
Abstract
The antimycotic drug caspofungin is known to alter the cell function of cardiomyocytes and the cilia-bearing cells of the tracheal epithelium. The objective of this study was to investigate the homeostasis of intracellular Ca2+ concentration ([Ca2+]i) after exposure to caspofungin in isolated human tracheal epithelial cells. The [Ca2+]i was measured using the ratiometric fluoroprobe FURA-2 AM. We recorded two groups of epithelial cells with distinct responses to caspofungin exposure, which demonstrated either a rapid transient rise in [Ca2+]i or a sustained elevation of [Ca2+]i. Both patterns of Ca2+ kinetics were still observed when an influx of transmembraneous Ca2+ ions was pharmacologically inhibited. Furthermore, in extracellular buffer solutions without Ca2+ ions, caspofungin exposure still evoked this characteristic rise in [Ca2+]i. To shed light on the origin of the Ca2+ ions responsible for the elevation in [Ca2+]i we investigated the possible intracellular storage of Ca2+ ions. The depletion of mitochondrial Ca2+ stores using 25 µM 2,4-dinitrophenol (DNP) did not prevent the caspofungin-induced rise in [Ca2+]i, which was rapid and transient. However, the application of caffeine (30 mM) to discharge Ca2+ ions that were presumably stored in the endoplasmic reticulum (ER) prior to caspofungin exposure completely inhibited the caspofungin-induced changes in [Ca2+]i levels. When the ER-bound IP3 receptors were blocked by 2-APB (40 µM), we observed a delayed transient rise in [Ca2+]i as a response to the caspofungin. Inhibition of the ryanodine receptors (RyR) using 40 µM ryanodine completely prevented the caspofungin-induced elevation of [Ca2+]i. In summary, caspofungin has been shown to trigger an increase in [Ca2+]i independent from extracellular Ca2+ ions by liberating the Ca2+ ions stored in the ER, mainly via a RyR pathway.
Similar content being viewed by others

Introduction
The mucociliary clearance of lower airways contributes to the removal of debris and pathogens that are trapped in the upper mucus layer from the lower airways. This complex mechanism is driven by cilia-bearing cells of the bronchiolar and tracheal epithelium. The kinocilia of these cells bear over 200 motor proteins as a propelling apparatus, including dynein, the ATP hydrolyzing enzyme1,2. Under basal conditions, cilia beat continuously without external stimulation but can beat faster when necessary. This elevated beating frequency depends on several interdepending signal transduction cascades, including changes in intracellular concentrations of Ca2+ ions ([Ca2+]i)3,4,5,6. However, most signal cascades in these cells depend on at least a transient [Ca2+]i to be activated in order to allow the cilia to beat faster for prolonged periods7,8. Thus, the regulation of [Ca2+]i is a cornerstone for many cellular signal cascades and cell functions. In epithelial cells in the airway, different canonical Ca2+ stores or Ca2+ influx pathways are involved in the regulation of [Ca2+]i. The known intracellular stores are the endoplasmic reticulum (ER) and the mitochondria. The ER can reduce [Ca2+]i via activation of the sarcoplasmic ATPase (SERCA)-carrying Ca2+ ions into the ER. The release of Ca2+ ions from the ER is mainly orchestrated by activating the ryanodine receptors (RyR) or IP3 receptors to enhance [Ca2+]i. There may also be alternative pathways that lead to Ca2+ leakage from the ER; however, these are still unknown. Mitochondria can buffer Ca2+ ions when [Ca2+]i exceeds a threshold of 500 nM and they slowly release Ca2+ into the cytosol when [Ca2+]i falls below the aforementioned threshold9. It is still not known whether further intracellular Ca2+ stores such as lysosomes contribute to the regulation of [Ca2+]i.
Different plasma membrane-bound Ca2+ channels are known as Ca2+ influx pathways, which include transient receptor potential (TRP) channels, store-operated calcium (SOC) channels or voltage-dependent calcium channels that can all enhance [Ca2+]i. Consequently, any disturbance or rise in [Ca2+]i in epithelial airway cells can be achieved by the activation of different Ca2+ pathways or by liberating them from internal stores. Elevated [Ca2+]i directly contribute to altered cell function, e.g., mucociliary clearance or mucus secretion, which can either lead to colonization of the lower airways by pathogens or can lead to improved clearance rates. When treating mycotic infections in critically ill patients, echinocandines represent the established first-line therapy of antimycotic substances including caspofungin. Caspofungin is used under clinical conditions, with a high capability to treat systemic or local Candida spp. infections. In healthy men, a mean maximum serum concentration Cmax = 9.1–11 µM was achieved after a loading dose of 70 mg caspofungin10.
In order to successfully treat mycotic infections in different organs, caspofungin has to reach therapeutic concentrations in many tissues or regions including the liver and the lower airways of the lungs that exceed plasma concentrations11,12. This distribution pattern depends on the physiology of specific organs13. In critically ill patients and in perfused isolated rat hearts, we have previously reported that caspofungin has severe effects on cardio-circulatory function14,15,16. Caused by impaired contractility of cardiomyocytes as a result of changes in [Ca2+]i levels17. It is therefore of interest to determine whether caspofungin may also interact with the homeostasis of [Ca2+]i in airway epithelial cells. [Ca2+]i is part of many cellular signal cascades that are a precondition to the altering of cell functions such as the beat rate of cilia. In order to find out, we investigated whether caspofungin also changes the [Ca2+]i in isolated human tracheal epithelial cells (HTEpC) and tried to elucidate the underlying regulatory mechanism.
We measured [Ca2+]i using the fluoroprobe FURA-2 AM. To determine the amount of Ca2+ ions in the ER, we used Mag-Fluo-4 AM. Pharmacological approaches were used to identify signal pathways that are involved in the regulation of [Ca2+]i. The data gathered by this study should expand our knowledge of the interactions of this antifungal drug in terms of the regulation of [Ca2+]i in airway epithelial cells, which can be considered to be part of the immune system.
Materials and methods
Calcium imaging in isolated epithelial cells
For calcium imaging, we used the human tracheal epithelial cell line (HTEpC, C12644, PromoCell, Heidelberg, Germany). Cells were cultivated in an Airway Epithelial Cell Growth Medium Kit (C-21160) containing the Airway Epithelial Cell Growth Medium Supplement Pack (C-39160, PromoCell, Heidelberg, Germany). The airway epithelial cells were kept in a humidified chamber at 37 °C with air containing 5% CO2. For the Ca2+ imaging, cells were seeded onto laminin-coated coverslips. Dye loading with 2.5 µM FURA-2 AM (Biotium, Fremont, CA, USA) in darkness was performed in a 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer for 30 min at 37 °C. For the contents of the HEPES buffer, see the section on drugs and buffer solutions. After the loading period, the cells were rinsed in fresh HEPES buffer and were transferred to the recording chamber of an upright fluorescence microscope equipped with 20 × immersion lenses (BX50 WI, Olympus, Hamburg, Germany), which contains 2 ml HEPES. The excitation light was provided by a 50 W xenon lamp. The microscope was equipped with a dichromatic excitation longpass mirror (400 nm).
The ratiometric dye, FURA-2 AM, was excited at 340 nm and 380 nm (± 8 nm) when equipped with bandpass excitation filters. The emitted fluorescence was directed through a dichromatic shortpass filter of 560 nm to a bandpass filter of 510 nm.
Imaging of luminal Ca2+ ions in the ER of tracheal epithelial cells
For calcium imaging in the ER, HTE cells were seeded on laminin-coated coverslips. To assess the intraluminal Ca2+ concentration ([Ca2+]ER) within the ER, the cells were loaded with 5 µM of the low-affinity calcium indicator Mag-Fluo-4 AM (Kd = 22 µM, Invitrogen, Paisley, UK) in darkness at 37 °C for 60 min. Afterward, the cells were rinsed in HEPES buffer for 20 min and were then transferred into the recording chamber of the fluorescence microscope equipped with 20 × immersion lenses (BX50 WI, Olympus, Hamburg, Germany), which also contains 2 ml HEPES. Experiments were performed in both Ca2+-containing and Ca2+-free HEPES buffer (for contents see below). As single wavelength indicators are difficult to calibrate, we used normalized Mag-Fluo-4 signals (F/Fo) to express changes in the luminal [Ca2+]ER. Pharmacological agents were applied with a blunt pipette into the recording chamber. The Mag-Fluo-4 was then excited at 490 nm (± 10 nm) and the emitted fluorescence was directed through a dichromatic shortpass filter of 560 nm to a bandpass filter of 510 nm.
Fluorometric ROS measurements in tracheal epithelial cells under exposure to caspofungin
As an indicator of reactive oxygen species (ROS), we measured hydrogen peroxide (H2O2) in collected cell culture supernatants by using the fluorometric Hydrogen Peroxide Assay Kit (PromoCell, Heidelberg, DE). HTE cells were seeded on laminin-coated coverslips and were incubated with 60, 90 or 120 µM caspofungin (CAS) in 1 ml HEPES solution for 30, 60 and 90 min. Additionally, we incubated cells without adding CAS as a control. The cell supernatant (250 µl) was collected and centrifuged at 1,200 G for 15 min to remove all cell particles. To remove the proteins, the sample was then filtered through a 10 kDa MW spin filter (CAT.No. PK-CA577-1997, PromoCell, Heidelberg, DE). Samples were subsequently prepared in duplicate as follows. 50 µl of the samples were mixed with 50 µl Reaction Mix (Horse Radish Peroxidase, OxiRed Probe solution, Assay Buffer) in a 96-well plate (Costar, Corning, NY, USA) and were incubated for 10 min. A H2O2 standard was also established in duplicate with 0, 0.1, 0.2, 0.3, 0.4, or 0.5 nm/well. Fluorescence was measured (Ex/Em = 535/587 nm) in a microplate reader (Tecan, Maennedorf, CH). To calculate the H2O2 quantity, a H2O2 standard curve was plotted after background subtraction using the equation:
where Sa = sample amount [pmol], and Sv = sample volume [µl]).
Drugs and buffer solutions
The following drugs were administered: caspofungin (60 µM or 120 µM diluted in 20 µl H2O, MSD, Kenilworth, USA), NiCl2 (50 µM, Roth, Karlsruhe, Germany). Caffeine (30 mM, Roth, Karlsruhe, Germany) was sufficient to completely deplete ER Ca2+ stores in these cells. 2,4-dinitrophenol (DNP, 25 µM, Sigma-Aldrich, St. Louis, USA) was used to deplete mitochondrial Ca2+ stores by depolarization of mitochondrial membrane potential, 2-Aminoethoxydiphenylborane (2-APB, 40 µM, TOCRIS Bioscience, Bristol, UK); a high ryanodine concentration (40 µM, TOCRIS Bioscience, Bristol, UK) was used to inhibit Ca2+ liberation from ER Ca2+ stores. Ryanodine, 2-APB, and DNP were diluted in Dimethylsulfoxide (DMSO), and end concentrations were achieved by applying the stock solution directly to the buffer solution in the recording chamber. The DMSO did not exceed volumes of 20 µl in 2 ml HEPES in order to prevent the induction of unspecific disturbances of the cilia-bearing cells.
All preparations and experiments were carried out in HEPES solution consisting of 20 mM HEPES, 4.5 mM KCl, 2.5 mM CaCl2, 11 mM glucose, 140 mM NaCl, and 1 mM MgCl2. The pH was adjusted to 7.4 using 4 M NaOH at 30 °C, or 4–8 °C for tissue preparation. For Ca2+-free solutions, CaCl2 was replaced by 1 mM ethylene glycol tetra-acetic acid (EGTA). A vitality test was performed at the end of each recording with KCl solutions (200 mM). Solutions containing 200 mM KCl also contained 2.5 mM CaCl2.
Statistical analysis
A Mann–Whitney U test was used to compare equivalent measuring points from different experiments, and a Wilcoxon rank-sum test was used to compare dependent variables. Statistical data evaluation and testing were performed using the GraphPad PRISM (Version 5.04) software (GraphPad Software, La Jolla, CA, USA).
Results
Changes in [Ca2+]i in tracheal epithelial cells
The [Ca2+]i was expressed as changes in the FURA-2-fluorescence ratio. For the controls, isolated tracheal epithelial cells were rested in Ca2+-containing or in Ca2+-free buffer solutions for more than 800 s. In the cells resting in Ca2+-containing buffer, the FURA-2-fluorescence ratio did not change significantly during this period (1.09 ± 0.08, p = 0.12, compared to initial FURA-2-fluorescence ratio). At the end of all the experiments, we applied a brief pulse of KCl (200 mM), and only the cells that responded to this vitality test by showing a significant increase in the FURA-2-fluorescence ratio were included for further statistical evaluation (Fig. 1). Cells resting in the Ca2+-free buffer medium also showed no changes in the FURA-2-fluorescence ratio throughout the entire exposure period (0.89 ± 0.03, p = 0.09, Wilcoxon rank-sum test, Fig. 1C, D). However, these cells also responded to a brief KCl pulse with an increase in the FURA-2-fluorescence ratio.

Prolonged incubation of tracheal epithelial cells in buffer solutions did not change [Ca2+]i. (A) The [Ca2+]i was indicated by a FURA-2 ratio of 340 nm/380 nm. As a control, isolated tracheal epithelial cells were kept under long-term incubation in Ca2+-containing 2.5 mM buffer. During this prolonged incubation period, the basal [Ca2+]i did not change. The vitality test at the end of each experiment was carried out by applying KCl (200 mM), inducing a rapid Ca2+-influx and an increase in [Ca2+]i. (B) The initial levels of [Ca2+]i had not changed when they were briefly measured at the end of the exposure period before the application of KCl. (C) The prolonged incubation of tracheal epithelial cells in Ca2+-free buffer solution did not change the [Ca2+]i. The application of KCl (200 mM, containing 2.5 mM CaCl2) precipitated an increase in [Ca2+]i. (D) In the Ca2+-free buffer solution, the initial [Ca2+]i also did not change when it was briefly measured at the end of the exposure period before the KCl application (n = number of individual experiments, ns = not significant, Wilcoxon rank-sum test).
When the tracheal epithelial cells were exposed to caspofungin (60 µM) in Ca2+-containing buffer, almost all of the investigated cells immediately showed an elevation in [Ca2+]i. We observed a majority of cells responding with two subsequent Ca2+-transients with a maximum of 2.46 ± 0.29 (FURA-2-fluorescence ratio, n = 30). The first rapid increasing Ca2+ transient returned to the baseline, while the second one showed a rapid increase in [Ca2+]i which did not return to the baseline during the remaining exposure period (Fig. 2A). Increasing the caspofungin concentration to 120 µM resulted in prolonged elevation of [Ca2+]i that remained above the baseline at the end of the exposure period. There was a caspofungin dose-dependency of maximum amplitudes of initial Ca2+ transients (Fig. 2B). The application of NiCl2 was used to inhibit Ca2+ entry via the plasma-membrane-bound Ca2+ channels. However, it did not prevent the peak rise in [Ca2+]i induced by caspofungin (60 µM, 2.47 ± 0.18, p = 0.093, n = 30, Fig. 2B, E).

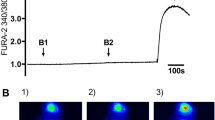
In tracheal epithelial cells, caspofungin triggers an increase in [Ca2+]i. (A) In the Ca2+-containing buffer solution (2.5 mM), the application of caspofungin (60 µM) induced a sudden rise in the FURA-2 ratio displaying [Ca2+]i. In the majority of the investigated cells (n = 21) we observed two transient prolonged peaks in [Ca2+]i, whereas the remaining cells (n = 4) showed a single transient rise of [Ca2+]i. (B) The amplitude of the initial Ca2+ increase induced by the caspofungin in the tracheal epithelial cells is concentration-dependent, with an EC50 of 55 µM. The dose–response curve fits a Hill equation. (C) Fluorescence images of the FURA-2 ratio converted into false colors that were taken at different time points from a prolonged recording of caspofungin application. I: Early response of cells to caspofungin application (50 s after caspofungin application); II: Further response of cells to caspofungin application with an increase in FURA-2 ratio (100 s after caspofungin application); III: Prolonged elevation of FURA-2 ratio (200 s after caspofungin application); IV: Prolonged elevated FURA-2 ratio (approx. 600 s after caspofungin application and just before KCl administration). (D) Ni2+ was applied in order to inhibit the influx of Ca2+ ions into the tracheal epithelial cells. The subsequent application of caspofungin still evoked a transient rise in [Ca2+]i that was independent of the Ca2+ influx. (E) A significant increase in [Ca2+]i was triggered when exposed to caspofungin (60 µM). Inhibition of membrane-bound Ca2+ channels by application of Ni2+ ions did not reduce the amplitudes of a transient rise in [Ca2+]i (scale bar in C: 20 µm. n = number of individual cells, ns = not significant, ***p < 0.001, Mann–Whitney U test).
Caspofungin induces a rise in [Ca2+]i independent of external Ca2+ ions
To further elucidate the caspofungin-induced rise in [Ca2+]i, we applied caspofungin to tracheal epithelial cells in Ca2+-free solutions. Exposure to caspofungin in the Ca2+-free solution evoked a rise in [Ca2+]i in all of the investigated cells (n = 30) which was biphasic, and which remained above the baseline level until the end of the experimental observation period (Fig. 3A). The peak rise in [Ca2+]i was significantly higher in the Ca2+-free buffer solutions (2.95 ± 0.72, n = 30) than the peak transients observed in the Ca2+-containing buffer solutions (2.27 ± 0.17, n = 30, p < 0.01; Fig. 3B).

The caspofungin-induced rise in [Ca2+]i is independent of external Ca2+ ions. (A) In the tracheal epithelial cells kept in Ca2+-free buffer solution, the application of caspofungin induced two consecutive elevations in [Ca2+]i. The second rise slowly decreased over time but remained above the baseline at the end of the exposure period. (B) The application of caspofungin in the Ca2+-free buffer medium induced a significant increase in [Ca2+]i. This rise in [Ca2+]i was also significantly higher than the rise triggered by caspofungin in Ca2+-containing buffer solution (n = number of individual cells, ns = not significant. The horizontal bars in the experimental recordings depict the exposure periods to the Ca2+-free buffer solution and caspofungin application ,**p<0.01. ***p < 0.001, Mann–Whitney U test).
Caspofungin liberates Ca2+ from internal stores
In order to determine the source of the Ca2+ ions that increase the levels of [Ca2+]i during caspofungin exposure, we pharmacologically depleted the known intracellular Ca2+ stores, e.g., DNP (25 µM) was used in Ca2+-free buffer solutions to deplete the mitochondrial Ca2+ stores. This uncoupling of the respiratory chain induces a complete depolarization of the mitochondrial membrane potential eliciting an efflux of Ca2+ ions from these organelles.
The Ca2+ efflux from the mitochondria was visible by brief Ca2+ transients (increase in FURA-2 ratio) that almost immediately returned to baseline levels (Fig. 4A). The following exposure to caspofungin (60 µM), still in the presence of DNP, evoked transient elevation in [Ca2+]i that always returned to the baseline. The transient increases in Ca2+ ions, when exposed to caspofungin in the presence of DNP, were significantly less than the transient increases in Ca2+ ions when the mitochondrial stores were not depleted (2.41 ± 0.07, n = 11, p < 0.01, Fig. 4B).

Caspofungin does not liberate Ca2+ ions from mitochondrial Ca2+ stores. (A) In a Ca2+-free buffer solution, mitochondrial Ca2+ stores were depleted by DNP (25 µM). DNP application resulted in a transient increase in [Ca2+]i before returning to the baseline level. Nevertheless, when in the presence of DNP, caspofungin induced a rapid transient rise in [Ca2+]i without a prolonged elevation. (B) The amplitude of the rise in [Ca2+]i induced by caspofungin in the presence of DNP was lower than the elevation of [Ca2+]i triggered by caspofungin without any depletion of mitochondrial Ca2+ stores (the horizontal bars in the experimental recordings depict the exposure periods of defined pharmacological agents. n = number of individual investigated cells, **p < 0.01, ***p < 0.001, Mann–Whitney U test).
Ca2+ stores in the ER were identified and depleted by cyclopiazonic acid (CPA, 10 µM), a reversible inhibitor of SERCA. In a Ca2+-free buffer solution, CPA induced a prolonged increase in [Ca2+]i that slowly returned to baseline levels (Fig. 5A). Cell vitality was verified by exposure to a depolarizing concentration of KCl (200 mM, Fig. 5A). The depletion of ER Ca2+-stores by caffeine (30 mM) led to a rapid but transient increase in [Ca2+]i. The subsequent application of CPA (10 µM) had no further effect, demonstrating that caffeine had already depleted the ER Ca2+ stores, which are identical to CPA-sensitive stores (Fig. 5B).

ER Ca2+ stores contribute to caspofungin-induced increases in [Ca2+]i. (A) The ER Ca2+ stores are identified by their sensitivity to the SERCA inhibitor CPA (10 µM). Exposure to CPA led to a slow increase in [Ca2+]i that returned to baseline levels while still in the presence of CPA. (B) Caffeine (30 mM) also depleted the ER Ca2+ stores leading to a rapid increase in [Ca2+]i that soon returned to baseline levels. The subsequent application of CPA did not lead to a further rise in [Ca2+]i, demonstrating that the caffeine had already depleted ER Ca2+ stores that are identical to CPA-sensitive Ca2+ stores. (C) Applying 30 mM of caffeine was enough to deplete the ER Ca2+ stores. The consecutive application of caspofungin while in the presence of caffeine did not change the [Ca2+]i. Under these conditions, no Ca2+ transients were observed. (D) The [Ca2+]i measured under caffeine application and subsequent caspofungin application was significantly lower than the rise in [Ca2+]i measured when the ER’s Ca2+ stores were not depleted (the horizontal bars in the experimental recordings depict the exposure periods of defined pharmacological agents. n = number of individual investigated cells, **p < 0.01, ***p < 0.001, Mann–Whitney U test).
In further experiments, exposing the cells to caffeine led to a brief Ca2+ transients of the FURA-2 fluorescence ratio that almost immediately returned to the baseline. The subsequent application of caspofungin (60 µM or 120 µM) had no effect on any tracheal epithelial cells investigated.
We observed no increase in [Ca2+]i in the presence of caspofungin (1.32 ± 0.02, n = 30, p = 0.1 when compared to the FURA-2 fluorescence ratio after the addition of caffeine but before caspofungin application). However, Ca2+ transients were significantly reduced vs. Ca2+ transients induced by caspofungin in Ca2+-free buffer solutions (p < 0.001, Fig. 5C, D.
Caspofungin enhances [Ca2+]i via the activation of IP3 and ryanodine receptors
To evaluate the route by which Ca2+ ions are liberated by caspofungin from the ER’s Ca2+ stores, we pharmacologically inhibited the known Ca2+ pathways. 2-APB (40 µM) was applied to Ca2+-free extracellular solutions in order to inhibit inositol-1,4,5-triphosphate (IP3) receptors prior to caspofungin application. 2-APB had no effect on the FURA-2 fluorescence ratio indicating transients of [Ca2+]i (1.04 ± 0.003, n = 30, p = 0.21, Fig. 6A, B). The subsequent application of caspofungin still induced single Ca2+ transients with a significantly reduced peak to 2.04 ± 0.08 (n = 30, p < 0.001) compared to Ca2+ transients induced by caspofungin alone (2.95 ± 0.72, Fig. 6B). However, we observed no prolonged elevation in the FURA-2 fluorescence ratio, which we observed when the Ca2+-free buffer solution was exposed to caspofungin (Fig. 3A). In a further series of experiments, we inhibited the ryanodine receptors using 40 µM ryanodine. This concentration of ryanodine did not induce an efflux of Ca2+ from ER stores, as demonstrated by the unaltered FURA-2 fluorescence ratio (Fig. 6C, D).

The caspofungin-induced rise in [Ca2+]i depends on ryanodine and IP3 receptor activity. (A) In the Ca2+-free buffer solution, IP3 receptors were inhibited using 2-APB (40 µM), which did not affect the baseline [Ca2+]i. The application of caspofungin evoked a transient rise in [Ca2+]i above the baseline. (B) The amplitude of the transient rise in [Ca2+]i induced by caspofungin was significantly lower than the rise in [Ca2+]i measured without the application of 2-APB (Mann–Whitney U test). The elevation in [Ca2+]i induced by caspofungin in the presence of 2-APB was significantly higher than the baseline values (Wilcoxon rank-sum test). (C) The application of ryanodine (40 µM) did not change the basal [Ca2+]I and the application of caspofungin did not lead to an increase in [Ca2+]i. The application of KCl at the end of the exposure period demonstrated the vitality of the cells. (D) The application of ryanodine did not change the baseline [Ca2+]i levels in the Ca2+-free buffer solution. The application of caspofungin did not lead to an increase in [Ca2+]i. No Ca2+ transients were observed and the [Ca2+]i was significantly lower than the rise in [Ca2+]i measured after caspofungin application in Ca2+-free solution without inhibition of the ryanodine receptors (the horizontal bars in the experimental recordings depict the exposure periods of defined pharmacological agents. n = number of individual investigated cells, ns = not significant, **p < 0.01, ***p < 0.001, Mann–Whitney U test).
The application of caspofungin in the presence of ryanodine did not evoke any increase in the FURA-2 fluorescence ratio, and no Ca2+ transients in any of the investigated cells were observed (Fig. 6C, D). The [Ca2+]i was no different to the values obtained prior to caspofungin application (1.1 ± 0.01, n = 30, p = 0.52). At the end of each experiment, Ca2+ transients were still elicited by applying KCl, demonstrating the vitality of the cells (Fig. 6C).
Caspofungin depletes ER Ca2+ stores in tracheal epithelial cells
For the controls, HTEpC-c cells loaded with Mag-Fluo-4 AM were rested in Ca2+-containing buffer (Fig. 7A), Ca2+ free HEPES buffer (Fig. 7B), and Ca2+-free HEPES buffer after prior loading with BAPTA-AM (Fig. 7C). None of these conditions reduced the Mag-Fluo-4 fluorescence signal displaying [Ca2+]ER, demonstrating that the ER Ca2+ stores were stable under resting conditions without significant leakage of Ca2+ ions into the cytosol (Fig. 7A). However, exposure to caspofungin rapidly altered the Mag-Fluo-4 fluorescence. In all the cells that were investigated, we observed a rapid decrease in fluorescence within a few seconds of exposure (Fig. 7D, E) that immediately recovered, before a slower and more prolonged decline in the fluorescence showed the [Ca2+]ER. The amplitude of the initial downward spike of Mag-Fluo-4 fluorescence and the degree of the prolonged reduction was concentration-dependent (Fig. 8A, B). A final exposure to the Ca2+-containing buffer solution with a depolarizing KCl concentration demonstrated a recovery of the Mag-Fluo-4 signal equivalent to the replenishment of Ca2+ stores within the ER (Fig. 7D).

Caspofungin depletes ER Ca2+ stores. (A) Tracheal epithelial cells resting in Ca2+-containing buffer showed no changes in Mag-Fluo-4 fluorescence. (B) Ca2+-free buffer solution did not change the Mag-Fluo-4 fluorescence after prolonged exposure. (C) Cells were loaded with BAPTA to buffer cytosolic free Ca2+ ions in order to reduce [Ca2+]i. These resting cells showed only minor changes in the Mag-Fluo-4 signal, assuming that the ER Ca2+ stores remained constantly filled. (D) Exposure to caspofungin led to a rapid decrease in Mag-Fluo-4-fluorescence (red box) that almost immediately recovered back to baseline levels. Prolonged exposure to caspofungin then led to a slow decline in Mag-Fluo-4-fluorescence to a stable lower plateau. (E) Enlarged section of the initial spike from (D) (red box) showing the rapid kinetic and amplitude (red arrow) of the initial response to caspofungin exposure. (F) In cells exposed to caspofungin that were preloaded with BAPTA, the initial downward spike in Mag-Fluo-4-fluorescence completely disappeared and a prolonged reduction in Mag-Fluo-4-fluorescence was recorded. Replenishing the stores with Ca2+ ions by depolarizing the cells using KCl had little effect because the BAPTA instantly buffered the cytosolic Ca2+ ions. The luminal ER Mag-Fluo-4-fluorescence remained low (the horizontal bars in the experimental recordings show the exposure periods of the defined pharmacological agents).

The effects of caspofungin effects on ER Ca2+ stores are concentration-dependent. (A) Caspofungin induced an immediate downward spike in Ca2+ ions. The amplitudes of these transient Ca2+ spikes were concentration-dependent. Low caspofungin concentrations (60 µM) already induced the Ca2+ transients that were significantly augmented by high caspofungin concentrations (120 µM). (B) Prolonged depletion of ER Ca2+-stores was noted for different caspofungin concentrations in the presence of BAPTA. The sustained minimum signals of Mag-Fluo-4-fluorescence induced by prolonged exposure to caspofungin were concentration-dependent. The degree of ER Ca2+-store depletion was most pronounced under high caspofungin concentrations (120 µM), (n = number of individual investigated cells, ns = not significant, **p < 0.01, ***p < 0.001, two-way ANOVA).
The initial sharp decrease in Mag-Fluo-4 fluorescence completely disappeared when the cells were loaded with BAPTA prior to caspofungin exposure; only the slow, prolonged phase of the declining Mag-Fluo-4 was still visible. In addition, replenishing the ER Ca2+ stores by Ca2+-containing buffer and depolarizing the cells with KCl only had a limited effect, assuming that the cytosolic Ca2+ ions were instantly buffered by BAPTA (Fig. 7F).
The total depletion of ER Ca2+ stores was achieved by the application of caffeine (30 mM). The stores were replenished after subsequent exposure to a buffer solution containing depolarizing KCl (200 mM) and Ca2+ (2.5 mM), inducing a maximum rise in Mag-Fluo-4 fluorescence (Fig. 9A). In the presence of caffeine, exposure to caspofungin had no further effect. Neither was the initial downward Ca2+ spike visible nor was the prolonged reduction of Mag-Fluo-4 fluorescence further diminished (Fig. 9B, C).

Caspofungin depletes caffeine-sensitive ER Ca2+ stores. (A) In the Ca2+-free buffer solution, exposure to caffeine (30 mM) led to a rapid and prolonged reduction in Mag-Fluo-4-fluorescence. The ER stores were replenished by a voltage-gated Ca2+ influx (exposure to 200 mM K+ ions). (B) Caffeine exposure induces the discharge of ER Ca2+ stores prior to caspofungin application. In addition, caspofungin did not induce a further reduction in Mag-Fluo-4-fluorescence, demonstrating that ER stores were already depleted by caffeine. (C) The addition of caffeine led to a significant reduction in the Mag-Fluo-4-fluorescence indicating that the Ca2+ stores in the ER were empty. Subsequent application of caspofungin in the presence of caffeine did not alter the Mag-Fluo-4-fluorescence any further. (D) Isolated mice tracheae were either exposed to caspofungin (120 µM) or kept as controls in buffer solutions. Under both conditions, we observed only minor alterations in ROS generation displayed by emission fluorescence at 590 nm. (the horizontal bars in the experimental recordings depict the exposure periods of defined pharmacological agents. n = number of individual investigated cells, ns = not significant, ***p < 0.001, two-way ANOVA).
ROS generation under caspofungin exposure in the tracheal epithelium
Here, we were able to determine whether the observed effects of caspofungin on intracellular Ca2+ stores were caused by cellular ROS generation. In freshly isolated mice tracheae, we measured ROS generation under exposure to caspofungin (120 µM) using a fluorescence dye. The results showed no significant ROS generation in the control experiment or when the tracheae were exposed to caspofungin for 30 min (Fig. 9D).
Discussion
In this study, which looked at isolated HTEpCs, we found evidence that caspofungin triggers a rise in [Ca2+]i by releasing Ca2+ ions from caffeine-sensitive ER stores, primarily via a RyR pathway and via an IP3 regulated cascade to a lesser extent. This increase in [Ca2+]i and the releasing of Ca2+ ions from the ER was slightly dependent on extracellular Ca2+ concentrations or transmembraneous Ca2+ influxes, even though we could not prove it by blocking transmembraneous Ca2+ channels by using NiCl2. It is possible that NiCl2 is not able to block all the transmembraneous Ca2+ channels of tracheal epithelial cells or that we used an insufficient concentration. However, the cells showed a reaction directly after the application of 500 µM NiCl2, implying that the dosage was sufficient. In most of the cells investigated, the rise in [Ca2+]i was biphasic, whereby an initial transient rise in [Ca2+]i was followed by a second prolonged rise in [Ca2+]i. This data is supported by our experiments using Mag-Fluo-4-fluorescence, which showed a rapid biphasic depletion of ER Ca2+ stores after exposure to caspofungin. These present findings are similar to the effect of acetylcholine on the tracheal ciliary beating rate, inducing a transient rise in [Ca2+]i that is followed by a prolonged increase in the beating rate8. We found no evidence that caspofungin induces ROS generation since ROS are known to interfere with calcium-signaling pathways.
The stimulation of cilia-bearing cells in order to enhance their beating rate is the main task to rapidly convey heavy loads of particles and pathogens. This mechanism depends on the liberation of Ca2+ ions from internal stores18. It is also in line with reports that evidenced the activation of the ciliary beat frequency following the kinetics of [Ca2+]i19. We were also able to demonstrate that caspofungin activates intracellular signal transduction cascades that lead to the release of Ca2+ ions from ER stores via RyR. We also demonstrated that caspofungin depleted the Ca2+ stores in the ER.
However, although we still cannot conclude that caspofungin directly affects RyR, we have excluded any interaction with membrane-bound receptors or with their downstream reaction cascades. Therefore, with the present data, we can assume that caspofungin can penetrate tracheal epithelial cells and to activate specifically RyR. This is similar to cardiomyocytes, in which caspofungin also triggers the liberation of Ca2+ ions from the ER via RyR17.
The permeability of caspofungin and its diffusion into mammalian tissues and cells
Caspofungin inhibits the synthesis of 1,3-β-D-glucan, which is an essential component of the fungal cell wall20,21. While caspofungin underlies very low excretion kinetics that result in high sustained plasma levels, it attains therapeutic concentrations in various tissues22. It is also characterized by its ability to diffuse into many organs and tissues in order to reach the concentrations necessary to treat invasive candidiasis; thus, its penetration into the central nervous system (CNS) is poor23. Even when the meninges are inflamed, the concentrations of caspofungin in the CNS are far below serum concentrations24. In contrast, caspofungin concentrations are highest in the liver, lungs, kidney, and spleen, where caspofungin can deploy its antifungal properties13. In the lungs, the concentration of caspofungin leads to favorable response rates of up to 62% in neutropenic patients suffering from pulmonary invasive fungal disease25.
In mammals, caspofungin mainly acts extracellularly against most Candida species. However, caspofungin, a membrane-anchored cyclic lipopeptide, is also able to penetrate mammalian cells and can reach high concentrations within the tracheal epithelium allowing it to act upon cilia-bearing cells. We can also conclude that caspofungin concentrations are also high in the epithelial lining fluid, and it may act from the luminal side onto epithelial cells.
Since caspofungin is used to treat pulmonary infections caused by Candida spp., it is able to diffuse via the epithelium into the airway lumen so that it can reach these therapeutic concentrations. We found evidence that caspofungin can reach intracellular concentrations in isolated lower airway cells that are high enough to liberate Ca2+ ions from cell organelles. So far, the effects of caspofungin on intracellular organelles have only been described for a few cell types. In stimulated mammalian macrophages, caspofungin has been shown to reach concentrations that will have an effect on trapped Candida glabrata26. Recently, we described a change in the contractility of isolated cardiomyocytes via the liberation of Ca2+ ions from internal Ca2+ stores15,17. These data support our present findings and lead us to postulate that the diffusion of caspofungin into mammalian cells is not restricted to particular cell types; it seems rather a general characteristic of this antimycotic substance.
The effects of caspofungin on intracellular Ca2+ stores
In our present study, we identified that caspofungin could trigger the release of Ca2+ ions from Ca2+ stores in the ER, mainly via a caffeine/ryanodine-sensitive pathway. We found no evidence for the liberation of Ca2+ ions stored in mitochondria, although these organelles do store Ca2+ ions under resting conditions (see Fig. 4). However, they primarily buffer Ca2+ ions when [Ca2+]i exceeds a threshold of approximately 500 nM9,27. In liver cells and cardiomyocytes, caspofungin is known to disturb the electron transport within the mitochondrial respiratory chain by inhibiting complex I and III. The effect on complex III is probably caused by its interference with cytochrome C28. However, it is not known whether this mechanism also depolarizes mitochondrial membrane potential, which is essential for Ca2+ buffering in these organelles9. Here we measured no effect of caspofungin onto mitochondrial Ca2+ stores.
When the mitochondrial Ca2+ stores were depleted by DNP, exposure to caspofungin had little effect (see Fig. 4). This small decrease in caspofungin response can be explained by the inhibition of ATP synthesis due to the application of DNP application29. ATP usually powers the sarco-endoplasmic calcium ATPase (SERCA) that refills the ER’s Ca2+ stores. Therefore, the interrupted mitochondrial ATP synthesis after exposure to DNP reduces SERCA activity, which then leads to a slow Ca2+ leakage from the ER stores via different pathways9,30. Eventually, this results in a slightly reduced Ca2+ response during subsequent caspofungin application, which was what was observed here. Our observations provide evidence that the mitochondria in tracheal epithelial cells store Ca2+ ions under resting conditions but are not affected by caspofungin.
Under steady-state conditions, the influx of Ca2+ ions into the cytosol from the ER stores is balanced on the one hand by RyR, IP3, or alternative leaks that drive the efflux from the ER, and on the other hand by SERCA, which regulates Ca2+ influx into the ER resulting in a zero net flux of Ca2+ ions into the cytosol31,32. Ca2+ sparks are transient elevations of the [Ca2+]i that are caused by a net efflux from the ER. This efflux is caused by an increased open probability of the RyR, which cannot immediately be offset by the SERCA-driven influx. Using caffeine to open the RyR pathway leads to a rapid and transient Ca2+ efflux that depletes the ER’s stores. The following balance of efflux and influx of Ca2+ ions to pre-caffeine levels while still in the presence of caffeine is caused by clearing the elevated [Ca2+]i by pumping Ca2+ ions out of the cells or into alternative stores while the RyR is still activated and open33. The refilling of the ER stores cannot be achieved under these conditions, which is the reason why we observed no further Ca2+ transients when caspofungin was applied after the ER stores were depleted by the addition of caffeine. When caspofungin is applied to cells that have not had their ER stores depleted by caffeine, we observed Ca2+ transients in most of the cells.
In our experiments using Mag-Fluo-4 as an indicator for ER luminal Ca2+ contents, we observed a rapid decline in Mag-Fluo-4-fluorescence followed by a partial recovery, which eventually led to a prolonged decline in Mag-Fluo-4-fluorescence. The partial recovery is most probably a response to the SERCA balancing the efflux and influx of Ca2+ ions from the ER, as is generally assumed for many cell types34. A similar mechanism of Ca2+ depletion from the ER has been described previously for smooth muscle cells derived from different organs in rats35,36. Here, the prolonged decline of Mag-Fluo-4-fluorescence was caused by the continuous presence of caspofungin activating RyR.
Under these conditions, SERCA was unable to balance the influx and efflux of Ca2+ ions from the ER stores. Most probably, this net efflux is provoked by caspofungin triggering the increase in open state probability of RyR, since the kinetics of Ca2+ transients are similar. The clearance of cytosolic Ca2+ transients is caused by alternative clearance pathways, e.g., the transmembrane efflux, buffering, or by an uptake in alternative Ca2+ stores like mitochondria. We interpret these findings such that caspofungin specifically activates ER-bound RyR and depletes these Ca2+ stores. Caspofungin now provokes an imbalance in favor of a net Ca2+ efflux that results in an immediate increase in [Ca2+]i, which is either transient or sustained. The rapid amplitude in [Ca2+]i under caspofungin exposure is caused by the steep function of Ca2+ content in the ER as has previously been described in cardiomyocytes37,38. This Ca2+ content contributes to the regulation of ciliary beat activity by the activation of different signal cascades8.
Alternatively, caspofungin may promote the dissociation of Ca2+ ions from cytosolic buffers as an alternative source for increasing [Ca2+]i. In many cell types, cytosolic Ca2+ is strongly buffered, and for every free cytosolic Ca2+ ion approximately 100 to 200 Ca2+ ions are bound to buffers39,40. In cardiomyocytes, the major buffers are troponin and SERCA, whereas in airway epithelial cells the Ca2+ is supposed to be buffered by calmodulin8,34. However, in this case, the kinetics of increased [Ca2+]i would be different.
We have not observed biphasic Ca2+ transients or sustained elevations of [Ca2+]i. This is because Ca2+ pumps on the plasma membrane, the ER and mitochondrial Ca2+ uptake would still be fully active and would be sufficient to immediately reduce elevated [Ca2+]i content.
Since the initial binding of Ca2+ ions to SERCA contributes significantly to Ca2+ buffering, it may be that caspofungin contributes to the dissocitaion of Ca2+ ions from these binding sites41. Nevertheless, in this case, we would observe Ca2+ transients while RyR was inhibited by ryanodine, or when the ER stores were depleted by caffeine. However, we did not observe these Ca2+ kinetics. These data argue against the assumption that caspofungin drives Ca2+ ions out of the buffering sites of the SERCA. Furthermore, the inhibition of SERCA by caspofungin would lead to a slow, sustained increase in [Ca2+]i which then returns to the baseline, as we observed for CPA (Fig. 5)9,30. In contrast, we did not observe a slow increase in [Ca2+]i under caspofungin exposure; the kinetic was always steep and rapid. In contrast, in many of the airway cells observed, the decay in the elevated [Ca2+]i levels was slow. This prolonged kinetic may be caused either by reduced ATP content and subsequently reduced activity of ATP-driven ion pumps or by the inhibition of ion transporters. However, it is questionable whether caspofungin reduces ATP generation or inhibits ion transport and this needs to be investigated further.
In our experiments, we noted a reduced response to caspofungin while using 2-APB, which binds to the IP3 receptor. Since phospholipase-C activation and liberation of Ca2+ ions from Ca2+ stores via the IP3 receptor contribute to Ca2+ wave propagation in airway epithelia, we investigated whether caspofungin interferes with this signal cascade42. The reduced response to caspofungin exposure when 2-APB was applied may be caused by the activation of these receptors by caspofungin in competition with 2-APB. This assumption is supported by the fact that the response to caspofungin was delayed in the presence of 2-APB. Therefore, 2-APB might only be effective when caspofungin concentration is high enough to prevent 2-APB from binding. Further findings revealed that ryanodine abolished the calcium signals elicited by Caspofungin. Controversially, IP3-mediated calcium release seems not to occur in parallel to RyR-mediated calcium release. Nevertheless, we observed a reduced response to caspofungin in the presence of 2-APB. 2-APB is also known to modulate TRP channel, gap junctions, STIM ORAI channel conductance and ICRAC in higher concentrations43,44. Owing to this lack of specificity for the IP3 receptor, 2-APB could also impair other calcium pathways and not particularly the activity of the IP3 receptor. On the other hand, our experiments were performed in calcium free buffer solution, so store-operated calcium entry (SOCE) could not occur. Further experiments are necessary to explore whether the IP3 receptor pathway is affected by using a more specific IP3 receptor blocker such as Xestospongin C or to examine SOCE pathways, which is more complicated due to diverse interactions and a finetuned orchestration of multiple targets. Regardless, we conclude that RyR is the principal target of caspofungin since we measured the main effects of caspofungin via this Ca2+ efflux pathway.
The observed sustained elevation of Ca2+ ions under caspofungin exposure may be due to several reasons, as a similar effect has been described when these cells are exposed to histamine or acetylcholine8,45. Most probably, this prolonged kinetic is caused by an imbalance between the influx and efflux of Ca2+ ions from the cytosol due to a slow clearance rate when reaching baseline levels of [Ca2+]i during the observation period. These findings imply that caspofungin has effects on other cell organelles. It may also interfere with mitochondrial ATP synthesis, as has been described for mammalian mitochondria28. Under these circumstances, the remaining ATP supply from mitochondria, glycolysis or phosphocreatine stores is too low to fully activate Ca2+ ATPase in the plasma membrane. This mechanism prevents a rapid return of [Ca2+]i to baseline levels. Beyond plasma membrane-bound, sodium-calcium exchangers are also activated when [Ca2+]i is high and may blunt the peak of observed the Ca2+ transients46.
We also found no evidence that caspofungin depolarizes mitochondrial membrane potential, which is the driving force behind Ca2+ storage in these organelles. This is supported by our findings that the number of Ca2+ transients was only slightly altered when mitochondria were depolarized using DNP. Therefore, in many tracheal epithelial cells, caspofungin may have additional effects on other cellular structures that cause a prolonged elevation of [Ca2+]i, with the slow return to the baseline level due to an energetic restriction.
HTE cells, which are isolated cells from the human tracheal epithelium, are suitable for investigating the different effects and signal pathways under stable conditions, whereas freshly isolated human tracheal cells should only be used in further studies to underline these observations. Additionally, a contribution of alternative Ca2+ influx pathways, e.g. store operated calcium entry (SOCE) or IP3 receptors in the intact tracheal epithelium, should also be considered.
Conclusion
Caspofungin liberates Ca2+ ions from internal ER stores and enhances [Ca2+]i either transiently or in a sustained kinetic. This mechanism likely occurs due to the activation of a RyR pathway. A sustained elevation in [Ca2+]i levels may be supported via energetic imbalances during caspofungin application. Further studies should focus on RyR activation by caspofungin, detecting its binding site to these receptors, and determining how cellular ATP synthesis is compromised by caspofungin.
References
Gibbons, I. R. Dynein family of motor proteins: present status and future questions. Cell Motil. Cytoskeleton 32, 136–144. https://doi.org/10.1002/cm.970320214 (1995).
Sanderson, M. J. & Sleigh, M. A. Ciliary activity of cultured rabbit tracheal epithelium: beat pattern and metachrony. J. Cell Sci. 47, 331–347 (1981).
Zagoory, O., Braiman, A. & Priel, Z. The mechanism of ciliary stimulation by acetylcholine: roles of calcium, PKA, and PKG. J. General Physiol. 119, 329–339. https://doi.org/10.1085/jgp.20028519 (2002).
Lansley, A. B., Sanderson, M. J. & Dirksen, E. R. Control of the beat cycle of respiratory tract cilia by Ca2+ and cAMP. Am. J. Physiol. 263, L232-242. https://doi.org/10.1152/ajplung.1992.263.2.L232 (1992).
Salathe, M., Pratt, M. M. & Wanner, A. Cyclic AMP-dependent phosphorylation of a 26 kD axonemal protein in ovine cilia isolated from small tissue pieces. Am. J. Respir. Cell Mol. Biol. 9, 306–314. https://doi.org/10.1165/ajrcmb/9.3.306 (1993).
Geary, C. A., Davis, C. W., Paradiso, A. M. & Boucher, R. C. Role of CNP in human airways: cGMP-mediated stimulation of ciliary beat frequency. Am. J. Physiol. 268, L1021-1028. https://doi.org/10.1152/ajplung.1995.268.6.L1021 (1995).
Zagoory, O., Braiman, A., Gheber, L. & Priel, Z. Role of calcium and calmodulin in ciliary stimulation induced by acetylcholine. Am. J. Physiol. Cell Physiol. 280, C100-109. https://doi.org/10.1152/ajpcell.2001.280.1.C100 (2001).
Salathe, M. & Bookman, R. J. Mode of Ca2+ action on ciliary beat frequency in single ovine airway epithelial cells. J. Physiol. 520(Pt 3), 851–865 (1999).
Henrich, M. & Buckler, K. J. Effects of anoxia and aglycemia on cytosolic calcium regulation in rat sensory neurons. J. Neurophysiol. 100, 456–473. https://doi.org/10.1152/jn.01380.2007 (2008).
Stone, J. A. et al. Single- and multiple-dose pharmacokinetics of caspofungin in healthy men. Antimicrob. Agents Chemother. 46, 739–745. https://doi.org/10.1128/aac.46.3.739-745.2002 (2002).
Petraitiene, R. et al. Antifungal efficacy of caspofungin (MK-0991) in experimental pulmonary aspergillosis in persistently neutropenic rabbits: pharmacokinetics, drug disposition, and relationship to galactomannan antigenemia. Antimicrob. Agents Chemother. 46, 12–23 (2002).
Hajdu, R. et al. Preliminary animal pharmacokinetics of the parenteral antifungal agent MK-0991 (L-743,872). Antimicrob. Agents Chemother. 41, 2339–2344 (1997).
Stone, J. A. et al. Disposition of caspofungin: role of distribution in determining pharmacokinetics in plasma. Antimicrob. Agents Chemother. 48, 815–823 (2004).
Koch, C. et al. Cardiac effects of echinocandins after central venous administration in adult rats. Antimicrob. Agents Chemother. 59, 1612–1619. https://doi.org/10.1128/AAC.04446-14 (2015).
Arens, C. et al. Effects of echinocandin preparations on adult rat ventricular cardiomyocytes. Preliminary results of an in vitro study. Anaesthesist 63, 129–134. https://doi.org/10.1007/s00101-014-2289-8 (2014).
Lichtenstern, C. et al. Cardiac effects of echinocandin preparations—three case reports. J. Clin. Pharm. Ther. 38, 429–431. https://doi.org/10.1111/jcpt.12078 (2013).
Koch, C. et al. Caspofungin modulates ryanodine receptor-mediated calcium release in human cardiac myocytes. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.01114-18 (2018).
Ma, W., Silberberg, S. D. & Priel, Z. Distinct axonemal processes underlie spontaneous and stimulated airway ciliary activity. J. Gen. Physiol. 120, 875–885 (2002).
Salathe, M. & Bookman, R. J. Coupling of [Ca2+]i and ciliary beating in cultured tracheal epithelial cells. J. Cell Sci. 108(Pt 2), 431–440 (1995).
Denning, D. W. Echinocandin antifungal drugs. Lancet 362, 1142–1151. https://doi.org/10.1016/S0140-6736(03)14472-8 (2003).
Cappelletty, D. & Eiselstein-McKitrick, K. The echinocandins. Pharmacotherapy 27, 369–388. https://doi.org/10.1592/phco.27.3.369 (2007).
Sandhu, P. et al. Disposition of caspofungin, a novel antifungal agent, in mice, rats, rabbits, and monkeys. Antimicrob. Agents Chemother. 48, 1272–1280 (2004).
Chen, S. C., Slavin, M. A. & Sorrell, T. C. Echinocandin antifungal drugs in fungal infections: a comparison. Drugs 71, 11–41. https://doi.org/10.2165/11585270-000000000-00000 (2011).
Strenger, V. et al. Low penetration of caspofungin into cerebrospinal fluid following intravenous administration of standard doses. Int. J. Antimicrob. Agents 50, 272–275. https://doi.org/10.1016/j.ijantimicag.2017.02.024 (2017).
Zhang, X. et al. Caspofungin treatment for pulmonary invasive fungal disease in hematology patients: a retrospective study in a clinical practice setting in China. Clin Ther 39, 1758–1768. https://doi.org/10.1016/j.clinthera.2017.07.013 (2017).
Baltch, A. L., Bopp, L. H., Smith, R. P., Ritz, W. J. & Michelsen, P. B. Anticandidal effects of voriconazole and caspofungin, singly and in combination, against Candida glabrata, extracellularly and intracellularly in granulocyte-macrophage colony stimulating factor (GM-CSF)-activated human monocytes. J. Antimicrob. Chemother. 62, 1285–1290. https://doi.org/10.1093/jac/dkn361 (2008).
Duchen, M. R. Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 529(Pt 1), 57–68 (2000).
Shirey, K., Stover, K. R., Cleary, J., Hoang, N. & Hosler, J. Membrane-anchored cyclic peptides as effectors of mitochondrial oxidative phosphorylation. Biochemistry 55, 2100–2111. https://doi.org/10.1021/acs.biochem.5b01368 (2016).
Henrich, M. & Buckler, K. J. Effects of anoxia, aglycemia, and acidosis on cytosolic Mg2+, ATP, and pH in rat sensory neurons. Am. J. Physiol. Cell Physiol. 294, C280–C294 (2008).
Henrich, M. & Buckler, K. J. Cytosolic calcium regulation in rat afferent vagal neurons during anoxia. Cell Calcium 54, 416–427 (2013).
Eisner, D. A., Trafford, A. W., Diaz, M. E., Overend, C. L. & O’Neill, S. C. The control of Ca release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc. Res. 38, 589–604 (1998).
Negretti, N., Varro, A. & Eisner, D. A. Estimate of net calcium fluxes and sarcoplasmic reticulum calcium content during systole in rat ventricular myocytes. J. Physiol. 486(Pt 3), 581–591 (1995).
Greensmith, D. J., Galli, G. L., Trafford, A. W. & Eisner, D. A. Direct measurements of SR free Ca reveal the mechanism underlying the transient effects of RyR potentiation under physiological conditions. Cardiovasc. Res. 103, 554–563. https://doi.org/10.1093/cvr/cvu158 (2014).
Eisner, D. A., Caldwell, J. L., Kistamas, K. & Trafford, A. W. Calcium and excitation-contraction coupling in the heart. Circ. Res. 121, 181–195. https://doi.org/10.1161/CIRCRESAHA.117.310230 (2017).
Shmigol, A. V., Eisner, D. A. & Wray, S. Simultaneous measurements of changes in sarcoplasmic reticulum and cytosolic. J. Physiol. 531, 707–713. https://doi.org/10.1111/j.1469-7793.2001.0707h.x (2001).
Ziomek, G., van Breemen, C. & Esfandiarei, M. Measurement of calcium fluctuations within the sarcoplasmic reticulum of cultured smooth muscle cells Using FRET-based confocal imaging. J. Vis. Exp. https://doi.org/10.3791/53912 (2016).
Trafford, A. W., Diaz, M. E., Negretti, N. & Eisner, D. A. Enhanced Ca2+ current and decreased Ca2+ efflux restore sarcoplasmic reticulum Ca2+ content after depletion. Circ. Res. 81, 477–484 (1997).
Bassani, J. W., Yuan, W. & Bers, D. M. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 268, C1313-1319. https://doi.org/10.1152/ajpcell.1995.268.5.C1313 (1995).
Berlin, J. R., Bassani, J. W. & Bers, D. M. Intrinsic cytosolic calcium buffering properties of single rat cardiac myocytes. Biophys. J. 67, 1775–1787. https://doi.org/10.1016/S0006-3495(94)80652-6 (1994).
Trafford, A. W., Diaz, M. E. & Eisner, D. A. A novel, rapid and reversible method to measure Ca buffering and time-course of total sarcoplasmic reticulum Ca content in cardiac ventricular myocytes. Pflugers Arch. 437, 501–503. https://doi.org/10.1007/s004240050808 (1999).
Higgins, E. R., Cannell, M. B. & Sneyd, J. A buffering SERCA pump in models of calcium dynamics. Biophys. J. 91, 151–163. https://doi.org/10.1529/biophysj.105.075747 (2006).
Hansen, M., Boitano, S., Dirksen, E. R. & Sanderson, M. J. A role for phospholipase C activity but not ryanodine receptors in the initiation and propagation of intercellular calcium waves. J. Cell Sci. 108(Pt 7), 2583–2590 (1995).
Varnai, P., Hunyady, L. & Balla, T. STIM and Orai: the long-awaited constituents of store-operated calcium entry. Trends Pharmacol. Sci. 30, 118–128. https://doi.org/10.1016/j.tips.2008.11.005 (2009).
Peinelt, C., Lis, A., Beck, A., Fleig, A. & Penner, R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J. Physiol. 586, 3061–3073. https://doi.org/10.1113/jphysiol.2008.151365 (2008).
Harris, R. A. & Hanrahan, J. W. Effects of EGTA on calcium signaling in airway epithelial cells. Am. J. Physiol. 267, C1426-1434. https://doi.org/10.1152/ajpcell.1994.267.5.C1426 (1994).
Reyes-Garcia, J., Flores-Soto, E., Carbajal-Garcia, A., Sommer, B. & Montano, L. M. Maintenance of intracellular Ca2+ basal concentration in airway smooth muscle (review). Int. J. Mol. Med. 42, 2998–3008. https://doi.org/10.3892/ijmm.2018.3910 (2018).
Acknowledgements
We would like to thank Mrs. Ilona Magel for her excellent technical assistance, and Mrs. Andrea Mohr and Dr. Matthias Hecker for their kind support while performing the ROS measurements.
Author information
Authors and Affiliations
Contributions
M.H. and S.M. participated in study design. Data acquisition was done by M.H., S.M. and C.K.; statistical analysis and interpretation was performed by M.H., S.M., C.K. and S.W. All authors participated in drafting the manuscript or revising it critically for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Müller, S., Koch, C., Weiterer, S. et al. Caspofungin induces the release of Ca2+ ions from internal stores by activating ryanodine receptor-dependent pathways in human tracheal epithelial cells. Sci Rep 10, 11723 (2020). https://doi.org/10.1038/s41598-020-68626-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-68626-7
This article is cited by
-
Effects of long-acting muscarinic antagonists on promoting ciliary function in airway epithelium
BMC Pulmonary Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
