
Abstract
Pathogenic variants (PVs) in multiple genes are known to increase the risk of early-onset renal cancer (eoRC). However, many eoRC patients lack PVs in RC-specific genes; thus, their genetic risk remains undefined. Here, we determine if PVs in DNA damage response and repair (DDRR) genes are enriched in eoRC patients undergoing cancer risk assessment. Retrospective review of de-identified results from 844 eoRC patients, undergoing testing with a multi-gene panel, for a variety of indications, by Ambry Genetics. PVs in cancer-risk genes were identified in 12.8% of patients—with 3.7% in RC-specific, and 8.55% in DDRR genes. DDRR gene PVs were most commonly identified in CHEK2, BRCA1, BRCA2, and ATM. Among the 2.1% of patients with a BRCA1 or BRCA2 PV, < 50% reported a personal history of hereditary breast or ovarian-associated cancer. No association between age of RC diagnosis and prevalence of PVs in RC-specific or DDRR genes was observed. Additionally, 57.9% patients reported at least one additional cancer; breast cancer being the most common (40.1% of females, 2.5% of males). Multi-gene testing including DDRR genes may provide a more comprehensive risk assessment in eoRC patients. Further validation is needed to characterize the association with eoRC.
Similar content being viewed by others

Introduction
Renal cancer (RC) often develops with no signs or symptoms and is referred to as the “silent disease”1. While factors including smoking, environment, obesity, and race have been linked to increased risk of RC, inherited factors are the most well-validated source of increased risk2,3,4. Hereditary RC syndromes, typically associated with early-onset disease and a clinically significant family history of cancer, result from germline pathogenic variants (PV) in high-penetrance ‘RC-specific’ genes including VHL, MET, FLCN, TSC1, TSC2, FH, SDH, PTEN and BAP15,6,7. A previous report of an early-onset RC (eoRC) cohort screened with an RC-specific panel found 6.1% of individuals had a PV in an RC-specific gene7. However, for most eoRC patients a PV in an RC-specific gene is not identified, leaving many eoRC genetically undefined. Thus, there is a need to identify additional genes related to eoRC risk. Currently, there are no National Comprehensive Cancer Network (NCCN) guidelines for detection, prevention, or risk reduction in individuals who present with an eoRC but lack a PV in a defined RC-specific gene8.
DNA damage response and repair genes (DDRR) play an important role in maintaining genome integrity, and when mutated in the germline can increase cancer risk for several types of cancers, including breast, colorectal, ovarian, and others9. Although PVs in DDRR genes are associated with increased risk of a variety of cancer types, they are not typically considered risk factors for RC. However, germline PVs in some DDRR genes have been observed in RC, including PVs in the DNA mismatch repair (Lynch syndrome) genes MSH2 and MLH1 in renal urothelial carcinoma, and PVs in CHEK2 in advanced renal cell carcinoma10,11,12,13,14,15,16,17,18,19,20,21. To address the hypothesis that PVs in additional DDRR genes may contribute to the missing heritability of eoRC, we analyzed germline sequencing data from a cohort of 844 individuals with RC.
Materials and methods
Ambry Genetics eoRC study cohort, and variant determination
De-identified data were requested from RC patients that were tested by Ambry Genetics (Konica Minolta, Aliso Viejo, California) using germline cancer testing panels. Ambry samples were selected for patients with RC, and de-identified data was obtained for all RC patients tested with multi-gene cancer panels (n = 844, ≤ 60 years at diagnosis, specimens collected between July 2012–December 2016). All genetic test results from germline testing of individuals diagnosed with RC at ≤ 60 during this time period were used in this study.
There is currently no standard definition specifying the age when RC is considered early-onset. Different models have been used to determine a specific age as a trigger for germline testing in patients with RC who lack family history of RC, including ages < 4622 or < 407 years. For this study, we selected individuals 60 years or younger as the cut-off for our cohort, which is substantially below the median age of RC diagnosis of 64 years in the general population as reported in SEER22,23, but considerably older than other suggested cut-offs. We did so because the main hypothesis of the study was that PVs in DDRR genes might be responsible for increased risk of RC. Variants in multiple DDRR genes are associated with early-onset colorectal cancer24,25, which typically manifests in patients at 50 years or younger. We considered that PVs in DDRR genes were most likely to impact repair of DNA damage induced during cell replication, leading to genetic instability and cancer; given renal cells turn over much less frequently than colon cells, we hypothesized that it may take longer for cancers associated with PVs in DDRR genes to manifest in RC, causing us to select a cut-off of ≤ 60 years old for assessment.
De-identified data included family history of cancer, genetic test results, personal history of cancers (apart from RC), presence of multifocal tumors, and RC-subtype/stage. The RC patients had been tested with CancerNext versions 1–4, and CancerNext-Expanded versions 1 and 2 (Table S1). De-identified patient information was analyzed for genetic test results, and personal and family medical histories. Classification of variants by Ambry Genetics is based on ACMG recommendations for standards for interpretation and reporting of sequence variations. These variants are also regularly deposited in ClinVar by Ambry Genetics. Variant classification was updated through March 31, 2018 for all data. Gene variants were classified as pathogenic variant (PV—see below for criteria), variant of uncertain significance (VUS) or inconclusive, or negative/indeterminate. Ambry Genetics follows strict criteria when classifying variants as PV, Variant Likely Pathogenic (VLP), VUS, Variant Likely Benign (VLB) and Benign (for details see https://www.ambrygen.com/clinician/our-scientific-excellence/variant-classification). Variants reported as PV and VLPs were grouped as PVs. All test results were used for this study. The analysis of VUS, which currently lack clinical significance, was beyond the scope of this study. Given updating of test panels by Ambry Genetics, not all patients were tested for all genes. Individuals were provided different versions of the panel over the course of the study (see below and also see Table S1).
Any de-identified personal or family history information including sex, ethnicity/race, age of cancer diagnosis, tumor histology, history of additional personal cancer, and history of family cancer and types was reported first as summarized data and later as de-identified individual case reports. For analysis comparing outcomes for RC-specific genes versus genes not typically associated with RC, we focused our statistical comparison on only those individuals who had CancerNext Expanded panel version 2 testing which analyzes all 49 genes including the RC-specific genes. 491 individuals who had the CancerNext Expanded version 2 test were used for this statistical comparison. For additional statistical test comparisons that analyzed the correlations between specific genes and categories such as tumor pathology or age, any individual who had been tested for that specific gene was included.
The Western IRB issued a regulatory opinion that the Genomic Data Sharing Policy for Ambry Genetics does not involve human subjects based on 45 CFR46.102(f) and associated guidance, thus the requirement to obtain written patient informed consent was waived. A Data Use Agreement, and Materials Transfer Agreement was established between Ambry Genetics and Fox Chase Cancer Center. The FCCC Institutional Review Board (IRB) provided study oversight and approval (protocol number 14831). Ambry Genetics provided de-identified patient medical and family history (where available), and genetic results for the patients. All methods were performed in accordance with the relevant guidelines and regulation of the approved study.
Genetic analysis with Ambry CancerNext and CancerNext Expanded panels
Individuals were provided different versions of the panel by their healthcare provider (see Table S1). The number of genes in the panels ranged from the smallest CancerNext panel Version 1 which include 22 genes (APC, ATM, BARD1, BRIP1, BMPR1A, CDH1, CHEK2, EPCAM, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD50, RAD51C, SMAD4, STK11, TP53) to the largest CancerNext Expanded Version 2 panel, which contained 49 genes (APC, ATM, BAP1,BARD1, BRCA1, BRCA2, BRIP1, BMPR1A, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, FH, FLCN, GREM1, MAX, MEN1, MET, MITF,MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, NF1,PALB2, PMS2, POLD1, POLE,PTEN,RAD50, RAD51C, RAD51D,RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, SMAD4, SMARCA4, STK11, TMEM127, TP53, TSC1, TSC2, VHL). The DDRRs identified in germline testing of this cohort are bolded26.
Ambry Genetics sequenced genomic DNA that was obtained from patient blood or saliva samples. DNA was evaluated by next generation sequencing (NGS) of all coding sequences, and ± 5 bases into the 5′ and 3′ ends of flanking introns and untranslated regions. In addition, sequencing of the promoter region was performed for the following genes: PTEN (c.− 1,300 to c.− 745), MLH1 (c.− 337 to c.− 194), and MSH2 (c.− 318 to c.− 65). Additional Sanger sequencing was performed for any regions missing or with insufficient depth of coverage for reliable heterozygous variant detection, and on potentially homozygous variants, variants in regions with complicated pseudogene interference, and when variant calls did not meet allele frequency quality thresholds. Additional details on specific testing methods are available at https://www.ambrygen.com/clinician/genetic-testing/28/oncology/cancernext-expanded.
Control population in ExAc and gnomAD
To compare the frequency of DDRR gene PVs found in the study to that in the general population, our results were compared to the Exome Aggregation Consortium (ExAc) dataset of largely unrelated ~ 60,000 whole exome sequencing results, and to the Genome Aggregation database (gnomAD) dataset consisting of ~ 125,000 exomes and ~ 15,000 genomes27,28. These datasets are the most commonly used genomic data at the population-level.
ClinVar analysis
ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), a database of medically relevant gene variants, was used to investigate all PVs in this study (retrieved on February 4, 2020). PVs that were not reported in ClinVar were noted as ‘not reported’. Associated conditions for each PV were categorized into hereditary cancer predisposing syndrome(s), condition(s) related to renal cancer, and any other condition(s). To further elucidate any PVs related to renal cancer, the search term “renal cancer” was queried, and the results were noted as “associated with ClinVar search term ‘Renal Cancer.’”.
Statistical analysis
To identify potential correlations between PVs and characteristics such as tumor pathology, additional primary tumor type, and age of diagnosis, genes were combined into pathways/groups of interest, histology’s were grouped, and cancer types were grouped. Each individual was categorized as having a variant in one of the genes within the group or no variant in the group. Gene categories were used for comparison of RC diagnosis with a DDRR or an RC-specific gene.
We also tested the hypothesis that different gene groups are associated with age at RC diagnosis. We used the median age of RC diagnosis in the study cohort (48 years), and studied PVs in patients < 48 years or ≥ 48 years of age. To test the association between the presence of PVs, and age of RC diagnosis, two-sided Fisher’s exact tests were used, and p-values ≤ 0.05 were considered significant. Odds ratios (OR) were calculated to determine the odds that an outcome had occurred given a particular variant, compared to the odds of the outcome occurring in the absence of that variant in the population tested. Finally, we queried the SEER database for patients under 60 years old with RC to compare the distribution of their clinical characteristics (where available) to those in our study cohort22.
Due to the evolving nature of the panels during the course of this study, each version included a different total number of genes, and analysis of each gene is based on the number of individuals whose test included that gene (Table S1). Only data from 491 individuals was considered for comparison of individuals with RC-specific genes compared to those with variants in genes not typically associated with RC, as the other individuals did not have all 49 genes analyzed. For statistical comparisons analyzing correlations between specific genes with various characteristics, all individuals who had been tested for that specific gene were included.
To identify potential correlations between PVs and characteristics such as tumor pathology, additional primary tumor type, and age of diagnosis, RC-specific genes, other cancer-associated genes, and DDRR genes were combined into groups, and histologies were grouped. The categories for comparison of PVs and patient characteristics are as follows:
-
1.
Known RC genes (BAP1, FH, FLCN, MEN1, MET, MITF, PTEN, SDHA, SDHAF2, SDHB, SDHC, SDHD, TSC1, TSC2, and VHL ) versus genes not typically associated with RC (APC, ATM, BARD1, BRCA1, BRCA2, BRIP1, BMPR1A, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, GREM1, MAX, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, NF1, PALB2, PMS2, POLD1, POLE, RAD50, RAD51C, RAD51D, RET, SMAD4, SMARCA4, STK11, TEMEM127, TP53) versus DDRR genes alone (ATM, BARD1, BRCA1, BRCA2, BRIP1, CHEK2, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, POLD1, POLE, RAD50, RAD51C, RAD51D).
-
2.
Histology categories combined from the original categories: (1) Chromophobe, (2) Papillary renal, (3) Clear cell, (4) Wilms, (5) Renal cell (likely clear cell but cannot be confirmed), (6) Unknown, (7) Mixed papillary [clear cell papillary type, papillary renal/chromophobe renal, and sarcomatoid/papillary/clear cell]29, (8) Mixed chromophobe [chromophobe/oncocytoma, chromophobe/renal cell, clear cell/chromophobe, and clear cell/oncocytoma/chromophobe]30, (9) Oncocytoma, (10) Mixed oncocytoma [clear cell/oncocytoma, oncocytoma/collecting duct, and renal cell/oncocytoma] 31, and (11) Others [included clear cell/sarcomatoid, collecting duct, mixed epithelial and stromal, mucinous tubular and spindle cell, multilocular cystic renal, neuroendocrine, renal cell/Wilms, renal cortical, sarcomatoid, transitional, urothelial and urothelial transitional]. Transitional, urothelial, urothelial and papillary transitional cases were not included in the analysis for counts of pathogenic variants. Renal oncocytomas, mixed epithelial and stromal tumors are considered benign tumors, and were not included in the analysis for counts of pathogenic variants 32,33.
Study approval
The Western IRB issued a regulatory opinion that the Genomic Data Sharing Policy for Ambry Genetics does not involve human subjects based on 45 CFR46.102(f) and associated guidance, thus the requirement to obtain written patient informed consent was waived. A Data Use Agreement, and Materials Transfer Agreement was established between Ambry Genetics and Fox Chase Cancer Center. The FCCC Institutional Review Board (IRB) provided study oversight and approval (protocol number 14831). Ambry Genetics provided de-identified results for the study. All methods were performed in accordance with the relevant guidelines and regulation of the approved study.
Results
Patient characteristics
We first benchmarked the eoRC study cohort to the reported incidence of RC in SEER data for the general US population to provide context. In the study cohort, 40% of cases were between 50–59 years of age, and median age of diagnosis was 48 years. As expected, a higher percentage of RC cases were diagnosed between 20–44 years of age as compared to patients ≤ 60 diagnosed with RC in the general US population (SEER) (35%, versus 21.9%) (Fig. 1A). The study cohort was 67.1% female and 32.9% male (Fig. 1B, Table 1), versus 34.8% female and 65.2% male for the general US population prevalence of RC diagnosed ≤ 60 (Fig. 1B). Race/ethnicities in study cohort were 65.6% Caucasian, 5.8% African American/Black, 5.3% Ashkenazi Jewish, 7.6% Hispanic, 0.5% other, and 5.5% unknown (Table 1).

Patient characteristics. (A) Age range of individuals diagnosed with RC ≤ 60 years in SEER cohort compared to the study cohort (n = 746; of the remaining individuals in the study, 26 were diagnosed < 19 years, 33 were diagnosed at 60 years, and 39 were excluded from the calculations as their age was reported as a wide range of years). (B) Percentage of males and females diagnosed with RC ≤ 60 years in SEER compared to the study cohort (n = 844). (C) The percentages of reported RC histology up to age 60 years in the SEER data (n = 97,805) compared to the study cohort (n = 844); not all histological subtypes reported in SEER were reported in the study cohort. (D) The percentage of cancer incidence (at ≤ 60 years) in the general SEER population versus the study cohort. The SEER data reflect individuals reporting the indicated cancer type, not individuals with RC in addition to the indicated cancer type. (E) Percentage of different primary cancers reported (≤ 60 years) in SEER (n = 97,795) versus the study cohort (n = 844). Less than 0.4% not reported for figure clarity.
The tumor pathologies reported varied (Fig. 1C and Table 1). Clear cell constitutes 44.5% of all RCs in SEER, and was the most commonly reported histology in the eoRC cohort (243/844 = 28.8%). Renal cell (not defined, but likely to predominantly reflect clear cell) was also common (168/844 = 19.9%, Fig. 1C and Table 1). Papillary and chromophobe histology were each identified in ~ 4–5% of the individuals (38/844 = 4.5% and 40/844 = 4.7%, respectively). Other histologies were identified rarely, but included Wilms tumor (19/844 = 2.3%) and oncocytoma (6/844 = 0.7%). For 34.7% of patients, the RC subtype was unknown.
High incidence of other cancers in study cohort
57.9% (n = 489/844) of the cases in the study cohort reported at least one additional primary cancer (Fig. 1D, Table 1, Table S2). Each of the primary cancer types is also represented at a higher level in the study cohort than in the general US population as reported by the SEER database (Fig. 1D). For female-specific cancers, 40.1% of females (227/566) also had breast cancer, in comparison to the 4.3% breast cancer rate in women ≤ 60 in the general population (SEER) (Fig. 1D and Table S2). The rate of additional primary cancer in the study cohort (57.9%) is much higher than the rate of each cancer type observed in SEER cases with eoRC (21.6%) (Fig. 1E). Finally, 784 patients out of 844 reported a family history of cancer, and of these 784 patients, 196 (24.7%) specifically reported at least one family member with RC (Table 1).
Multi-gene cancer panel testing identifies PVs in DDRR genes in the study cohort
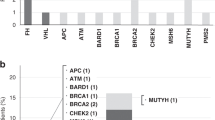
The most common gene with PVs identified in the eoRC patients was the DDRR gene CHEK2 (19/844, 2.25%, Fig. 2A, Table S3 and S4), consistent with a recent report by Carlo et al.16 Of patients with CHEK2 PVs, 47.3% (n = 9/19) had a common, highly damaging variant (c.1100delC, p.Thr367Metfs) that is known to be associated with an increased risk for breast, prostate, colorectal and thyroid cancers (Table S4)34,35,36,37.

Enrichment of PVs in DDRR genes. (A) Cases with germline PVs in the entire cohort (n = 844). (A) Red bars; DDRR genes, yellow bars; other-cancer associated genes; blue bars; RC genes. APC variants identified in this study were all the moderate risk c.3920T>A, p.I1307K variant, and 5 of the 19 CHEK2 variants were the moderate risk c.470T>C, p.I157T variant. *The individual with MUTYH was a compound heterozygote with two PVs. The data is presented as percent rather than ‘n’ due to the fact that not all 49 genes were tested for all patients in the full study cohort of 844 individuals. The percent adjusts for the number of individuals that were tested for each gene. The ‘n’ values are listed in Supplemental Table 3. (B,C) Odds of finding PVs in ATM (pink circle), BRCA1 (black circle), BRCA2 (orange circle), and CHEK2 (blue circle) from study cohort versus control population, ExAc (B) and gnomAD (C). Data is presented as log10 odds ratio (OR), and log10 confidence intervals. Dotted black line; association with outcome i.e. OR > 0 is enrichment in study cohort, OR = 0 no difference in cohorts. PVs not found in gnomAD or ExAc are indicated by the absence of any data or PVs not listed from Supplemental Table 4 were not found in the control population. Note: Computation of proportion or burden of individuals with all PVs in a specific gene(s) in the control population cannot be accurately performed as all PVs have not been defined, and while there is some agreement on which variants in a specific gene(s) are currently considered PVs, this is not true for all variants in that gene (as referenced in ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/). (D) Cases with germline PVs in the cohort tested for all genes in the study (n = 491, 49 genes). The total ‘n’ is listed in Supplemental Table 6. (E) Individuals with germline PVs who were diagnosed with only RC (n = 230/491). The total ‘n’ is shown above the bar as PV per gene. (F) Individuals with germline PVs who were diagnosed with RC plus at least one additional primary cancer type (n = 61/491). The total ‘n’ shown above the bar as PV per gene. The color scheme as in (A). In (D–F), to remain consistent between graphs, the data is presented as percent rather than ‘n’ even though all 49 genes were tested for all individuals represented in these graphs.
After CHEK2, PVs were most frequently observed in the DDRR genes BRCA2 (10/815, 1.23%), ATM (9/844, 1.07%) and BRCA1 (7/815, 0.86%) (Table S3). We compared the overall frequency of PVs in CHEK2, BRCA1, BRCA2, and ATM to the control population in ExAc and gnomAD, representing individuals sequenced for disease-specific and population genetic studies27,28. Overall, PVs in each of these genes were more common in the study cohort versus the control populations (Fig. 2B,C, Table S5A). An outlier was the moderate risk CHEK2 c.470T>C p. I157T PV38 identified in 5 individuals in the study cohort, which was higher in the controls (gnomAD- OR, 0.60; 95% CI, 0.234–1.433; ExAc- OR, 0.72; 95% CI, 0.282–1.74). We compared the prevalence of all PVs in DDRR genes presented in Table S4, from 844 cases, to controls from gnomAD23. We found ~ 4.8-fold enrichment of PVs in DDRR genes in the study cohort versus the controls in gnomAD (8.4% vs. 1.8% respectively, Table S5B; each DDRR gene was corrected for number of patients assessed).
Cancer risk with MUTYH (DDRR gene) has only been defined for individuals with homozygous or compound heterozygous PVs, but not for heterozygous carriers39. We identified 17/844 individuals with MUTYH PVs, out of which 16/17 were heterozygous carriers and only 1/17 was compound heterozygous. Only the individual with compound heterozygous MUTYH PVs was counted in the full study cohort (n = 844, Table S3 and Fig. 2A). Similar to MUTYH, cancer risk from the FH (RC-specific gene) c.1431_1433dupAAA, p.K477DUP variant is currently considered to be pathogenic only in the compound heterozygous or homozygous state40. We identified 2 RC patients who were heterozygous carriers of this specific FH variant (Tables S3 and S4).
The overall gene variation rate in the full study cohort (n = 844) is presented in Table S3. The full study cohort was not tested for all 49 genes. The largest panel was tested in the sub-cohort of 491 cases, and consisted of 49 genes, which included 15 RC-specific genes, and 34 other-cancer associated genes including 19 DDRR genes (Table S1). Here, 12.8% (63/491) of cases had PVs. PVs were identified in one or more of the 16 genes not typically associated with RC in 9.16% cases (n = 45/491, Table S6), versus 3.7% (n = 18/491) with a PV in RC-specific genes (Fig. 2D, Table S6). Of the 16 genes not typically associated with RC, 12 were in DDRR genes (8.55%, n = 42/491 or 66.7%, n = 42/63). Among the 491 patients, 2 patients were found to have PVs in two genes. One patient had PVs in two DDRR genes (BRCA1 and MUTYH het), and the other patient in a RC-specific gene and a DDRR gene (SDHB and MUTYH het) (Table S4). There was no MUTYH or FH compound heterozygous or homozygous PV in the sub-cohort of 491 cases.
DDRR genes PVs are similarly enriched in patients diagnosed with eoRC alone, or with eoRC and other cancers
Individuals who were tested for all 49 genes (n = 491) could be further separated into two sub-cohorts: those with eoRC as their only diagnosis (n = 230/491, 46.8%), and those with eoRC and one or more additional types of cancer (n = 261/491, 53.2%). To test the hypothesis that DDRR gene PVs might be associated with eoRC, we first analyzed PVs in eoRC cases with no additional primary cancer diagnosis. Among the 230 patients who only presented with eoRC, PVs were identified in 13% of cases (n = 30/230, Fig. 2E), which is approximately twice the reported frequency of PVs in RC-specific genes7. Among this 13%, 8.7% (n = 20/230) of PVs were in one of 10 DDRR genes (ATM, BRCA1, BRCA2, BRIP1, CHEK2, MLH1, MRE11A, NBN, PALB2, RAD51C), 3.5% (n = 8/230) were in one of 4 RC-specific genes (BAP1, FLCN, SDHB, VHL), and the remaining cases bore PVs in non-DDRR genes associated with cancers other than RC (Fig. 2E).
Next, we performed similar analysis as described above for patients who presented with eoRC plus one or more additional cancers. Among the 261 patients who presented with eoRC and at least one additional cancer, PVs were identified in 12.7% cases (33/261, Fig. 2F). Among these 12.7% of cases, PVs in other-cancer associated genes, including DDRR genes, were found in 8.8% of cases (n = 23/261), versus 3.8% (n = 10/261) of cases with PVs in RC-specific genes. This population was also enriched for PVs in 7 DDRR genes (8.4%, n = 22/261, ATM, BRCA1, BRCA2, CHEK2, MSH6, PALB2, PMS2), versus PVs in 7 RC-specific genes (BAP1, FLCN, MET, MITF, PTEN, SDHB, VHL).
Overall, these data suggest that DDRR gene PVs are enriched similarly in individuals diagnosed with eoRC alone or eoRC plus at least one additional primary cancer, but that the frequency of PVs in DDRR genes, in either group, exceeded that in the control populations tested (gnomAD/ExAc) (Fig. 2, Table S5A). The specific PVs identified were similar in frequency to those identified in the full patient cohort (n = 844), with CHEK2 the most represented DDRR genes (Fig. 2). To gain additional insight into the prevalence of these PVs in cancer patients, we surveyed ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and found that multiple PVs from this study (Table S4) have been reported in hereditary cancer predisposing syndromes (HCPS, summarized in Table S7). HCPS reflects a pattern of cancers in a family characterized by earlier onset, with individuals not necessarily having the same tumor and/or having more than one primary tumor, and having tumors that are more likely to be multicentric.
RC patients with BRCA1 or BRCA2 PVs
Notably, 1.2% (10/815) of the eoRC cases had a BRCA2 PV, and 0.9% (7/815) RC cases had a BRCA1 PV (Table 2, Table S3). This included 1.7% (n = 6/355, Table 2) of the cases who presented with only eoRC. Interestingly, despite the fact that the cohort was 67.1% female, 47.1% (8/17) of the detected BRCA1 and BRCA2 PVs were in males (Table 2). Of the 17 RC cases with a BRCA1 or BRCA2 PV, 8 (47.1%, 8/17) had an additional cancer associated with hereditary breast and ovarian cancer (HBOC) syndrome (breast, ovarian, prostate, pancreatic or melanoma), 3 had an additional non-HBOC cancer (17.6%, 3/17), and 6 presented with only eoRC (35.3%, 6/17) (Table 2). Family history was reported for 16 cases, and of those, 14/16 (87.5%) indicated that at least one family member had an HBOC-associated cancer. Of those with a BRCA2 PV, 7/10 (70%) reported that at least one family member had RC (Table 2).
No correlation between age of RC diagnosis and type of PV in RC
To determine if identification of specific classes of germline PV correlated with age of diagnosis in this cohort, genes were divided into four broad (overlapping) categories: all genes in the panel, RC-specific genes, non-RC genes (including DDRR genes) and DDRR genes (see “Methods”). The groups were compared to median age at first RC diagnosis of < 48 or ≥ 48 years. Given the invariable early-onset of Wilms tumor, the 20 individuals with this diagnosis were excluded from the analysis. Within this eoRC cohort, there was no significant association between age at diagnosis of RC and the type of PV for any of the four broad categories above (Fig. 3A).

Identified PVs compared to age of RC diagnosis, and histology of RC. (A) Statistical comparison of PVs in all genes, RC-specific genes, non-RC and DDRR genes in younger individuals (< 48) versus older individuals (≥ 48). N = 485, these cases were tested for all 49 genes; all cases with Wilms tumor were removed. The results were non-significant (p > 0.05) using two-sided Fisher’s exact tests. (B) Counts of PVs by RC histology: clear cell (blue bar) and renal cell (orange bar) carcinoma in the study cohort. The' renal cell' subtype, is likely clear cell, but this cannot be confirmed. (C) Counts of PVs by all RC histology observed in the study cohort. DDRR genes (maroon bars), other-cancer associated genes (yellow bars), RC-specific genes (blue bars). The total number of individuals with a PV and percent PVs per gene category is shown above the bar. (B,C) Includes counts from both homozygous and heterozygous carriers of MUTYH, and carriers of a FH variant that is currently considered to be pathogenic only in the compound heterozygous or homozygous state.
Correlation of renal histologies with PVs in specific genes
Of the 243 clear cell cases in this cohort, 13.6% (33/243) had a PV, of which 2.9% were RC-associated PVs. Similar findings were observed for the cases described as renal cell carcinoma, 13.1% (22/168) had a PV, of which 2.4% were RC-associated. DDRR gene PVs were found in 24/243 (~ 10%) of clear cell cases, and in 16/168 (9.52%) of renal cell cases. Figure 3B,C contrast the findings in clear cell and renal cell histology with the other non-clear cell histologies.
Discussion
This study for the first time demonstrates that PVs in multiple DDRR genes occur in patients with eoRC. Importantly, this study found that DDRR gene PVs were represented both in cases diagnosed with eoRC and additional cancers, and also cases diagnosed with eoRC alone. Comparison with a large control population indicated that germline PVs in DDRR genes were more common in this study cohort than in the control population, although further studies are required to confirm this finding and estimate the penetrance of PVs in DDRR genes for eoRC. We also found that germline testing using an RC-specific panel would have identified only 3.7% (18/491) of the RC cases with actionable PVs according to the NCCN recommended screening or management guidelines, compared to the 9.16% (45/491) additional cases identified with the expanded panels.
The most common gene with PVs identified in the patients in this study was the DDRR gene, CHEK2 (19/844, 2.25%). This is consistent with recent reports by Carlo et al. and Huszno et al.15,16. While evidence is mounting that CHEK2 PVs may increase risk for RC, in this study we did not consider CHEK2 as a gene typically associated with RC as it is not currently included on RC panels and would fail to be included in testing in many cases. In addition, limitations of the previous studies and the analysis reported here together indicate that larger studies with appropriate controls are needed before confirming that CHEK2 indeed confers a risk for RC.
Identification of germline DDRR gene PVs can have specific implications for the proband and the family. For example, 1.7% of cases diagnosed with eoRC alone had PVs in BRCA1 or BRCA2, but not all of these cases had a family history strongly indicative of HBOC syndrome. This is important because identification of a BRCA PV can potentially change medical management; for instance, PARP inhibitor therapy is effective in tumors with BRCA PVs, including non-breast tumors41,42. Also, screening and prevention of HBOC-syndrome cancers would likely be increased significantly in the proband and in family members found to have the same PV. Further, many of the specific PVs identified in this study have been annotated as relevant to various HCPS, emphasizing their role in the development of multiple cancer types. Our results support broader panel testing as a way to identify unexpected high-penetrant PVs in eoRC patients, when there is a personal or family history of additional cancers (especially an HBOC-syndrome cancer).
In RC, a number of germline PVs have been associated with treatment response. For example, bevacizumab with everolimus or erlotinib were added as treatment options for RC cases with germline PVs in FH8,43. Currently, the clinical significance of PVs in DDRR genes is not clinically defined for RC. There is an urgent need to study the biological impact of PVs in DDRR genes in renal tissue. Such work may also lead to improved understanding of RC pathogenesis. Studies are in progress to assess cancer risk in different tissue types, and response to treatment due to a germline defect in DDRR genes44. A recent study showed that VHL inactivation in RC led to reduced expression of DDRR genes (such as BRCA1 and BRCA2), and thereby increased sensitivity to PARP inhibitors45. These results indicate that RC tumors with DDRR gene vulnerabilities may be responsive to PARP or other DDRR gene inhibitors under development. Ongoing clinical trials are assessing the effect of PARP inhibitor, olaparib, in patients with somatic DDRR gene variants in the setting of metastatic RC (NCT03786796). Finally, it is also important to estimate the penetrance of DDRR gene PVs to clinically define RC risk. These studies will assist in genetic counseling of RC patients and their families.
The limitations of this study include the following: this is a relatively small cohort, and not all cases were tested for all 49 genes. The cohort is not representative of all individuals with RC, as the individuals reported in this study likely had clinical characteristics (e.g. high rate of additional primary cancers) or family history that led to the expanded panel testing. In the study cohort, females were overrepresented, even though more males are typically diagnosed with RC (Fig. 1B)46. This difference may reflect the observation that women are more likely to pursue genetic testing than men, or the fact that 34.4% of cases also had a diagnosis of breast, ovarian, or uterine cancer. Alternatively, men diagnosed with RC might be considered high-risk due to smoking or other environmental factors that lead their physician to be less suspicious of a hereditary component. A large percentage (34.7%) of tumors from the study cohort was listed as “unknown subtypes”, limiting comparison of PVs and RC histology types. Finally, matched (such as age and gender) comparisons were not possible using the large publicaly available control population (ExAC and gnomAD databases), and we made no adjustment for population stratification. Differences in the study cohort, and the large publicly available control population (ExAC and gnomAD databases) in ascertainment strategies and data collection (i.e. bioinformatic pipeline for variant calling/filtering, sequence coverage, race/ethnicities) prevent us from making any conclusions about the relationship between the PVs and RC risk28,47,48,49. The comparisons performed in this manuscript were not adjusted for multiple testing.
Conclusions
This study is the first to indicate a role for PVs in multiple DDRR genes in eoRC. These results need to be validated in other large data sets. Additionally, to fully elucidate the biological relevance of DDRR genes to RC, family and functional studies are needed as a next step to quantify the associated risks.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary files].
References
Shaw, G. The silent disease. Nature537, S98–S99. https://doi.org/10.1038/537S98a (2016).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin.70, 7–30. https://doi.org/10.3322/caac.21590 (2020).
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer136, E359–E386. https://doi.org/10.1002/ijc.29210 (2015).
Znaor, A., Lortet-Tieulent, J., Laversanne, M., Jemal, A. & Bray, F. International variations and trends in renal cell carcinoma incidence and mortality. Eur. Urol.67, 519–530. https://doi.org/10.1016/j.eururo.2014.10.002 (2015).
Coleman, J. A. & Russo, P. Hereditary and familial kidney cancer. Curr. Opin. Urol.19, 478–485. https://doi.org/10.1097/MOU.0b013e32832f0d40 (2009).
Rosner, I., Bratslavsky, G., Pinto, P. A. & Linehan, W. M. The clinical implications of the genetics of renal cell carcinoma. Urol. Oncol.27, 131–136. https://doi.org/10.1016/j.urolonc.2008.11.001 (2009).
Nguyen, K. A. et al. Advances in the diagnosis of hereditary kidney cancer: initial results of a multigene panel test. Cancer123, 4363–4371. https://doi.org/10.1002/cncr.30893 (2017).
Motzer, R. J. et al. NCCN Guidelines Insights: Kidney Cancer, Version 2.2020. J. Natl. Compr. Cancer Netw.17, 1278–1285. https://doi.org/10.6004/jnccn.2019.0054 (2019).
Brown, J. S., O’Carrigan, B., Jackson, S. P. & Yap, T. A. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov.7, 20–37. https://doi.org/10.1158/2159-8290.CD-16-0860 (2017).
Costa, W. H., Jabboure, G. N. & Cunha, I. W. Urological cancer related to familial syndromes. Int. Braz. J. Urol.43, 192–201. https://doi.org/10.1590/S1677-5538.IBJU.2016.0125 (2017).
Aarnio, M. et al. Uroepithelial and kidney carcinoma in Lynch syndrome. Fam. Cancer11, 395–401. https://doi.org/10.1007/s10689-012-9526-6 (2012).
Win, A. K. et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J. Clin. Oncol.30, 958–964. https://doi.org/10.1200/JCO.2011.39.5590 (2012).
Bonadona, V. et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA305, 2304–2310. https://doi.org/10.1001/jama.2011.743 (2011).
Stratton, K. L. et al. Outcome of genetic evaluation of patients with kidney cancer referred for suspected hereditary cancer syndromes. Urol. Oncol.34(238), e231–e237. https://doi.org/10.1016/j.urolonc.2015.11.021 (2016).
Huszno, J. & Kolosza, Z. Checkpoint kinase 2 (CHEK2) mutation in renal cell carcinoma: a single-center experience. J. Kidney Cancer VHL5, 19–23. https://doi.org/10.15586/jkcvhl.2018.101 (2018).
Carlo, M. I. et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol.4, 1228–1235. https://doi.org/10.1001/jamaoncol.2018.1986 (2018).
Baglietto, L. et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J. Natl. Cancer Inst.102, 193–201. https://doi.org/10.1093/jnci/djp473 (2010).
Ten Broeke, S. W. et al. Cancer risks for PMS2-associated Lynch syndrome. J. Clin. Oncol.36, 2961–2968. https://doi.org/10.1200/JCO.2018.78.4777 (2018).
Varley, J. Li–Fraumeni syndrome. Atlas Genet. Cytogenet. Oncol. Haematol.5, 76–77. https://doi.org/10.4267/2042/37718 (2001).
Mork, M. et al. Lynch syndrome: a primer for urologists and panel recommendations. J. Urol.194, 21–29. https://doi.org/10.1016/j.juro.2015.02.081 (2015).
Barrow, P. J. et al. The spectrum of urological malignancy in Lynch syndrome. Fam. Cancer12, 57–63. https://doi.org/10.1007/s10689-012-9573-z (2013).
Shuch, B. et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J. Clin. Oncol.32, 431–437. https://doi.org/10.1200/JCO.2013.50.8192 (2014).
Clemmensen, T. et al. Pathologic and clinical characteristics of early onset renal cell carcinoma. Hum. Pathol.74, 25–31. https://doi.org/10.1016/j.humpath.2017.11.005 (2018).
Arora, S. et al. Genetic variants that predispose to DNA double-strand breaks in lymphocytes from a subset of patients with familial colorectal carcinomas. Gastroenterology149, 1872-1883 e1879. https://doi.org/10.1053/j.gastro.2015.08.052 (2015).
Jasperson, K. W., Tuohy, T. M., Neklason, D. W. & Burt, R. W. Hereditary and familial colon cancer. Gastroenterology138, 2044–2058. https://doi.org/10.1053/j.gastro.2010.01.054 (2010).
Knijnenburg, T. A. et al. Genomic and molecular landscape of DNA damage repair deficiency across the Cancer Genome Atlas. Cell Rep.23, 239-254 e236. https://doi.org/10.1016/j.celrep.2018.03.076 (2018).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature536, 285–291. https://doi.org/10.1038/nature19057 (2016).
Karczewski, K. et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv https://doi.org/10.1101/531210 (2019).
Kocher, N. J., Rjepaj, C., Lehman, E. & Raman, J. D. Incidence and histologic features of mixed renal tumors. J. Surg. Oncol.117, 430–433. https://doi.org/10.1002/jso.24879 (2018).
Amin, M. B. et al. Chromophobe renal cell carcinoma: histomorphologic characteristics and evaluation of conventional pathologic prognostic parameters in 145 cases. Am. J. Surg. Pathol.32, 1822–1834. https://doi.org/10.1097/PAS.0b013e3181831e68 (2008).
Wobker, S. E. & Williamson, S. R. Modern pathologic diagnosis of renal oncocytoma. J. Kidney Cancer VHL4, 1–12. https://doi.org/10.15586/jkcvhl.2017.96 (2017).
Williams, G. M. & Lynch, D. T. in StatPearls (2020).
Williamson, S. R. Renal cell carcinomas with a mesenchymal stromal component: what do we know so far?. Pathology51, 453–462. https://doi.org/10.1016/j.pathol.2019.04.006 (2019).
Jalilvand, M. et al. An association study between CHEK2 gene mutations and susceptibility to breast cancer. Comp. Clin. Pathol.26, 837–845. https://doi.org/10.1007/s00580-017-2455-x (2017).
Hale, V., Weischer, M. & Park, J. Y. CHEK2 (*) 1100delC mutation and risk of prostate cancer. Prostate Cancer2014, 294575. https://doi.org/10.1155/2014/294575 (2014).
Meijers-Heijboer, H. et al. The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am. J. Hum. Genet.72, 1308–1314. https://doi.org/10.1086/375121 (2003).
Wojcicka, A. et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer53, 516–523. https://doi.org/10.1002/gcc.22162 (2014).
West, A. H. et al. Clinical interpretation of pathogenic ATM and CHEK2 variants on multigene panel tests: navigating moderate risk. Fam. Cancer17, 495–505. https://doi.org/10.1007/s10689-018-0070-x (2018).
Win, A. K. et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int. J. Cancer139, 1557–1563. https://doi.org/10.1002/ijc.30197 (2016).
Ezgu, F., Krejci, P. & Wilcox, W. R. Mild clinical presentation and prolonged survival of a patient with fumarase deficiency due to the combination of a known and a novel mutation in FH gene. Gene524, 403–406. https://doi.org/10.1016/j.gene.2013.03.026 (2013).
Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol.5, 387–393. https://doi.org/10.1016/j.molonc.2011.07.001 (2011).
Shroff, R. T. et al. Rucaparib monotherapy in patients with pancreatic cancer and a known deleterious BRCA mutation. JCO Precis. Oncol. https://doi.org/10.1200/PO.17.00316 (2018).
Park, I., Shim, Y. S., Go, H., Hong, B. S. & Lee, J. L. Long-term response of metastatic hereditary leiomyomatosis and renal cell carcinoma syndrome associated renal cell carcinoma to bevacizumab plus erlotinib after temsirolimus and axitinib treatment failures. BMC Urol.19, 51. https://doi.org/10.1186/s12894-019-0484-2 (2019).
Arora, S. et al. Existing and emerging biomarkers for immune checkpoint immunotherapy in solid tumors. Adv. Ther. https://doi.org/10.1007/s12325-019-01051-z (2019).
Scanlon, S. E., Hegan, D. C., Sulkowski, P. L. & Glazer, P. M. Suppression of homology-dependent DNA double-strand break repair induces PARP inhibitor sensitivity in VHL-deficient human renal cell carcinoma. Oncotarget9, 4647–4660. https://doi.org/10.18632/oncotarget.23470 (2018).
Woldrich, J. M., Mallin, K., Ritchey, J., Carroll, P. R. & Kane, C. J. Sex differences in renal cell cancer presentation and survival: an analysis of the National Cancer Database, 1993–2004. J Urol179, 1709–1713. https://doi.org/10.1016/j.juro.2008.01.024 (2008) (discussion 1713).
Karczewski, K. J. et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res.45, D840–D845. https://doi.org/10.1093/nar/gkw971 (2017).
Kobayashi, Y. et al. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med.9, 13. https://doi.org/10.1186/s13073-017-0403-7 (2017).
Walsh, R. et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med.19, 192–203. https://doi.org/10.1038/gim.2016.90 (2017).
Acknowledgements
We express gratitude to Dr. Margie Clapper, for many helpful discussions and advice with this work. We thank members of the FCCC Risk Assessment Program and Department of Clinical Genetics, and Yan Zhou and Elizabeth Handorf in the FCCC Bioinformatics and Biostatistics Facility, for assisting with this work. Lisa Bealin (Risk Assessment Program, FCCC) for assisting with the IRB protocol, and the FCCC Office of Research and Developmental Alliances for executing Data Use and License Agreements for work with Ambry Genetics. Grant Support: All Fox Chase Cancer Center (FCCC) authors and the Biostatistics Facility was supported by NCI Core Grant P30 CA006927 (to FCCC), DOD W81XWH-18-1-0148 (to S.A.), a subsidy of the Russian Government to support the Program of Competitive Growth of Kazan Federal University (to E.V.D.), NIH R01 DK108195 and support from the Colorectal Cancer Alliance (to E.A.G).
Author information
Authors and Affiliations
Contributions
T.R.H., performed most studies, data analysis, writing; E.V.D., data analysis, figure preparation; R.W.L, ClinVar analysis; V.S., L.H., M.R., sequence data, discussion; E.A.G., M.J.H., M.B.D., A.F., L.K., discussion and writing; S.A., study conception, discussion, planning and writing.
Corresponding author
Ethics declarations
Competing interests
A.F. has consulted for Invitae Corporation, and received speaker fees from AstraZeneca, M.J.H. performs collaborative research (with no funding) with the following: Myriad Genetics, Invitae Corporation, Ambry Genetics, Foundation Medicine, Inc. He also performs collaborative research (with no funding) and is part of a Precision Oncology Alliance funded by Caris Life Sciences (cover travel and meals at meetings), L.H., M.R. and V.S. are full-time salaried employees of Ambry Genetics. All other authors report no conflicts or disclosures.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hartman, T.R., Demidova, E.V., Lesh, R.W. et al. Prevalence of pathogenic variants in DNA damage response and repair genes in patients undergoing cancer risk assessment and reporting a personal history of early-onset renal cancer. Sci Rep 10, 13518 (2020). https://doi.org/10.1038/s41598-020-70449-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70449-5
This article is cited by
-
Candidate variants in DNA replication and repair genes in early-onset renal cell carcinoma patients referred for germline testing
BMC Genomics (2023)
-
Identification of specific susceptibility loci for the early-onset colorectal cancer
Genome Medicine (2023)
-
CHEK2 is a potential prognostic biomarker associated with immune infiltration in clear cell renal cell carcinoma
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
