
Abstract
Rye is used as food, feed, and for bioenergy production and remain an essential grain crop for cool temperate zones in marginal soils. Ergot is known to cause severe problems in cross-pollinated rye by contamination of harvested grains. The molecular response of the underlying mechanisms of this disease is still poorly understood due to the complex infection pattern. RNA sequencing can provide astonishing details about the transcriptional landscape, hence we employed a transcriptomic approach to identify genes in the underlying mechanism of ergot infection in rye. In this study, we generated de novo assemblies from twelve biological samples of two rye hybrids with identified contrasting phenotypic responses to ergot infection. The final transcriptome of ergot susceptible (DH372) and moderately ergot resistant (Helltop) hybrids contain 208,690 and 192,116 contigs, respectively. By applying the BUSCO pipeline, we confirmed that these transcriptome assemblies contain more than 90% of gene representation of the available orthologue groups at Virdiplantae odb10. We employed a de novo assembled and the draft reference genome of rye to count the differentially expressed genes (DEGs) between the two hybrids with and without inoculation. The gene expression comparisons revealed that 228 genes were linked to ergot infection in both hybrids. The genome ontology enrichment analysis of DEGs associated them with metabolic processes, hydrolase activity, pectinesterase activity, cell wall modification, pollen development and pollen wall assembly. In addition, gene set enrichment analysis of DEGs linked them to cell wall modification and pectinesterase activity. These results suggest that a combination of different pathways, particularly cell wall modification and pectinesterase activity contribute to the underlying mechanism that might lead to resistance against ergot in rye. Our results may pave the way to select genetic material to improve resistance against ergot through better understanding of the mechanism of ergot infection at molecular level. Furthermore, the sequence data and de novo assemblies are valuable as scientific resources for future studies in rye.
Similar content being viewed by others

Introduction
Rye (Secale cereale L.) is a widely popular cereal crop, especially in northern Europe, due to its high nutritional value and tolerance to unfavorable environmental conditions. Rye remains an important grain crop for cool temperate zones under marginal soil conditions, particularly the ones not suitable for wheat1. Rye is used as food, feed and for bioenergy production worldwide1. Rye flour is used for making bread and consumed widely in North and Central Europe2. The genome of rye consists of seven chromosomes (2n = 14) with a total estimated genome of size of ≈ 8000 Mb3. Until recently, compared to sequencing information of other closely related cereals, the least number of genomic DNA and cDNA sequences was found for rye.
Ergot caused by Claviceps purpurea is a noxious disease of rye and has been a problematic for rye farmers throughout the history of mankind and agriculture. Ergot is known to cause severe problems in cross-pollinated rye by contamination of harvested grains4. Ergot is a serious threat to bread production as milling industry accepts only limited amounts of sclerotia (< 0.05%) from C. purpurea in the harvested grains for bread making4. The mycotoxins produced by ergot are ubiquitously present and contamination levels exceeding the EU maximum levels can lead to significant economic losses to farmers5. In addition, C. purpurea represents an interesting biotrophic model system, as no visible signs of plant defense reactions occur in infected plant tissue. Interestingly, during the infection process, the fungus is able to keep the host organism alive for an extended period of time and the fungus is thus classified as true biotroph6. C. purpurea infects exclusively young unfertilized ovaries. However, the basis of the organ specificity is not clear and until recently no resistance genes were known in cereals. Ergot infection has a complex infection pattern and the molecular response of underlying mechanisms of this disease is still largely unknown, hence gene expression profiling after infection will be useful.
Next Generation sequencing of RNA (RNAseq) can provide astounding details about the transcriptional landscape of an organism. It is the preferred method for cataloguing and quantifying the complete set of transcripts for a specific tissue, developmental stage or physiological condition in response to a specific stress7. However, the major challenge of this technology is to assemble a high quality reference transcript set from short read data8. RNAseq is a preferred technology to understand morphology9, physiology10 and microbe-plant interaction11. However, there has been limited application of this technology in case of S. cereal. There is no universal accepted pipeline to deal with the vast amount of data generated in RNAseq12,13. Plant researchers design experiments and adopt different analysis strategies depending on the species being studied and their research goals. Many variations of RNAseq protocols and analyses have been published, making it challenging for researchers to identify best practice for conducting an RNAseq study12,13. The challenge starts by dealing with the processing of raw data and then continue with identification of genes in expression profiling and interpretation of the obtained information. The straightforward analysis of RNA sequencing reads is based on an existing reference genome and annotated gene models. However, rye is a non-model plant species and only a draft genome is available3. Trimmomatic, which is an efficient tool that can correctly handle paired-end reads was used for the preprocessing of raw data14. After preprocessing, reads were assembled using Trinity, which produces good-quality assemblies at single k-mer15. The Trinity assembler was selected as it has been shown to be better than other de novo assemblers at reconstructing full-length transcriptome16,17. The de novo transcriptome assembly in plants is complex due to sequence similarity of transcripts that are isoforms, paralogs and orthologs that may lead to imperfect assemblies in the form of bubbles or extra branches in de Bruijn graphs 18. We employed the CD-HIT tool to refine de novo assemblies by removing the shorter redundant sequences based on sequence similarity19. Numerous methods were applied to assess the overall quality, accuracy, contiguity, and completeness of the de novo assembled transcriptomes. The most common is through measuring the proportion of RNA-seq reads used to generate each assembly that map back to the assembled transcriptome. This method is common for RNA-seq analysis, when no reference genome is available. It has the advantages that assemblies are species specific and mapping to transcript assemblies is usually contiguous instead of spliced. The completeness of the de novo assemblies were assessed through Benchmarking Universal Single-Copy Orthologs (BUSCO) that indicate the number of conserved orthologs present in transcriptome20. After de novo assembly and quality assessments, we performed functional annotations and determined differentially expressed genes (DEGs). Overall, we presented a stringent workflow in this study for processing of RNAseq data, obtaining de novo assembly, assessment of assembly, functional annotation and differential gene expression from spikes of the non-model plant S. cereale.
In this Genomic era, knowledge about the ergot disease has enhanced considerably but the fundamental underlying mechanisms are still poorly understood. Considerable variation in ergot resistance exists among rye germplasms21,22,23. However, little genetic information are available due to the complex unknown infection pattern and huge genome size of rye15,16. Hence, an understanding of the underlying molecular mechanisms will assist in breeding of ergot resistant S. cereale hybrids. RNA sequencing is a highly efficient method, but false positives still occur due to sensitivity of this technology. Keeping this drawback in mind, we used both a de novo assembled and the draft reference genome of rye to count the differentially expressed genes in a moderately ergot resistant compared to an ergot susceptible hybrid with and without inoculation. When using a draft reference genome, sequence divergence between reads and the rye reference genome may compromise results; nucleotide mismatches are more likely to decrease the number of mapped reads. However, indels are usually better tolerated with gapped alignments24. Similarly, in case of de novo assembly a large number of erroneous contigs produced by the assemblers might result in an unreliable estimation. Hence, it remains challenging to analyze the quantification reliability of the assembled contigs generated from de novo assembly. However, utilization of de novo assemblies avoids the mapping issues raised in case of draft genome and captures divergent and novel genes useful for species-specific discovery of new functions. Hence, we performed read quantification based on rye draft genome and de novo assembly generated in this study. These two different datasets are used to support the validity of the method and to DEGs. This analysis method has been shown to produce comparable gene expression profiles in other systems but has not been applied in RNAseq studies of plants25,26. Enrichment analysis of gene ontology (GO) terms and gene set enrichment analysis (GSEA) were applied to identify potential pathway responsible to cause ergot infection. Moreover, the behavior and expression profiles of orthologs of identified genes in Triticum aestivum were explored. To the best of our knowledge, this is the first attempt with proper experimental considerations to unravel genes involving in ergot infection mechanism in rye hybrid.
Material methods
Plant material
In a field experiment, we identified moderately ergot resistant and susceptible hybrids. Both hybrids consist of same genetic components except the restorer component. We presented the data as the average of 2-year i.e. 2018 and 2019 (Fig. 1). In both years, we had two replicates of each hybrid. Each hybrid was cultivated in a plot of two m2. A triticale guard on each side of hybrids also cultivated on the plot of two m2 to avoid pollination with neighboring hybrid. We measured the number of ergot sclerotia per 100 spikes and percent weight of ergot. The hundred spikes were selected randomly from five different places in group of 20 spikes per place from the plot area of specific hybrid and then total number of ergot sclerotia were counted. The percent weight of ergot sclerotia were determined through weighing the sclerotia in the total grain weight of specific rye hybrid harvested from 1m2 area. On average, the frequency of ergot in the ears was 3 times higher in DH372 compared with Helltop (Fig. 1A), whereas percent weight of sclerotia in the grains of DH372 was four-fold higher than Helltop (Fig. 1B). Based on our data, we found out that Helltop was moderately ergot resistant and DH372 was highly susceptible.

Field trial data of Helltop and DH372 after ergot inoculation. (A) Number of ergots per 100 spikes. (B) Percentage weight of ergot out of total grain weigh. Data is the average of two years i.e. 2018 and 2019.
Plant material for transcriptome study
The experiment to obtain plant material for comparative transcriptome was conducted in environment-controlled growth chambers. Seeds of the two hybrids were sown in peat pots (diameter, 15 cm) in greenhouse. After two weeks, plants were placed in cold room for vernalization for 8 weeks. The conditions were maintained at 4 °C, 8/16 h day/night cycle, 120 μE m−2 s-1 light intensity and 80% humidity. After vernalization, plants were moved into two different growth chambers to avoid cross contamination designated as control or treated. The latter were inoculated with spore solution of C. purpurea. Both chambers were set at same growth conditions i.e. 16 h light (8,000 lx; 18 °C) and 8 h darkness (15 °C). During growth, nutrients were supplemented once a week during watering. At flowering initiation stage, individual spikes were inoculated with either sterilized water (mock control) or with freshly prepared spore suspension of C. purpurea. Spore suspension for inoculation were produced on wheat grains as described by Engelke with slight modifications27,28. Briefly, fresh cultures were propagated on Potato Dextrose Agar (PDA, Difco) medium incubated at room temperature. Conidia were produced on autoclaved wheat grains. The inoculated kernels were placed in incubation room at 17–19 °C for 21 days. Conidial suspensions from mycelium growth kernels were harvested in sterile water, filtered through cheesecloth, and concentrations determined with a hemocytometer by microscopy. Spore concentration at 1 × 106 conidia/ml was used for inoculation.
After spray, both treatments were covered with plastic bags to maintain humidity and avoid cross contamination. Spikes were collected in triplicate 5 days post inoculation and immediately frozen in liquid nitrogen and stored at − 80 °C until RNA extraction.
RNA extraction and sequencing
Total RNA was extracted from 50 mg from each of twelve individual spikes using a CTAB method29 with slight modifications. Briefly, spikes were grounded separately using Geno/Grinder (SPEX Sample prep. Stanmore. UK) for 45 s at 1.500 Hz. The fine tissue powder was suspended in 1 ml of pre-heated (65 °C) CTAB extraction and 2% β-mercaptoethanol and mixed carefully. The suspension was incubated in thermomixer at 65 °C for 15 min to permit the complete dissociation of nucleoprotein complexes and centrifuge at 12,000×g for 5 min at 4 °C. Later, supernatant was transferred to a new 2 mL tube and added 400 μl of chloroform per 1 mL of CTAB. The tubes were shaken vigorously for 15 s and centrifuged at 12,000×g for 10 min at 4 °C. Following centrifugation, aqueous phase was transferred into a new micro-centrifuge tube, and step of centrifugation was repeated. The aqueous phase was collected and precipitated with one third volume of 8 M LiCl and incubated overnight at 4 °C, followed by centrifugation at 16,000 rpm for 30 min at 4 °C. Supernatant was removed and pellet was washed with 200 µL70% ETOH, followed by centrifugation at 13,000 rpm for 10 min at 4 °C. RNA quality was verified using a spectrophotometer (Nanodrop 3,300. Wilmington. USA) and Bioanalyzer (Agilent 2,100. Santa Clara. USA). Samples that showed higher RIN value were treated with DNAse Ambion DNA-free DNase Treatment and Removal kit (Cat #AM1906, California, USA) following the manufacturing protocol (https://www.thermofisher.com/order/catalog/product/AM1906#/AM1906). After DNAse treatment, RNA quality was analyzed again using Bioanalyzer. The mRNA of selected samples was fragmented and transformed to 100 bp short insert strand specific cDNA libraries for sequencing on DNBseq PE100 from BGI (Europe). The raw reads from this library has been deposited in sequence read archive (SRA) with submission ID “PRJNA612415” (https://dataview.ncbi.nlm.nih.gov/object/PRJNA612415?reviewer=r5fg70lms7oandmb7aid7p0mib).
Assembly normalization and quality assessment
The raw reads of twelve individual assemblies were filtered to remove low quality reads and reads containing adapter sequence before performing the assembly using the software Trimmomatic (version 0.36) with default settings14. The raw reads with a Phred score of base quality below 30 containing only “N” and/or length below 80 bp were removed. These were normalized using a kmer size of 2030. This step progressively removed high coverage reads from short read data sets and decreased the sampling variation, discarding redundant data, and removing the majority of errors. Furthermore, the assembly procedure was a combination of different methods. First, the Trinity pipeline (version 2.4) was used with a minimum contig length of 200 bp and a minimum kmer covariance of 2. The longest isoform for each gene was selected (perl script by Brian Haas on https://groups.google.com/forum/#!topic/trinityrnaseq-users/cXM1KiJe7dU). After selection of the longest isoform, contigs were merged according to a similarity criterion of 90% in CD-HIT-EST (version 4.6.3)19. The contigs were then translated to coding protein sequences using Transdecoder (version 2.0.1) following identification of the longest ORFs31. Through merging all reads from individual libraries, we also created a combined dataset belonging to each hybrid. The non-redundant assemblies specific to S. cereale were used in downstream functional annotation using Kyoto Encyclopedia of Genes and Genomes (KEGG) and GO enrichment analysis.
The quality of the assemblies was assessed using several methods. First, the assemblies were evaluated against a database of single copy orthologous genes for plants as implemented in BUSCO (version 1.161)20. After BUSCO assessment, the processed RNAseq reads were mapped to the combined transcriptome assembly (pRNAseq-2-CTA) to determine percentage of mapped reads, properly paired reads and singleton. Similarly, the processed RNAseq reads were mapped to own transcriptome assembly (pRNAseq-2-OTA). The information obtained in both analysis were used to assess the quality of de novo assembly as described by Zhao et al.32.
Functional annotation KEGG and GO enrichment
The assemblies were aligned against the NCBI non-redundant protein sequence using BLASTX searches database in the OmicsBox program and only the best homologue was reported. GO mapping was performed against the Gene Ontology database implemented in OmicsBox (version 1.2.4)33. Sequences that shared similarities with known protein sequences in BLASTX searches with significant similarity (E < 1e−10) were identified using the online tool InterProScan 5.0. The OmicBox program was used to assign Gene Ontology (GO) terms to the annotated sequences to predict the functions of the unique sequences with an e-value hit filter set to 1 × 10−3, annotation cutoff of 55 and evidence code set to 0.8 for the different categories as implemented in OmicsBox. Furthermore, KEGG analysis was used to identify potential pathways represented in the transcriptomes of DH372 and Helltop34. The OmicBox program was used to assign GO terms to the annotated sequences to predict the functions of the unique sequences and encoded translated proteins.
Identification of differentially expressed genes, annotation and gene ontology
The raw reads were cleaned following the procedure as described in section above and then these clean reads were aligned to the de novo assemblies to count reads. Similarly, these reads were also aligned to the available draft reference genome of rye3. Differential gene expression analysis was performed in OmicsBox (Version 1.2.4) using the package EdgeR (Version 3.11) with FDR correction ≤ 0.05, P value ≤ 0.01, fold change ≤ 2 or ≥ 235. EdgeR was also used to normalize the expected counts for relative expression and effective library size using the Trimmed Mean of M-values (TMM) normalization method. Genes with at least 1 count per million (CPM) were selected for further differential expression analysis. Differentially expressed genes (DEG) with FDR ≤ 0.05 and log fold change (logFC) of two were extracted for further analysis for gene ontology and enrichment analysis.
Expression profile and behavior of wheat orthologs
The orthologues of identified genes involved in pectinesterase activity and cell wall modification were extracted from reference genome of T. aestivum using sequence similarity searches. All the identified accession numbers of orthologs were analyzed manually and validated through BLAST search using NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The behaviors of these genes under various experimental conditions and in different plant parts were explored using the Genevestigator (www.genevestigator.com) at p < 0.05 and fold change > 2. The Genevestigator is a manually curated and well-annotated database of expression profiling from 11 different plant species with more than 26,889 exclusive plant samples (August. 2019).
Results
Sequencing, de novo assembly and assessment of assembled transcriptome
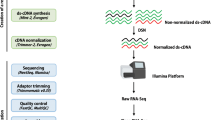
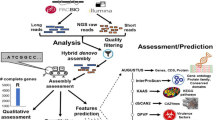
The clean reads from two S. cereale hybrids each containing three independent biological replicates with and without inoculation of C. purpurea, respectively (Fig. 2A) were assembled to generate de novo assembled transcriptome (Fig. 2B). The graphical representation of the experimental set up and bioinformatics analysis are shown in Fig. 2. We obtained 18 to 26 × 106 high quality paired-end reads in individual libraries, whereas 137 to 150 × 106 clean reads were assembled into contigs (Table 1). The de novo assembly of these libraries resulted in 82,404 to 98,494 contigs in individual assemblies and 240,689 to 248,337 contigs in pooled assemblies (Table 1). After clustering with CD-HIT-EST, the contigs were assembled into 50,616 to 78,418 transcripts in the individual transcriptomes and 192,116 to 208,690 transcripts of Helltop and DH 372 in the combined transcriptome, respectively. The combined transcriptome assembly of DH372 contains contig lengths between 201 and 16,695 bp and 31,574 contigs are equal or longer than 1000 bp. Similarly, the combined transcriptome assembly of Helltop encompasses contigs between 201 and 14,790 bp, and 25,842 contigs are equal to or longer than 1000 bp. Finally, we identified unigenes in non-redundant combined transcriptome assemblies through blasting against the rye reference genome. This analysis resulted in 18,615 unigenes in combined assembly of DH372, and 18,786 unigenes in Helltop assembly that are used for downstream functional analysis (Table 1).

Graphical representation of experimental set up and bioinformatics analysis. (A) Experimental set up for RNA extraction from DH372 and Helltop, (B) Bioinformatics analysis from raw reads to de novo assembly leading to identification of genes linked to ergot infection.
BUSCO assessment of the quality of transcriptome assemblies revealed that individual transcriptome assemblies have low gene representation (41.4 to 60%), low completeness (34.88 to 53.94%), higher fragmentation (27.21 to 38.84%) and higher missing sequences (11.16 to 26.74%) compared with the combined assemblies. The combined assemblies of DH372 and Helltop possess high percentage of gene representation (91.16 to 91.86%), high completeness (71 to 72%), low percentage of fragmented (7.21 to 7.44%) and very low missing sequences (0.7 to 1.63%) (Table 2). In the assessment using the pRNAseq-2-OTA, percentage of clean reads mapped back to own transcriptome is in range of 82.6 to 89.1% in case of individual assemblies and 91.9 to 92.3% of clean reads mapped to their respective combined assemblies (Table 2). Similarly, in pRNAseq-2-CTA, clean reads mapped to individual assemblies were in the range of 93.8 to 96.2%, whereas clean reads mapped to the respective combined assemblies were in the range of 96.1–96.3%. In the assessments of pRNAseq-2-CTA, we observed higher percentage of mapped and properly paired reads compared to pRNAseq-2-OTA, whereas the % of singletons are low. After all the redundancy reduction steps and obtaining good quality of assembly specific to S. cereale, we decided to dissect the annotation of these assembled sequences in the respective combined assemblies.
Functional annotation of non-redundant combined de novo assemblies of DH372 and Helltop
In the combined transcriptome assemblies, most sequences got hit to Tritucum asestivum (87%, 16,378 unigenes in DH372 and 16,644 in Helltop), followed by Hordeum vulgare (5%, 1030 in DH372 and 1,034 in Helltop) and Brachypodium distachyon. The top 10 organisms all belong to the Poaceae family and 92% of the contigs got a top hit to Triticeae (Supplementary Figs. 1A and 1B). Both assemblies had a similar percentage of hits to Triticum aestivum and Hordeum vulgare. However, a difference was observed in case of Sorghum bicolor and Eragrostis curvula, where the share of contigs in the assembly of Helltop are higher than in DH372 (Supplementary Fig. 1C).
In the GO analysis, a total of 9282, 13639 and 6825 unigenes of DH372 were assigned to biological processes (BP), molecular functions (MF) and cellular components (CC) GO categories, respectively. Similarly, unigenes of Helltop were assigned to 9921, 13,805 and 6915 of BP, MF and CC categories, respectively (Fig. 3A). Top categories of BP, MF and CC of the hybrids were found very similar in both assemblies (Supplementary Fig. 2). In both assemblies, highest number of unigenes in BP was associated with oxidation reduction process (9%), protein phosphorylation (8%) and regulation of transcription (5%). In MF, unigenes of both assemblies were predominantly assigned to ATP binding (10%), DNA binding (4%), metal ion binding (4%) and protein kinase activity (3%). Similarly, in the CC category, unigenes were abundant as integral component of membranes (42%), nucleus (13%) and cytoplasm (3%) (Supplementary Fig. 2). Overall, comparison of functional annotations between the assemblies of Helltop and DH372 showed that these are rather similar in their GO annotations. As both hybrids have differential responses towards ergot, we decided to identify unigenes exclusively found in each hybrid and linked them to various GO terms. We identified 1161 unigenes exclusive to DH372 and 1489 unigenes found only in Helltop de novo assembly (Fig. 3B). In the top GO terms, difference in BP, MF and CC of exclusive genes of DH372 and Helltop were more pronounced compared to all unigenes in the both hybrid assemblies. In the BP category, there was a higher number of Helltop genes was associated to proteolysis, biosynthetic process, ubiquitin dependent protein catabolic process, cell wall organization, defense response and recognition of pollen, whereas genes associated with oxidation reduction process, cellular oxidant detoxification, oxidative stress and hydrogen peroxide catabolic process was predominant in DH372 (Fig. 4). In MF category, higher number of genes found in Helltop were related to DNA binding, oxido-reductase activity, transferase and mono-oxegenase activity, transporter activity and ribonuclease activity. Similarly, in CC category, Helltop had higher number of genes linked to integral component of membrane, localized in nucleus, plasma membrane, extra cellular space and endoplasmic reticulum (Fig. 4).

Distribution of the Unigenes sequences (A) and gene ontology (B) in the assembly of DH372 and Helltop. (A) Count of unigenes in the transcriptome assemblies of Helltop and DH372 (Helltop = unigenes identified within transcriptome assembly of Helltop; DH372 = unigenes identified within transcriptome assembly of DH372). (B) The number of GO terms linked to unigenes identified in the transcriptome assemblies of Helltop, DH372 and unigenes exlusively found in either of transcriptome assembly.

Distribution of Gene ontology terms in genes exclusively present in the assemblies of DH372 and Helltop. (A) Biological process, (B) Cellular component, (C) Molecular function.
DH372 and Helltop have 2613 and 2634 unigenes sequences that have enzyme codes and these enzyme coding sequences represented 14% of the respective assemblies. The highest number of these genes were associated with hydrolases followed by transferases and oxidoreductases in the both assemblies (Fig. 5). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was used to identify potential pathways most represented in assembly of Helltop and DH372. The top ten KEGG pathways based on sequences number were compared between assemblies of Helltop and DH372. All assemblies have similar pathways in their top ten such as important metabolisms pathways (purine, thymine, starch and sucrose, Porphyrin and chlorophyll) and biosynthesis pathways (biosynthesis of antibiotics, gluconeogenesis, aminoacyl tRNA biosynthesis), pentose and glucoronate interconversions and drug metabolism. These most represented pathways are important basic pathways necessary for cell life and therefore naturally found in our assemblies. Purine metabolism was the pathway with the most sequences in each individual assembly with 348 and 338 unigenes in DH372 and Helltop, respectively. In case of unigenes exclusively assembled in each hybrid, thymine metabolism was the pathway with more genes. The major differences were observed in drug metabolism and pentose and glucoronate interconversions pathways. In the pentose and glucoronate interconversions, 8 unigenes were exclusively found in Helltop compared to 2 in DH372 (Fig. 5B). Similarly, in drug metabolism pathway, 7 unigenes were exclusively found in Helltop and 6 in DH372 (Fig. 5B).

Distribution of unigenes sequences of DH372 and Helltop to enzyme classes and KEGG (Kyoto Encyclopedia of Genes and Genome) pathways. The major classes of enzymes (A) and top 10 most highly represented KEGG pathways (B) are shown. Analysis was performed using the OmicsBox and the KEGG database, and unigenes were assigned to biological pathways in the KEGG pathway (https://www.genome.jp/kegg/).
Differential expression and enrichments analysis
Reads were counted by mapping to de novo assembled transcriptomes and the rye reference genome for the identification of differentially expressed genes (DEGs) between the two hybrids. The number of DEGs identified in the different comparisons can be found in Table 3. However, here onwards, we will describe only the genes that were common in both genomes to support the validity and reliability of our analysis. A higher number of DEGs were identified when comparison was made between control and inoculated samples of hybrids than the comparison between controls of the both hybrids or between infected plants of both hybrids (Table 3). In case of Helltop after ergot infection, in total 394 DEGs were identified, where 324 DEGs were up-regulated and 70 DEGs were down-regulated. Interestingly, a large number of DEGs were identified in DH372 (Table 3). Furthermore, the comparison of identified DEGs in Helltop and DH372 after ergot infection were presented in Venn diagram (Fig. 6). In this comparison, a far greater number of DEGs were specific to the ergot-susceptible DH372 than to Helltop, consistent with increased activity of ergot-activated transcriptional regulators in DH372 (Fig. 6). The genes that were differentially expressed in both hybrids after ergot infection were used for gene enrichment analysis. We identified the significantly enriched GO terms of these DEGs that were significantly associated to carbohydrate metabolic processes, hydrolase activity, pectinesterase activity, cell wall modification, pollen development and pollen wall assembly (Fig. 6). After GO terms enrichment analysis, we decided to use gene set enrichment analysis (GSEA) to identify the most significant pathways. The GSEA analysis identified pectin catabolic process and cell wall modification in biological process category, whereas pectinesterase activity and Rho GTPase binding were significant pathways in molecular function (Fig. 7). After identifying these pathways, we decided to dissect the genes involved in these pathways in detail in both hybrids after ergot infection and the list of genes are presented in Table 4. In case of cell wall modification and pectinesterase activity, in total 12 and 8 genes are differentially upregulated in response to ergot infection in both hybrids, respectively (Table 4). Among those genes, six genes named “XLOC_059237” “XLOC_1003867”, “XLOC_118963”, “XLOC_1220465”, “XLOC_1387037” and “XLOC_386424” are present in cell wall modification and pectinesterase pathways (Table 4). The COBRA-like 10 (XLOC_1432429) and probable pectinesterase inhibitor 21 (XLOC_118963) involved in cell wall modification were topmost differentially expressed with 2465- and 2261-fold changes, respectively. Interestingly, pectinesterase inhibitor 21 was also found in pectinesterase activity pathways. These two genes (XLOC_1432429, XLOC_118963) were also listed in top 30 differentially expressed genes in both hybrids after ergot infection (Supplementary Table 1). In cell wall modification pathway, three DEGs named “XLOC_1343481”, “XLOC_1343482” and “XLOC_145869” were found to be involved in Polygalacturonase. These results prompted us to dissect the orthologs of these genes in T. aestivum in order to identify their expression and behavior under various conditions using publicly available transcriptome data.

Comparison and Gene Ontology enrichment analysis of differentially expressed genes. (A) Counts of DEGs identified in Helltop and DH372 after ergot infection (Helltop = DEGs identified within Helltop; DH372 = DEGs identified within DH372 after ergot infection (B) Significant gene enriched terms found in DEGs identified in both hybrids after ergot infection.

Gene Set Enrichment Analysis (GSEA) of DEGs to identify pathways in Biological processes (A) and Molecular functions (MF). Pectin catabolic process and cell wall modification were significant pathway in GESA in biological process and pectinesterase activity and Rho GTPase binding were significant pathways in molecular function.
Expression profile and behavior of wheat orthologs
The majority of wheat orthologs have very similar expression pattern with higher expression during flower initiation stage (Supplementary Fig. 3A). In the quest to understand the function of these genes, we found out that majority of the genes in wheat were not highly expressed under biotic stress category. Orthologues of COBRA-like 10 (TraesCS7D02G136500), probable pectinesterase inhibitor 21 (TraesCS2D02G091800) and Polygalacturonase (TraesCS5B02G468900, TraesCS5A02G459000 and TraesCS1B02G274500) did not exhibit differential expression under various biotic stresses. Notably, orthologues of “XLOC_386424” (TraesCS1B02G274500) had significant up-regulation under various biotic stress (Supplementary Fig. 3B) and orthologs of “XLOC_149812” (TraesCS6A02G351000) exhibit down-regulation after F. graminearum infection.
Discussion
Ergot infections of rye is a severe problem of food security. Although several studies have investigated the infection mechanisms by which C. purpurea penetrate spikes, not much is known about the molecular mechanisms of the infection. One obvious reason is the lack of sequence resources for rye during ergot infection, which impose serious limitations to studies the molecular mechanism. In this study, we generated transcriptomic resources, and created two de novo assemblies of rye hybrids with known differential responses to ergot infection. These high-quality transcriptome assemblies may constitute an important step forward for understanding of this highly complex disease and complements the resources recently made available for other fungal diseases in Triticeae36,37,38. In a previous transcriptome study, de novo assembly had 115,400 contigs arising from five rye inbreeding lines using Roche/454 GS FLX technology39. However, no functional annotation of the aforementioned transcriptome was available for comparison. In this study, we had used Illumina technology to develop transcriptome assemblies of DH372 (ergot susceptible) and Helltop (ergot moderate resistant) during ergot infection and provided annotations (Supplementary Fig. 2 and Fig. 3). Moreover, we provided a comprehensive transcriptome analysis pipeline to deal with vast amount of data due to huge genome size of rye that suited well to decipher molecular mechanism during ergot infection. We employed an assembler, which produces good quality assembly at single k-mer. Numerous methods were applied to assess the overall quality, accuracy, contiguity, and completeness of a de novo assembled transcriptome. BUSCO assessments revealed that the combined transcriptome assemblies have good representation and can serve purpose of perfect de novo assembled transcriptome of rye spikes (Table 2). However, different levels of BUSCO completeness were observed when comparing different individuals of the same hybrids. Thus, indicating that hybrid specific transcripts may account for different coverages in various individual transcriptome assemblies. The quality assessments using the pRNAseq-2-CTA also revealed that the individual assemblies are well represented in the respective combined assembly of DH372 and Helltop. Nonetheless, BUSCO completeness observed in our de novo assemblies were comparable to the completeness of other cereals transcriptomes40,41. In crux, all three assessment methods (BUSCO, pRNAseq-2-OTA and pRNAseq-2-CTA) indicate that de novo assembled transcriptomes are of very high quality and will be very useful addition to the molecular toolbox of rye.
Lack of a well annotated rye reference genome and the differential response of hybrids towards ergot prompted us to perform functional annotations on the combined transcriptome assemblies of DH372 and Helltop. In the combined transcriptome assemblies, majority of contigs got top hit to Triticeae, which is in line with previous high quality de novo assemblies of wheat, barley and other cereals42,43,44,45. We also observed that a number of contigs in the de novo assembly of Helltop that got hit to Sorghum bicolor and Eragrostis curvula. This led us to do GO of both assemblies for the unigenes exclusive to each hybrid. Pronounced differences were observed as in case of Helltop genes associated with proteolysis, biosynthetic process, ubiquitin dependent protein catabolic process, cell wall organization, defense response and recognition of pollen, whereas genes exclusive to DH372 associated with oxidation reduction process, cellular oxidant detoxification, oxidative stress and hydrogen peroxide catabolic process (Fig. 4). Similarly, we observed major differences in drug metabolism pathway and pentose and glucoronate interconversions pathways (Fig. 5). Drug metabolism constitutes genes that belonging to the family of Cytochrome P450s, GSTs, glycosyl transferases and ABC transporters. These genes are known to play a role in plant-pathogen interactions in various plant species. Hence, this is in agreement with the notion that plant defense depends on metabolism of pathogenesis related proteins at pathogen contact site46,47,48,49. Another predominant pathway was pentose and glucoronate interconversions and this pathway is directly related to the target prediction of UDP-glucuronosyltransferase that can increase the strength of cell walls eventually leading to resistance to pathogens50. The enrichment analysis of DEGs revealed that DEGs were associated with carbohydrate metabolic processes, hydrolase activity, pectinesterase activity, cell wall modification, pollen development and pollen wall assembly. Similarly, gene set enrichment analysis (GSEA) lead us to identify cell wall modification and pectinesterase activity as candidate pathways for ergot resistance. These results are very interesting and in line with earlier findings that inhibition of pectin may be a possible route to control ergot in cereals51. Pectinesterase activity is known to enhance resistance against different pathogens in pepper52, wheat53, Arabidopsis thaliana54,55 , cotton56 and pearl millet57. Therefore, our transcriptome analysis is very useful to formulate hypothesis that can bring further understanding of underlying genetics of ergot resistance. The identified pathways in our transcriptome analysis are interlinked with each other and hence provide a starting point to explore the genes that cause resistance. In our analysis, we found six genes that are common between two pathways (Pectinesterase activity and cell wall modification). In earlier studies, polygalacturonase is known as a pathogenicity factor in the C. purpurea/rye interaction58. In cell wall modification pathway, three genes were differentially expressed (Table 4). The esterification through pectinesterase activity affect the cell wall structure leading to cell wall modification and ultimately resistance against pathogens59. Cell wall-associated plant defense is critical in basal resistance against fungal pathogens60. Plant cell walls are the first essential component of defense and is believed to be a critical factor in plant response to fungal infection by providing a physical impediment between pathogens and the internal contents of the plant cells61,62. Based on our transcriptome results, we assumed that rye plants initiate several mechanisms in response to ergot infection and findings of this study provide a good basis to design future approaches to unravel those mechanism and gain a deeper understanding.
Although RNA sequencing is a highly efficient method, false positives still occur because of the sensitivity of this technology. Hence, we had used reference as well as de novo assembled transcriptome datasets to count the DEGs and discussed only those that were found in both (Table 3). The majority of RNAseq experiments involved estimating transcript expression level via read counts, this results in several specific challenges that must be addressed. The abundance of transcripts due to huge genome size of rye makes it likely that transcripts belonging to parts of different genes will assemble, making the measurement of a gene’s transcript counts prone to large errors. Similarly, in case of using only a draft genome, many short reads, particularly those from Illumina-based RNA-seq experiments, are likely to align to identical sequence fragments at several loci and thus may be discarded from transcript counts affecting the estimated expression level for a given gene. The current study utilizes a draft reference genome as well as de novo assembled transcriptome for quantification of gene expression and we obtained comparable expression profile using both approaches. Hence, we believe that the results obtained using this approach are more reliable than using only a draft genome or a de novo assembled transcriptome. Therefore, we assume that the pipeline devised for this RNA-seq experiment in this study for rye are applicable and valuable to most plant RNA-seq experiments having huge genome size. However, we cannot rule out false positive completely as problems of transcriptional responses not always happening at the exact time they are measured or being highly tissue specific leading to "averaging" when bulk tissues are sampled. In future, this issue can be addressed through single cell transcriptome studies at multiple time points after ergot infection.
Among the genes in enriched pathways, The COBRA-like 10 (XLOC_1432429) was one of the top most expressed gene and known to mediate directional growth of pollen tubes in Arabidopsis thaliana63. It is common notion that C. purpurea pathogen cause fungal infection through mimicking pollen-tube growth64,65. Knockout of the COBRA-like 10 gene caused gametophytic male sterility63. Based on available information regarding COBRA-like 10, we can speculate that ergot conidia might not be able to infect the stigmas of flowers in presence of higher expression of this gene. However, at this point its mere speculation and it will be an interesting area to explore further, which might lead to discovery of underlying mechanism of infection and ergot resistance in rye. Moreover, pollen development and pollen wall assembly are significantly enriched terms in our GO term enrichment analysis. The infection process of C. purpurea is tissue specific as only flowers of blossoming rye spikes are infected. Probably, it requires specific recognition of the stigmatic surface. However, experimental proof of which specific gene or genes are missing. Early stages of infection show striking parallels to the plant fertilization process, and especially growth of pollen tubes in the pistil tissue6. The fungus evidently mimics pollen tube growth and might use specific signal components of the pollen–stigma interaction to avoid recognition by the plant6. Nevertheless, all these facts point out a potential role of efficient communication between female tissues and growing pollen tubes during ergot infection. Consequently, candidate genes identified in this study could be valuable assets for future studies.
The expression profiling of wheat orthologues of these genes revealed that they were highly expressed at flower initiation stage. This is again pointing towards significance of flower specificity in ergot infection and a probable role of these genes in ergot resistance during flower development stage. However, under various biotic stresses, majority of these genes remains unaffected and did not exhibit significant differential expression in wheat. This might be due to specific involvement of these genes during ergot infection and resistance in rye but not in wheat. Wheat is a self-fertile plant and flowers are closed, which reduce the risk for ergot infection. Wheat might have different mechanism of ergot infection and resistance due to specific self-pollination strategy and therefor have a different set of candidate genes than those found in rye in this study. There is also lack of expression data during ergot infection in wheat and which could be one reason for non-significant expression in publicly available transcriptome data of wheat. Focusing on gene families whose functional annotation is relevant to the mechanism of ergot infection and phenotype, we dissect their differential expression patterns and identify candidates for the genetic basis of the observed differences. Overall in this study, we provided a strong evidence of involvement of specific set of genes in ergot infection mechanism. We cannot say for certain that these DEGs are related to ergot resistance. However, this group of genes remain interesting candidate genes for future studies such as CRISPR-Cas to validate their function in ergot resistance. Functional annotations of two de novo assemblies along with enrichments analysis of DEGs emphasize the role of cell wall and flower morphology to evolve resistance against ergot in cross pollinated rye.
Conclusion
Our transcriptomic exploration demonstrates, how ergot influence transcript expression in two hybrids, which has allowed us to identify and annotate transcripts associated with ergot infection. The resource generated in this study will help to lay the foundation to understand the precise nature and origin of ergot infection in rye. This is an indispensable pre-requisite for the development of gene-based strategies that can exploit untapped genetic diversity in breeding materials and ex situ gene banks to improve resistance in rye hybrids. The identification of pathways and set of candidate genes is an important forward step toward a better understanding of the complex mechanism that can be utilized to devise novel strategies of ergot management in the future. In crux, availability of additional transcriptome sets from rye hybrids under specific conditions as presented here will serve as valuable resources for rye breeders and to the research community for further functional and comparative genomics studies.
References
Geiger, H. & Miedaner, T. Rye breeding. Cereals3, 157–181 (2009).
Bushuk, W. Rye: Production, Chemistry, and Technology (American Association of Cereal Chemists, Saint Paul, 2001).
Bauer, E. et al. Towards a whole-genome sequence for rye (Secale cereale L.). Plant J.89, 853–869 (2017).
Tudzynski, P., Correia, T. & Keller, U. Biotechnology and genetics of ergot alkaloids. Appl. Microbiol. Biotechnol.57, 593–605 (2001).
Streit, E. et al. Current situation of mycotoxin contamination and co-occurrence in animal feed—focus on Europe. Toxins4, 788–809 (2012).
Tudzynski, P. & Scheffer, J. Claviceps purpurea: molecular aspects of a unique pathogenic lifestyle. Mol. Plant Pathol.5, 377–388 (2004).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet.10, 57–63 (2009).
Brown, J. W., Calixto, C. P. & Zhang, R. High-quality reference transcript datasets hold the key to transcript-specific RNA-sequencing analysis in plants. New Phytol.213, 525–530 (2017).
Watson-Lazowski, A. et al. Plant adaptation or acclimation to rising CO2? Insight from first multigenerational RNA-Seq transcriptome. Glob. Change Biol.22, 3760–3773 (2016).
O’Rourke, J. A. et al. An RNA-Seq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants. Plant Physiol.161, 705–724 (2013).
Chakraborty, S., Britton, M., Martínez-García, P. & Dandekar, A. M. Deep RNA-Seq profile reveals biodiversity, plant–microbe interactions and a large family of NBS-LRR resistance genes in walnut (Juglans regia) tissues. Amb. Express6, 12 (2016).
Costa, V., Angelini, C., De Feis, I. & Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. BioMed Res. Int. https://doi.org/10.1155/2010/853916 (2010).
McIntyre, L. M. et al. RNA-seq: technical variability and sampling. BMC Genom.12, 293 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics30, 2114–2120 (2014).
Wang, S. & Gribskov, M. Comprehensive evaluation of de novo transcriptome assembly programs and their effects on differential gene expression analysis. Bioinformatics33, 327–333 (2017).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol.29, 644–652 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat. Protoc.8, 1494 (2013).
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. M. & Kelly, S. TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res.26, 1134–1144 (2016).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics28, 3150–3152 (2012).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics31, 3210–3212 (2015).
Mirdita, V. & Miedaner, T. Resistance to Ergot in Self-incompatible Germplasm Resources of Winter Rye. J. Phytopathol.157, 350–355 (2009).
Mirdita, V., Dhillon, B., Geiger, H. & Miedaner, T. Genetic variation for resistance to ergot (Claviceps purpurea [Fr.] Tul.) among full-sib families of five populations of winter rye (Secale cereale L.). Theor. Appl. Genet.118, 85–90 (2008).
Anna, K. et al. Ergot infection in winter rye hybrids shows differential contribution of male and female genotypes and environment. Euphytica216, 65 (2020).
Lindner, R. & Friedel, C. C. A comprehensive evaluation of alignment algorithms in the context of RNA-seq. PLoS ONE7, e52403 (2012).
Nookaew, I. et al. A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae. Nucleic Acids Res.40, 10084–10097 (2012).
Benjamin, A. M., Nichols, M., Burke, T. W., Ginsburg, G. S. & Lucas, J. E. Comparing reference-based RNA-Seq mapping methods for non-human primate data. BMC Genom.15, 570 (2014).
Engelke, T. Ansätze für eine integrierte Bekämpfung des Mutterkorns (Claviceps purpurea [Fr.] Tul.) im Roggen (Cuvillier, Göttingen, 2002).
Miedaner, T., Mirdita, V., Rodemann, B., Drobeck, T. & Rentel, D. Genetic variation of winter rye cultivars for their ergot (Claviceps purpurea) reaction tested in a field design with minimized interplot interference. Plant Breed.129, 58–62 (2010).
Smart, M. & Roden, L. C. A small-scale RNA isolation protocol useful for high-throughput extractions from recalcitrant plants. S. Afr. J. Bot.76, 375–379 (2010).
30Brown, C. T., Howe, A., Zhang, Q., Pyrkosz, A. B. & Brom, T. H. A reference-free algorithm for computational normalization of shotgun sequencing data. arXiv preprint https://arxiv.org/abs/1203.4802 (2012).
Pevzner, P. A., Tang, H. & Waterman, M. S. An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci.98, 9748–9753 (2001).
Zhao, Q.-Y. et al. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinform.12, S2 (2011).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics21, 3674–3676 (2005).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res.28, 27–30 (2000).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics26, 139–140 (2010).
Biselli, C. et al. Comparative transcriptome profiles of near-isogenic hexaploid wheat lines differing for effective alleles at the 2DL FHB resistance QTL. Frontiers Plant Sci.9, 37 (2018).
Ma, X., Keller, B., McDonald, B. A., Palma-Guerrero, J. & Wicker, T. Comparative transcriptomics reveals how wheat responds to infection by Zymoseptoria tritici. Mol. Plant Microbe Interact.31, 420–431 (2018).
Li, Y. et al. Transcriptome analysis identifies candidate genes and functional pathways controlling the response of two contrasting barley varieties to powdery mildew infection. Int. J. Mol. Sci.21, 151 (2020).
Haseneyer, G. et al. From RNA-seq to large-scale genotyping-genomics resources for rye (Secale cereale L.). BMC Plant Biol.11, 131 (2011).
Hill, C. B. et al. De novo transcriptome assembly and analysis of differentially expressed genes of two barley genotypes reveal root-zone-specific responses to salt exposure. Sci. Rep.6, 31558 (2016).
Liu, Y.-J. et al. Transcriptome analysis of wheat seedling and spike tissues in the hybrid Jingmai 8 uncovered genes involved in heterosis. Planta247, 1307–1321 (2018).
Duan, J., Xia, C., Zhao, G., Jia, J. & Kong, X. Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genom.13, 392 (2012).
Vicente, R. et al. De novo transcriptome analysis of durum wheat flag leaves provides new insights into the regulatory response to elevated CO2 and high temperature. Frontiers Plant Sci.10, 1605 (2019).
Kang, S.-H., Lee, J.-Y., Lee, T.-H., Park, S.-Y. & Kim, C.-K. novo transcriptome assembly of the Chinese pearl barley, adlay, by full-length isoform and short-read RNA sequencing. PLoS ONE13, e0208344 (2018).
Fox, S. E. et al. De novo transcriptome assembly and analyses of gene expression during photomorphogenesis in diploid wheat Triticum monococcum. PLoS ONE9, e96855 (2014).
Gullner, G., Komives, T., Király, L. & Schröder, P. Glutathione S-transferase enzymes in plant-pathogen interactions. Frontiers in plant science9, 1836 (2018).
Bienert, M. D., Baijot, A. & Boutry, M. Plant ABC Transporters 137–162 (Springer, Berlin, 2014).
Xin, M. et al. Cloning and characterization of a novel UDP-glycosyltransferase gene induced by DON from wheat. J. Integr. Agric.14, 830–838 (2015).
Oh, B.-J. et al. A cytochrome P450 gene is differentially expressed in compatible and incompatible interactions between pepper (Capsicum annuum) and the anthracnose fungus Colletotrichum gloeosporioides. Mol. Plant-Microbe Interact.12, 1044–1052 (1999).
Silveira, R. L., Stoyanov, S. R., Gusarov, S., Skaf, M. S. & Kovalenko, A. Plant biomass recalcitrance: effect of hemicellulose composition on nanoscale forces that control cell wall strength. J. Am. Chem. Soc.135, 19048–19051 (2013).
Volpi, C. et al. Claviceps purpurea expressing polygalacturonases escaping PGIP inhibition fully infects PvPGIP2 wheat transgenic plants but its infection is delayed in wheat transgenic plants with increased level of pectin methyl esterification. Plant Physiol. Biochem.73, 294–301 (2013).
An, S. H. et al. Pepper pectin methylesterase inhibitor protein CaPMEI1 is required for antifungal activity, basal disease resistance and abiotic stress tolerance. Planta228, 61–78 (2008).
Volpi, C. et al. The ectopic expression of a pectin methyl esterase inhibitor increases pectin methyl esterification and limits fungal diseases in wheat. Mol. Plant Microbe Interact.24, 1012–1019 (2011).
Lionetti, V. et al. Three pectin methylesterase inhibitors protect cell wall integrity for Arabidopsis immunity to Botrytis. Plant Physiol.173, 1844–1863 (2017).
Lionetti, V. et al. Overexpression of pectin methylesterase inhibitors in Arabidopsis restricts fungal infection by Botrytis cinerea. Plant Physiol.143, 1871–1880 (2007).
Tundo, S. et al. Pyramiding PvPGIP2 and TAXI-III but not PvPGIP2 and PMEI enhances resistance against Fusarium graminearum. Mol. Plant Microbe Interact.29, 629–639 (2016).
Prabhu, S. A. et al. Immuno-affinity purification of PglPGIP1, a polygalacturonase-inhibitor protein from pearl millet: studies on its inhibition of fungal polygalacturonases and role in resistance against the downy mildew pathogen. Mol. Biol. Rep.42, 1123–1138 (2015).
Oeser, B., Heidrich, P. M., Müller, U., Tudzynski, P. & Tenberge, K. B. Polygalacturonase is a pathogenicity factor in the Claviceps purpurea/rye interaction. Fungal Genet. Biol.36, 176–186 (2002).
Lionetti, V., Cervone, F. & Bellincampi, D. Methyl esterification of pectin plays a role during plant–pathogen interactions and affects plant resistance to diseases. J. Plant Physiol.169, 1623–1630 (2012).
Malinovsky, F. G., Fangel, J. U. & Willats, W. G. The role of the cell wall in plant immunity. Frontiers Plant Sci.5, 178 (2014).
Bacete, L., Mélida, H., Miedes, E. & Molina, A. Plant cell wall-mediated immunity: cell wall changes trigger disease resistance responses. Plant J.93, 614–636 (2018).
Hückelhoven, R. Cell wall–associated mechanisms of disease resistance and susceptibility. Annu. Rev. Phytopathol.45, 101–127 (2007).
Li, S. et al. Arabidopsis COBRA-LIKE 10, a GPI-anchored protein, mediates directional growth of pollen tubes. Plant J.74, 486–497 (2013).
Tudzynski, P., Tenberge, K. & Oeser, B. Claviceps purpurea. Pathog. Host Specif. Plant Dis. Histopathol. Biochem. Genet. Mol. Bases2, 161–187 (1995).
Miedaner, T. & Geiger, H. H. Biology, genetics, and management of ergot (Claviceps spp.) in rye, sorghum, and pearl millet. Toxins7, 659–678 (2015).
Acknowledgements
This work was funded by Innovation Fund Denmark; 7039-00016B and Pajbjerg Foundation. The authors thankful to Torben Asp and Istvan Nagy for allowing access to FLseq cluster and advice during bioinformatics analysis. We are also thankful to Hanne-Birgitte Christiansen for the assistance in preparation of inoculum of C. purpurea.
Author information
Authors and Affiliations
Contributions
K.M. performed the experiment and did bioinformatics analysis. A.J., J.O. and L.N.J. supervised the project. K.M., J.O., A.J., P.S.K. and L.N.J. designed the study. K.M., P.S., J.O., P.S.K., L.N.J. and A.J. participated in writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mahmood, K., Orabi, J., Kristensen, P.S. et al. De novo transcriptome assembly, functional annotation, and expression profiling of rye (Secale cereale L.) hybrids inoculated with ergot (Claviceps purpurea). Sci Rep 10, 13475 (2020). https://doi.org/10.1038/s41598-020-70406-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70406-2
This article is cited by
-
Leaf rust (Puccinia recondita f. sp. secalis) triggers substantial changes in rye (Secale cereale L.) at the transcriptome and metabolome levels
BMC Plant Biology (2024)
-
Comparative transcriptomics analysis pipeline for the meta-analysis of phylogenetically divergent datasets (CoRMAP)
BMC Bioinformatics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
