
Abstract
The aims of this study were to explore whether DNA methylation at INSR and IGF2 mediated the association of prenatal exposure to the Chinese great famine with adulthood waist circumference (WC) and BMI. A total of 235 subjects were selected into the present study from severely affected province and a neighbor province with less severely affected famine in China through multi-stage clustered random sampling. DNA methylation at the INSR and IGF2 gene promoter regions was detected by the Sequenom’s MassARRAY system. The “mediation” package of R was used to evaluate the mediation effect of DNA methylation on the association between prenatal exposure to the famine and adult WC and BMI. The results showed that prenatal famine exposure was significantly associated with higher overall methylation level of the INSR gene (d = 3.6%; 95% CI 1.2–6.0; P = 0.027) and larger adulthood WC (d = 2.72 cm; 95% CI 0.20–5.24; P = 0.034). Furthermore, famine significantly increased methylation levels at four CpG sites. Methylation of the CpG7 site mediated 32.0% (95% CI 5.0–100.0%, P = 0.029) of the association between prenatal exposure to the Chinese great famine and adulthood WC. In conclusion, Epigenetic changes to the INSR might mediate the adverse effect of prenatal famine exposure on WC in adulthood.
Similar content being viewed by others

Introduction
Obesity is a major risk factor for cardiometabolic disorders including cardiovascular disease, stroke, and diabetes1,2. According to the World Health Organization, over 600 million adults are obese worldwide3. In China, more than 38% of the total population were overweight or obese in 2011, which translates to 24.35 billion Yuan annual cost4.
Over the past four decades, epidemiological studies have observed that prenatal exposure to adverse environment was associated with higher risk of diseases in later life5. Epigenetic changes such as DNA methylation induced by the prenatal adverse environmental exposure might be a potential mechanism underlying the association6. DNA methylation features affect the transcription of gene(s) in nearby regions. Once changed, the methylation features can persist and lead to long term consequences7. Animal studies found that maternal exposure to malnutrition during gestation modified DNA methylation of certain genomic regions in offspring, and the methylation was subsequently associated with poor phenotypes in later life. Furthermore, the changes in DNA methylation level even continued to the third generation8,9,10. In human, prenatal exposure to the six months’ Dutch famine was associated with changes of DNA methylation features in genes, including INSR, CPT1A, and IGF2, which are involved in growth and metabolism11,12. A recent genome-wide epigenetic study of the Dutch famine reported that DNA methylation mediated associations between prenatal exposure to the famine and body mass index (BMI) in adulthood13. However, these findings are only from European, lacking replication in other races.
As the largest famine that has been well documented in human history, the 1959–1961 Chinese great famine was featured by its long duration (3 years) and extreme severity. As reported in our previous publication, 11.6% of the middle-aged and older Chinese adult had immediate family member(s) starved to death during 1959–196114. The Chinese great famine provided an opportunity to explore the mediation effect of DNA methylation in the association between prenatal famine exposure and adverse phenotypes in later life. Previously, we have reported that early-life severe famine exposure might increase methylation level in the IGF2 and INSR genes, and the change in the IGF2 gene was also linked with higher level of total cholesterol in adulthood15,16.
Waist circumference (WC) reflects central obesity, and has been suggested to be superior to body mass index (BMI), particularly among Asian populations, in predicting cardiovascular disease risk17. Thus, WC may be a better indicator to reflect the effect of early-life famine exposure on adult risks of non-communicable chronic disease. In this context, we explore the impact of the Chinese great famine on central obesity and the potential mediation effect of gene methylation level on the famine-obesity association among participants of the Genomic Research of the Chinese Famine (GRECF) study.
Results
Characteristics of study participants are presented in Table 1. A total of 75 participants had prenatal famine exposure, and 160 were not exposed to famine during gestation. Mean age of the non-exposed group was similar to the prenatal famine exposed group (P = 0.149). Compared with the non-exposed group, participants in the famine exposed group had a higher waist circumference (85.86 vs. 82.97 cm, P = 0.029). However, we did not observe significant differences in BMI, physical activity, smoking, drinking, or frequencies of meat, vegetable, fruit, and milk consumption between the two groups.
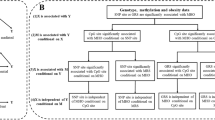
Multiple adjusted associations between prenatal famine exposure, DNA methylation, and waist circumference and BMI in adulthood were presented in Tables 2 and 3. Participants with prenatal famine exposure had 2.72 cm [95% confidence interval (CI) 0.20–5.24, P = 0.034] longer waist circumference compared to those without famine exposure after adjustment of gender, province, smoking, drinking, physical activity, and frequencies of meat, vegetable, fruit, and milk consumption. The effect of prenatal famine exposure on adulthood waist circumference is similar between men and women (Table S1). Meanwhile, prenatal famine exposure was positively associated with the overall DNA methylation level and that at four CpG sites (CpG1, CpG4, CpG5, and CpG7) in the INSR gene. Prenatal famine exposure had the greatest impact at the CpG7 site, with the exposed group having 11.2% (95% CI 5.5–16.9%, P = 1.4 × 10−3) higher methylation level compared to those without famine exposure. DNA methylation at the CpG7 site was also positively associated with waist circumference (Table 3: β = 10.88, 95% CI 1.82–19.94, P = 0.020). Mediation analysis demonstrated that the mediation path through the CpG7 explained 32.0% (95% CI 5.0–100.0%, P = 0.029) of the association between prenatal famine exposure and waist circumference in adulthood (Fig. 1). None of the other CpG sites both in INSR and IGF2 was associated with waist circumference and BMI (Table 3). In addition, the overall DNA methylation differences between prenatal famine exposure group and non-exposed group were larger in the more severely affected province than that in the less affected one (Table S2).

The mediation analysis of DNA methylation at CpG 7 in association between prenatal famine exposure and adult waist circumference. Path (a) was defined as the effect of the independent variable (prenatal famine exposure) on the mediator (DNA methylation of CpG 7 at INSR). Path (b) was the effect of the mediator on the dependent variable by controlling the effect of the prenatal famine exposure. Path (c) was the total effect of the independent variable on the dependent variable. Path (c′) shows the direct effect from independent variable to dependent variable. All the analysis adjusted for bisulfite batch, province, gender, physical activity level, the frequencies of meat, vegetables, fruit, milk, smoking and alcohol use.
Discussion
We found that DNA methylation in INSR might mediate the association between prenatal exposure to the Chinese great famine and waist circumference in adulthood. To our knowledge, epigenetic study of the Chinese great famine and waist circumference in later life is limited. Our findings suggest that maternal malnutrition during gestation may permanently change DNA methylation among offspring and link with the offspring’s waist circumference in adulthood. Which provided further evidence for hypotheses that epigenetic factors mediated the associations between exposure to an adverse environment during early development and adulthood health outcomes. Indicating that adequate nutrition supply to pregnant women before and during pregnancy seems a promising strategy to prevent central obesity in the next generation.
In the current study, we observed that individuals who were exposed to the Chinese great famine in fetal stage had a higher waist circumference in later life than those who did not experience the famine. The finding is similar to that of a study among 2,414 people born around the Dutch famine, in which, prenatal famine exposure increased waist circumference at age 50 in women but not in men18. Further analysis stratified by sex in the current study demonstrated that there was no statistically significant difference between men and women in the famine-waist circumference association. This indicates that the impact of short- and long-term famine exposure during fetal stage may differ in men. It is possible that the current study cannot detect gender difference due to smaller sample size and hence lower statistical power. However, the Chinese great famine lasted much longer than the Dutch famine, and the majority participants of the current study were recruited from Anhui province, which experienced the most severe famine during 1959–1961. The impact of famine should be more easily detected. Future large-scale studies are warranted to explore gender differences in the Chinese great famine’s impact on health conditions.
A significant association between prenatal exposure to the Chinese great famine and DNA methylation in INSR gene was found in the present study. Compared with the non-exposed group, those with prenatal exposure to the Chinese great famine had an average of 5.0% higher methylation level in INSR. This effect is larger than that reported in the Dutch famine study in similar DNA region, in which prenatal exposure to 6 months’ famine was associated with 3.8% higher methylation level in the INSR gene12. Again, this difference may be attributed to the much longer duration and more extreme severity of the Chinese great famine. In China, there are also regions with various severity of famine exposure during the 1959–1961. Future studies exploring a dose–response relationship between famine severity and DNA methylation level in the INSR and other genes are warranted.
The current study also identified that DNA methylation at the CpG7 site of the INSR gene was positively associated with waist circumference. In the Dutch famine study, prenatal malnutrition-associated DNA methylation regions in the INSR was correlated with birth weight, but not waist circumference12. Given the larger effect of the Chinese great famine on INSR gene methylation, it is not surprising to detect such association between famine-associated methylation and adult waist circumference in our study. Although further studies are needed to validate the finding in an independent Chinese sample, our finding may have a biological relevance. The CpG 7 unit lies in the promoter region of INSR, previous study had found that methylation of this unit may have reduced the expression of INSR12. The INSR encodes a member of the receptor tyrosine kinase family of proteins, which is processed to generate alpha and beta subunits of a heterotetrametric receptor. This receptor binds insulin or other ligands to activate the insulin signaling pathway, and subsequently regulates glucose uptake and release and is involved in lipids synthesis and storage. Loss-of-function mutations in this gene cause severe insulin resistance syndromes manifested by several studies19,20.
Finally, differential methylation at the CpG 7 unit of INSR associated with famine exposure contributed to a fairly large proportion (32.0%) of the association between prenatal famine exposure and adult waist circumference. Still, a large proportion of the famine-waist circumference association remains unexplained. Future genome-wide epigenetic studies are warranted to identify more methylation regions underlying such association.
Our study represents epigenetic research on the prenatal exposure to the Chinese great famine and central obesity in adulthood. The Chinese great famine lasted for 3 years and affected the entire main land China. It provides an opportunity to study the impact of long-term and severe malnutrition on epigenetic changes in human genome. In addition, a representative sample of residents from a famine-stricken region and a region with moderate famine exposure has been used. This enhanced the generalizability of our findings. Several limitations should be mentioned. Firstly, the percentage of mediation reported in the current study was estimated by the statistical model, which might overestimate the real effect of DNA methylation changes on WC. Secondly, DNA methylation data was measured from peripheral whole blood, which might be not the most relevant tissues to study for waist circumference. However, previous DNA methylation data from multiple tissues from the same donors showed a broad agreement in DNA methylation patterns of the CpG units in whole blood and various fat deposits21,22. Thirdly, the current study only selected a candidate locus, and did not conduct a genome-wide DNA methylation difference study, nor did perform a blood cell count, which limited the interpretation of the data. Forth, the statistically significant mediation observed in the current study did not clarify the mechanism, but it does provide possible pathways to explore in future. Fifth, age difference between prenatal famine exposure and non-exposed groups might be another important limitation in the current study. Due to the independent variable (famine-exposed/non-exposed groups) was defined by participants’ birthdates, which determines the age of participants. As a result, the ages of participants with prenatal famine exposure did not overlap the ages of participants without prenatal famine exposure. However, in the current study, we combined the pre-famine exposed group and post-famine exposed group as the non-exposed group. Thus, the mean age of exposed group and non-exposed group was similar. Sixth, the sample size is limited, and the level of statistical evidence is moderate. Thus, findings from the current study need to replicate in another Chinese famine cohort. Additionally, the information of smoking status was raw in the current study, which could not control totally the effect of smoking status on the association.
Methods
Study participants
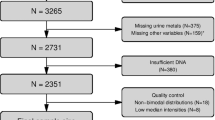
The GRECF study was designed to investigate the impact of the Chinese great famine on the human genome and its subsequent impact on chronic conditions in later adulthood. The study has been introduced in details in a previous publication15. Briefly, the GRECF study recruited 790 participants from two neighboring provinces, Anhui and Jiangxi, in China through multi-stage clustered random sampling. Famine was most intense in Anhui province, while Jiangxi province was moderately affected23, 24. A random sample of 264 subjects were selected for DNA methylation assay. After excluding 29 subjects with extreme values of the mean DNA methylation level (> 3 standard deviation), 235 subjects were enrolled into the final analysis.
Famine exposure
Food shortage gradually occurred in late 195825. For the purpose of this study, we selected the start date of the Chinese great famine as January 1st, 1959 and the end date as December 31st, 196114. Thus, subjects who were born between October 1st, 1959 and September 30th, 1961 were included as the prenatal famine exposed group, those who born between October 1st, 1962 and September 30th, 1964 were defined as post-famine exposed group, and those who born between January 1st, 1958 and December 31st, 1958 were categorized as the pre-famine exposed group. We combined the pre-famine exposed group and post-famine exposed group as the non-exposed group. Due to the Chinese great famine lasted from autumn of 1958 to autumn of 1962, especially during 1959–1961, subjects who born between October 1st, 1961 and September 30th, 1962 were excluded from the current study because they might also exposure to famine during second or third trimester of pregnancy26.
Waist circumference, BMI and covariates
Waist circumference was measured to the nearest 0.1 cm by trained staff using a tape at the level of the umbilicus. Waist circumference was measured twice for each participant, and the average of the two measurements was used for the current analysis. BMI was calculated by dividing weight in kilograms by square of the height in meters (kg/m2).
Age was obtained from the National Resident Registration System. Other demographic and health behavioral information, including gender, smoking, and drinking, were collected based on self-report. Smoking status was categorized as ‘smoking less than 400 cigarettes’ or ‘smoking ≥ 400 cigarettes’ in the past year. Drinking status was classified as ‘current drinking’ and ‘not current drinking’. Physical activity was measured by the International Physical Activity Questionnaire Short Form (IPAQ-SF), and participants were categorized as having ‘vigorous’ or ‘non-vigorous’ physical activity27. Dietary intake of meat, vegetable, fruit, and milk was collected by the Chinese Food Frequency Questionnaire28. The intake frequency was divided into ‘< 1 time/day’ and ‘≥ 1 times/day’ for each food item.
DNA methylation of the INSR and IGF2 genes
Genomic DNA was extracted from peripheral white blood cells using the salting-out method29. The EZ 96- methylation kit (Zymo Research) was used to bisulfite converted DNA. The conversion rate is greater than 99%. The Sequenom’s MassARRAY system (Sequenom, San Diego, CA) was used to quantify DNA methylation levels at CpG sites of the INSR and IGF2 genes using the manufacturers’ protocol on a 384-well plate. Primers of the INSR and IGF2 genes were designed using the Epidesigner online application (https://epidesigner.com/start3.html). Details of the primers and the CpG sites are presented in supplementary Tables S3 and S4.
DNA methylation of the INSR and IGF2 genes in the current study was measured by the EpiTYPER, which is a mass spectrometry-based bisulfite sequencing method that enables region-specific DNA methylation analysis in a quantitative and high-throughput fashion30. We totally followed the protocol of EpiTYPER to perform the measure the methylation of INSR and IGF2 genes. Sequenom’s EpiTYPER protocol includes the method and procedure of methylation data measurement31. To control the quality of measurement, we included a positive control (100% methylated) and a negative control (0%-methylated) for each run. All the samples were assayed in triplicate, if the triplicate methylation measurements had an extreme value ≥ 3 standard deviation, all data for the sample involved were discarded. The call rates over 85% for all CpG units. Among the 9 CpG sites at INSR, 7 CpG sites were measured individually, and two sites (CpG1 and CpG4) were combined and measured, because their fragment masses were so similar that they could not be distinguished. Among the 8 CpG sites at IGF2, 2 CpG sites (CpG6 and CpG7) were measured simultaneously because they could not be resolved due to their close proximity, and the others were measured individually. The overall methylation level is the arithmetic average of all CpG sites.
Statistical analysis
The SPSS 20.0 and R software were used to perform statistical analyses. Continuous variables are presented as means ± standard deviations, and categorical variables are presented as percentages. Comparisons between famine exposed and non-exposed groups were conducted by Chi-square tests for categorical variables and two-independent samples t-tests for continuous variables. The normality test showed in Table S5.
Association between famine exposure and waist circumference was assessed by a multivariate linear regression model. A linear mix effect model was used to evaluate the association of DNA methylation with famine exposure, and the bisulfite patch was treated as a random effect in the model. Such analysis was performed for the overall methylation level and the methylation level at each CpG site. A Bonferroni corrected p value of 5.56 × 10−3 (correcting for 9 tests: 0.05/9 = 5.56 × 10−3) was used to determine significant CpG sites associated with prenatal famine exposure. Significant CpG site was further tested for association with waist circumference using the linear mix effect model. The “mediation” package of R software was used to analyze the mediation effect of DNA methylation at the significant CpG sites on the association between prenatal famine exposure and waist circumference in adulthood32. All the regression analyses controlled for gender, province, smoking, drinking, physical activity, and frequencies of meat, vegetable, fruit, and milk consumption. The detailed procedures for mediation model were listed below:
A power analysis was performed to assess the statistical power for examining the difference of DNA methylation between the prenatal famine exposed group and non-exposed group. The mean methylation level of INSR was 0.434 [standard deviation (SD) = 0.092) in the non-exposed group and 0.467 (SD = 0.070) in the prenatal exposed group. At a two-tailed alpha of 0.05, we had 85.5% power to detect the difference.
Ethics statement
This study was approved by the Institute Review Board at the Peking University Health Science Center. Informed consent was obtained from all study participants. We confirmed that all methods were performed in accordance with relevant guidelines and regulations.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Finegood, D. T. Obesity, inflammation and type II diabetes. Int. J. Obes. Relat. Metab. Disord. 27(Suppl 3), S4-5. https://doi.org/10.1038/sj.ijo.0802490 (2003).
Caterson, I. D. et al. Prevention conference VII: Obesity, a worldwide epidemic related to heart disease and stroke: Group III: worldwide comorbidities of obesity. Circulation 110, e476-483. https://doi.org/10.1161/01.cir.0000140114.83145.59 (2004).
Mancini, M. C. & de Melo, M. E. The burden of obesity in the current world and the new treatments available: focus on liraglutide 3.0 mg. Diabetol. Metab. Syndr. 9, 44. https://doi.org/10.1186/s13098-017-0242-0 (2017).
Qin, X. & Pan, J. The medical cost attributable to obesity and overweight in China: Estimation based on longitudinal surveys. Health Econ. 25, 1291–1311. https://doi.org/10.1002/hec.3217 (2016).
Barker, D. J. The fetal and infant origins of adult disease. BMJ 301, 1111 (1990).
Bogdarina, I., Haase, A., Langley-Evans, S. & Clark, A. J. Glucocorticoid effects on the programming of AT1b angiotensin receptor gene methylation and expression in the rat. PLoS ONE 5, e9237. https://doi.org/10.1371/journal.pone.0009237 (2010).
Jaenisch, R. & Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl), 245–254. https://doi.org/10.1038/ng1089 (2003).
Morgan, H. D., Sutherland, H. G., Martin, D. I. & Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 23, 314–318. https://doi.org/10.1038/15490 (1999).
Burdge, G. C. et al. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br. J. Nutr. 97, 435–439. https://doi.org/10.1017/S0007114507352392 (2007).
Lillycrop, K. A. et al. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br. J. Nutr. 97, 1064–1073. https://doi.org/10.1017/S000711450769196X (2007).
Heijmans, B. T. et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci USA 105, 17046–17049. https://doi.org/10.1073/pnas.0806560105 (2008).
Tobi, E. W. et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 5, 5592. https://doi.org/10.1038/ncomms6592 (2014).
Tobi, E. W. et al. DNA methylation as a mediator of the association between prenatal adversity and risk factors for metabolic disease in adulthood. Sci Adv 4, eaao4364. https://doi.org/10.1126/sciadv.aao4364 (2018).
Li, C. et al. Early-life exposure to severe famine and subsequent risk of depressive symptoms in late adulthood: the China Health and Retirement Longitudinal Study. Br. J. Psychiatry 213, 579–586. https://doi.org/10.1192/bjp.2018.116 (2018).
Shen, L. et al. Early-life exposure to severe famine is associated with higher methylation level in the IGF2 gene and higher total cholesterol in late adulthood: The Genomic Research of the Chinese Famine (GRECF) study. Clin. Epigenetics 11, 88. https://doi.org/10.1186/s13148-019-0676-3 (2019).
Wang, Z. et al. Early-life exposure to the chinese famine is associated with higher methylation level in the INSR gene in later adulthood. Sci. Rep. 9, 3354. https://doi.org/10.1038/s41598-019-38596-6 (2019).
Amirabdollahian, F. & Haghighatdoost, F. Anthropometric indicators of adiposity related to body weight and body shape as cardiometabolic risk predictors in British young adults: Superiority of waist-to-height ratio. J. Obes. 2018, 8370304. https://doi.org/10.1155/2018/8370304 (2018).
Roseboom, T., Painter, R. & Rooij, S. D. Prenatal Famine Exposure and Long-Term Consequences for Anthropometry and Adult Health (Springer, New York, 2012).
Hosoe, J. et al. Structural basis and genotype-phenotype correlations of INSR mutations causing severe insulin resistance. Diabetes 66, 2713–2723. https://doi.org/10.2337/db17-0301 (2017).
Takasawa, K. et al. Clinical characteristics of adolescent cases with Type A insulin resistance syndrome caused by heterozygous mutations in the beta-subunit of the insulin receptor (INSR) gene. J. Diabetes https://doi.org/10.1111/1753-0407.12797 (2018).
Relton, C. L. et al. Data resource profile: Accessible resource for integrated epigenomic studies (ARIES). Int. J. Epidemiol. 44, 1181–1190. https://doi.org/10.1093/ije/dyv072 (2015).
Slieker, R. C. et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 6, 26. https://doi.org/10.1186/1756-8935-6-26 (2013).
Huang, C. et al. Malnutrition in early life and adult mental health: evidence from a natural experiment. Soc. Sci. Med. 97, 259–266. https://doi.org/10.1016/j.socscimed.2012.09.051 (2013).
Huang, C., Li, Z., Wang, M. & Martorell, R. Early life exposure to the 1959–1961 Chinese famine has long-term health consequences. J. Nutr. 140, 1874–1878. https://doi.org/10.3945/jn.110.121293 (2010).
Smil, V. China’s great famine: 40 years later. BMJ 319, 1619–1621 (1999).
Wang, Z., Li, C., Yang, Z., Ma, J. & Zou, Z. Fetal and infant exposure to severe Chinese famine increases the risk of adult dyslipidemia: Results from the China health and retirement longitudinal study. BMC Public Health 17, 488. https://doi.org/10.1186/s12889-017-4421-6 (2017).
Craig, C. L. et al. International physical activity questionnaire: 12-country reliability and validity. Med Sci. Sports Exerc. 35, 1381–1395. https://doi.org/10.1249/01.MSS.0000078924.61453.FB (2003).
Zhang, H. et al. Reproducibility and relative validity of a semi-quantitative food frequency questionnaire for Chinese pregnant women. Nutr. J. 14, 56. https://doi.org/10.1186/s12937-015-0044-x (2015).
Chacon-Cortes, D., Haupt, L. M., Lea, R. A. & Griffiths, L. R. Comparison of genomic DNA extraction techniques from whole blood samples: A time, cost and quality evaluation study. Mol. Biol. Rep. 39, 5961–5966. https://doi.org/10.1007/s11033-011-1408-8 (2012).
Suchiman, H. E. et al. Design, measurement and processing of region-specific DNA methylation assays: The mass spectrometry-based method EpiTYPER. Front. Genet. 6, 287. https://doi.org/10.3389/fgene.2015.00287 (2015).
van den Boom, D. & Ehrich, M. Mass spectrometric analysis of cytosine methylation by base-specific cleavage and primer extension methods. Methods Mol. Biol. 507, 207–227. https://doi.org/10.1007/978-1-59745-522-0_16 (2009).
Dustin, T. et al. Mediation: R Package for Causal Mediation Analysis. J. Stat. Softw. 59, 1–38 (2014).
Acknowledgements
We thank all subjects who participated in the current study, staff from the Center for Disease Control and Prevention for their help in physical examination and blood samples collection. We thank Mr. Kevin Spiegel for editing the manuscript. This work was supported by the National Science Foundation of China (NSFC 81402692, 81773454).
Author information
Authors and Affiliations
Contributions
Z.Z. and J.M. designed this study and supervised the data analysis. Z.W., J.S. and Y.L. carried out the methylation measurements. C.L., B.D. and L.S. carefully reviewed this study and supervised the data analysis. All the authors involved in writing the paper and had final approval of the submitted and published version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Z., Song, J., Li, C. et al. DNA methylation of the INSR gene as a mediator of the association between prenatal exposure to famine and adulthood waist circumference. Sci Rep 10, 12212 (2020). https://doi.org/10.1038/s41598-020-69120-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-69120-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
