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc. Natl. Acad. Sci. USA 103, 11766–11771 (2006).
Zhou, D. & Yang, R. Molecular Darwinian evolution of virulence in Yersinia pestis. Infect. Immun. 77, 2242–2250 (2009).
Sun, Y. C., Jarret, C. O., Bosio, C. F. & Hinnebusch, B. J. Retracing the evolutionary path that led to flea-borne transmission of Yersinia pestis. Cell Host Microbe 15, 578–584 (2014).
Cavalcanti, Y. V. N., Leal, N. C. & De Almeida, A. M. P. Typing of Yersinia pestis isolates from the state of Ceará, Brazil. Lett. Appl. Microbiol. 35, 543–547 (2002).
Filippov, A. A., Solodovnikov, N. S., Kookleva, L. M. & Protsenko, O. A. Plasmid content in Yersinia pestis strains of different origin. FEMS. Microbiol. Lett. 67, 45–48 (1990).
Garcia, E. et al. Pestoides F, an atypical Yersinia pestis strain from the former Soviet Union. Adv. Exp. Med. Biol. 603, 17–22 (2007).
Achtman, M. et al. Microevolution and history of the plague bacillus, Yersinia pestis. Proc. Natl. Acad. Sci. USA 101, 17837–17842 (2004).
Rajanna, C. et al. Characterization of pPCP1 plasmids in Yersinia pestis strains isolated from the former Soviet Union. Int. J. Microbiol. https://doi.org/10.1155/2010/760819 (2010).
Gueneau de Novoa, P. & Williams, K. P. The tmRNA website: reductive evolution of tmRNA in plastids and other endosymbionts. Nucl. Acids. Res. 32, D104–D108. https://doi.org/10.1093/nar/gkh102 (2004).
Keiler, K. C. Biology of trans-translation. Annu. Rev. Microbiol. 62, 133–151 (2008).
Dulebohn, D., Choy, J., Sundermeier, T., Okan, N. & Karzai, A. W. Trans-translation: the tmRNA-mediated surveillance mechanism for ribosome rescue, directed protein degradation, and nonstop mRNA decay. Biochemistry 46, 4681–4693 (2007).
Okan, N. A., Bliska, J. B. & Karzai, A. W. A role for the SmpB-SsrA system in Yersinia pseudotuberculosis pathogenesis. PLoS Pathog. 2, e6. https://doi.org/10.1371/journal.ppat.0020006 (2006).
Baumler, A. J., Kusters, J. G., Stojiljkovic, I. & Heffron, F. Salmonella typhimurium loci involved in survival within macrophages. Infect. Immun. 62, 1623–1630 (1994).
Julio, S. M., Heithoff, D. M. & Mahan, M. J. ssrA (tmRNA) plays a role in Salmonella enterica serovar Typhimurium pathogenesis. J. Bacteriol. 182, 1558–1563 (2000).
Okan, N. A., Mena, P., Benach, J. L., Bliska, J. B. & Karzai, A. W. The smpB-ssrA mutant of Yersinia pestis functions as a live attenuated vaccine to protect mice against pulmonary plague infection. Infect. Immun. 78, 1284–1293 (2010).
Tilko, S. Apbedīšanas tradīcijas Doma viduslaiku kapsētā [Mortuary Practices in the Dom Medieval Cemetery] (in Latvian). Senā Rīga: Pētījumi pilsētas arheoloģijā un vēsturē [Ancient Riga: Studies in the archaeology and history of the city]. Rīga: Latvijas vēstures institūta apgāds. 273- 289 (1998).
Gerhards, G. Traumas un ievainojumi Rīgas 13.-18. gadsimta iedzīvotājie [Trauma and Injuries of Riga 13th–18th Century Population] (in Latvian). Senā Rīga: pētījumi pilsētas arheoloģijā un vēsturē [Ancient Riga: Studies in the archaeology and history of the city] (in Latvian). Rīga: Latvijas vēstures institūta apgāds. 128–148 (2012).
Petersone-Gordina, E., et al. Investigating dietary life histories and mobility in children buried in St Gertrude Church cemetery, Riga, Latvia (15th–17th centuries AD). Archaeometry 62, 3–18 (2020).
Krause-Kyora, B. et al. Ancient DNA study reveals HLA susceptibility locus for leprosy in medieval Europeans. Nat. Commun. 9, 1569. https://doi.org/10.1038/s41467-018-03857-x (2018).
Vågene, Å. J. et al. Salmonella enterica genomes from victims of a major sixteenth-century epidemic in Mexico. Nat. Ecol. Evol. 2, 520–528 (2018).
Huson, D. H., Auch, A. F., Qi, J. & Schuster, S. C. MEGAN analysis of metagenomic data. Genome Res. 17, 377–386 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Peltzer, A. et al. EAGER: efficient ancient genome reconstruction. Genome Biol. 17, 60. https://doi.org/10.1186/s13059-016-0918-z (2016).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Bos, K. I. et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 514, 494–497 (2014).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Ronquist, F. et al. Mrbayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Rambaut, A. FigTree version 1.4.3. https://tree.bio.ed.ac.uk/software/figtree/.
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 11, e0163962. https://doi.org/10.1371/journal.pone.0163962 (2016).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F. & Orlando, L. MapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics. 29, 1682–1684 (2013).
Briggs, A. W. et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 104, 14616–14621 (2007).
Lazaridis, I. et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–413 (2014).
Haak, W. et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211 (2015).
Mathieson, I. et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528, 499–503 (2015).
Lamnidis, T. C. et al. Ancient Fennoscandian genomes reveal origin and spread of Siberian ancestry in Europe. Nat. Commun. 9, 5018. https://doi.org/10.1038/s41467-018-07483-5 (2018).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190. https://doi.org/10.1371/journal.pgen.0020190 (2006).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Acknowledgements
J.B. was funded by the International Max Planck Research School for Evolutionary Biology. This study was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the following projects: numbers 2901391021 (CRC 1266) and 390870439 (EXC 2150 - ROOTS) to A.N. and B.K.-K.
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
B.K.-K., G.G. and E.P. conceived and designed the research. J.S., A.I. and B.K.-K. generated and analyzed the ancient DNA data. J.S., J.B., E.P., G.G., A.N. and B.K.-K. interpreted the findings. J.S., J.B., E.P., G.G., A.N. and B.K.-K. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Susat, J., Bonczarowska, J.H., Pētersone-Gordina, E. et al. Yersinia pestis strains from Latvia show depletion of the pla virulence gene at the end of the second plague pandemic. Sci Rep 10, 14628 (2020). https://doi.org/10.1038/s41598-020-71530-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-71530-9
This article is cited by
-
A melting pot of Roman dogs north of the Alps with high phenotypic and genetic diversity and similar diets
Scientific Reports (2023)
-
Ancient Clostridium DNA and variants of tetanus neurotoxins associated with human archaeological remains
Nature Communications (2023)
-
Overall frailty gauged in victims of the Italian plague (Imola, 1630–1632): was plague an indiscriminate killer?
Archaeological and Anthropological Sciences (2022)
-
Ancient DNA analysis
Nature Reviews Methods Primers (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
