
Abstract
To develop a reproducible and stable closed chest model of ischemic cardiogenic shock in sheep, with high survival rate and potential insight into human pathology. We established a protocol for multi-step myocardial alcoholisation of the left anterior descending coronary artery by percutaneous ethanol injection. A thorough hemodynamic assessment was obtained by invasive and non-invasive monitoring devices. Repeated blood samples were obtained to determine haemoglobin and alcohol concentration, electrolytes, blood gas parameters and cardiac troponin I. After sacrifice, tissue was excised for quantification of infarction and histology. Cardiogenic shock was characterized by a significant decrease in mean arterial pressure (− 33%), cardiac output (− 29%), dP/dtmax (− 28%), carotid blood flow (− 22%), left ventricular fractional shortening (− 28%), and left ventricle end-systolic pressure–volume relationship (− 51%). Lactate and cardiac troponin I levels increased from 1.4 ± 0.2 to 4.9 ± 0.7 mmol/L (p = 0.001) and from 0.05 ± 0.02 to 14.74 ± 2.59 µg/L (p = 0.001), respectively. All haemodynamic changes were stable over a three-hour period with a 71% survival rate. The necrotic volume (n = 5) represented 24.0 ± 1.9% of total ventricular mass. No sham exhibited any variation under general anaesthesia. We described and characterized, for the first time, a stable, reproducible sheep model of cardiogenic shock obtained by percutaneous intracoronary ethanol administration.
Similar content being viewed by others


Introduction
Five to 10% of patients presenting with acute myocardial infarction (AMI) develop cardiogenic shock (CS). This population has a high mortality rate (40%–50%)1 , despite the improvement in survival due to primary reperfusion therapy, optimal pharmacological treatment, intra-aortic balloon pumping and the use of short-term mechanical circulatory support (STMCS)1. Insights into the pathophysiology of CS remain crucial to the development of new approaches, either pharmacological or mechanical, to reduce local and systemic effects of CS, and to appraise safety and efficacy of novel procedures prior to clinical translation. In this context, large animal models may provide a heuristic methodology to test pathophysiological hypotheses and new therapeutic procedures. These models should mimic human pathology, be reproducible, allow for control of the extent of ischemia, rely on minimally invasive techniques, require short preparation time and easy animal management, lead to impaired cardiac function and be cost-effective. Several models of myocardial infarction exist and are based on coronary artery ligation, coronary artery occlusion (by angiography balloon catheter or embolization), or intra-coronary alcoholisation. However, the current large animal models are suboptimal for several reasons, e.g. high mortality rate2, inconsistency in infarct size3 and the invasive approach itself4. Development of a consistent and stable model of CS, is needed to more fully evaluate the interplay between hemodynamic and heart mechanics on one hand and STMCS on the other. Moreover, the current models do not fully mimic the clinical scenario of CS induced by selective coronary occlusion for several reasons. First, systemic or selective coronary hypoxemia5 is responsible for a generalized diminished cardiac function due to the hibernating myocardial condition: in this setting, cellular mechanisms of adaptation are not the same than in distinct infarcted myocardium. Similar concerns should be taken into account in the case of global toxicity, induced by carbon monoxide6. The use of microspheres could overcome this difficulty7, but the injection in the left main coronary artery has a generalized effect on the left ventricle and is associated with a jeopardized cellular death in the whole myocardial wall thickness. Furthermore, the set-up of several studies requires STMCS activation soon after6,7 or even before8 CS induction in order to avoid premature death of animals, or an open-chest procedure for coronary ligatures9, which completely modifies the heart-pericardium and heart–lung interactions. In recent studies suggesting that early mechanical percutaneous support prior to revascularization might reduce the size of myocardial necrosis10, a condition of mild systemic hypo-perfusion is obtained rather than an effective hemodynamic CS.
The present study aimed to describe a reproducible closed chest model of acute CS due to myocardial infarction, mimicking the clinical setting and taking into account some specific aspects of human pathology, such as myocardial cell death, in a precise delineated coronary territory involving the whole myocardial wall thickness. This model was intended to be relatively stable in time, reproducible and have a high rate of survival after the induction of CS.
Methods
A total of thirty-two female Dorset sheep were used in this study. Their mean body weight was 49.0 ± 1.5 kg and they were 35 ± 6 months old. All animals received humane care in compliance with “European Community Standards on the Care and Use of Laboratory Animals” published on September 2010 (Directive 2010/63/EU on the protection of animals used for scientific purposes). The ethical committee N°050 of the French Ministry for Research and Innovation approved the project on 25 of September 2017.
Anaesthesia
Animals were fasted overnight with free access to water, and premedicated with intramuscular injection of ketamine (10–20 mg kg−1), acepromazine 0.1 mg kg−1 (5 mg mL−1) and buprenorphine (0.03 mL kg−1). Surface electrocardiographic leads were applied and a single lumen peripheral catheter (18 G) was then placed in a peripheral vein of the front limb. Propofol (2 mg kg-1) was given for induction of anaesthesia while its maintenance was ensured by isoflurane (inspired fraction 0.5 to 1.2%) and continuous infusion of midazolam (0.1 mg kg−1 h−1) after endotracheal intubation. Sufentanyl was administered for analgesia and animal comfort (0.15 µg kg−1 h−1). Anaesthesia depth was adjusted during animal preparation by monitoring vital signs and pupil position, then kept stable during the procedure. Myorelaxation was ensured by a bolus dose of rocuronium (0.6 mg kg−1) followed by a continuous intravenous infusion (0.3 mg kg−1 h−1). Volume controlled mechanical ventilation was used to maintain gas exchange and relied on an Aysis CS2 station (GE Healthcare SAS, Boston, MA, USA) set on controlled volume mode (Vt: 8 mL kg−1, Peep: 6–8 cmH20). The ventilation was adjusted to maintain an ETCO2 between 35 and 40 mmHg, and the FiO2, initially set to 0.5, was titrated to keep the PaO2 over 80 mmHg. 8 mL kg−1 of balanced crystalloids solution (Ringer Lactate Viaflo, Baxter, Deerfield, IL, USA) was administrated hourly. If intravascular volume expansion was required, hydroxyethyl starch was administered. 20 mg kg−1 of cefuroxime was given at the beginning of the experiment followed by 10 mg kg−1 re-injection every 2 h. Heart rhythm irregularities were prevented by using continuous infusion of amiodarone (1 mg kg−1 h−1), magnesium sulphate (35 mg kg−1 h−1) and xylocaine (5 mg kg−1 h−1). A unfractioned heparin bolus (100 UI kg−1) was given after neck incision, followed by a continuous intravenous infusion (100 UI kg−1 h−1). A three-lumen catheter (Arrow Medical Ltd, Kington, UK) placed in the left jugular vein was used for these purposes. Body temperature was monitored by a thermal probe and stabilized between 36 and 37 °C by an external heating system. Gastric and bladder tubes were inserted to empty respectively the stomach and monitor urine outflow.
Haemodynamic monitoring
All animals were monitored as follows:
An intracavitary ventricular electrocardiogram was obtained by Millar probe-associated electrodes (Millar Inc, Houston, TX, USA) and used to monitor heart rhythm and identify any abnormalities. Surface electrocardiogram was used to synchronize echocardiographic imaging.
A 6-F arterial catheter was placed in an artery of the anterior limb for continuous arterial pressure monitoring. TrueWave transducers (Edwards Lifesciences Corp, Irvine, CA, USA) were used for this purpose.
A Doppler probe (Transonic Systems Inc., Ithaca, NY, USA) was positioned around the left carotid artery.
A CCOmbo pulmonary artery catheter (Edwards Lifescience) connected to a Vigilance monitor (Edwards Lifescience) was inserted in the left jugular vein through a 9-F introducer allowing for continuous monitoring of pulmonary and central venous pressures, cardiac index by thermodilution, venous oxygen saturation (SvO2) and core body temperature.
A conductance catheter (Millar Inc) with 7-mm-spaced segments was inserted through an 8-F introducer in the right carotid artery. The probe was positioned in the left ventricle to collect ventricular pressures and volumes. The maximal positive (dP/dtmax) and negative (dP/dtmin) left ventricular pressure derivatives were calculated from the left ventricular pressure signal. The probe position was checked with radioscopy, transthoracic echocardiography and analysis of the collected signals. Volume calibration of the Millar conductance catheter was obtained by calculation of parallel conductance after distal pulmonary artery injection of a 15%-hypertonic saline solution. A 9-F introduction kit was placed in the right femoral vein and used to introduce a tip-ballooned 8F Fogarty catheter (Edwards Lifescience) up to the inferior vena cava junction with the right atrium, in order to control venous blood flow and allow end-systolic pressure–volume relationship calculation (ESPVR or maximal elastance or Emax). During dynamic manoeuvres, a respiratory pause was performed to limit the cardio-respiratory interactions induced by mechanical ventilation.
Data acquisition
A 16-bits Powerlab acquisition system (ADInstruments Ltd, Oxford, UK) was used for data collection and Labchart 8 (ADInstruments Ltd) for data analysis. Systems and captors were calibrated at the beginning of each experiment. A drift of the signals from the conductance catheter was researched before each point of intervention.
Echocardiography
Transthoracic echocardiographic examination (Vivid E95 Vet, GE, Boston, MA, USA) was performed concomitant to hemodynamic data collection at each point of intervention. Loops were recorded and a blinded-analysis was conducted. In the parasternal mid-papillary short axis plane, endocardial borders were traced in end-diastolic and end-systolic frames: LV end-diastolic (LVEDA) and end-systolic (LVESA) areas were used to calculate the fractional area change (FAC = [LVEDA − LVESA)/LVEDA] × 100).
Blood gas and electrolytes measurements
At each haemodynamic reading, blood samples were simultaneously obtained via the peripheral and pulmonary arterial catheters for blood gas analysis with lactate (Handheld Blood Analyzer iSTAT-1, Abbott Point of Care Inc., Princeton, NJ, USA). Blood samples were also taken to determine concentration of haemoglobin (UniCel DxH 800 Coulter Cellular Analysis System, Beckman Coulter, Inc., Brea, CA, USA), cardiac troponin I by immuno-assay, ethanol and key electrolytes (Na+, K+, Cl-, Ca2+) (AU2700 chemistry analyzer and DXI600 Access Immuno-assay system, Beckman Coulter) before infarct induction and prior to sacrifice.
Cardiogenic shock induction
As previously reported, an intra-coronary injection of ethanol was used to induce acute myocardial infarction11. We modified the technique by performing a sequential, multi-step myocardial “alcoholisation” procedure to obtain hemodynamically stable CS. An 8-F introduction kit was inserted in the right femoral artery and allowed the positioning of a 6F JL-guide catheter in the ostium of the main left coronary artery; after which a coronary angiography was performed. According to the distribution and segmentation of the left anterior descending coronary artery (LAD), an over-the-wire OPT 12 mm balloon catheter (Emerge OTW, Boston Scientific, Marlborough, MA, USA) was placed through a guide wire at different sites, progressing from the most distal to the most proximal. At each site, a sufficient volume of 99.9% ethyl alcohol was injected in order to completely block the blood flow distal to balloon inflation. Complete occlusion of the coronary artery was defined as TIMI (thrombolysis in myocardial infarction) flow ≤ 1 after alcohol injection. The injected alcohol volume depended on distal flow exclusion and rhythm disorder manifestations. The number of injected sites depended on the systemic hemodynamic consequences of necrosis extension. In accordance with previous animal models5,8,9,12, CS obtention was defined as a drop of more than 30% in mean arterial pressure and/or cardiac index associated to a rise of lactate concentration above 2.5 mmol L−1 in the hour following the last alcoholisation. Twenty-four animals underwent intracoronary alcoholisation (SHOCK group) and eight sham animals underwent interventricular coronary artery catheterization without any alcoholisation (SHAM group). A complete set of haemodynamic data (including standard hemodynamic, conductance catheter, echocardiographic derived parameters) was first collected after a 10 min stabilization period following the complete animal technical conditioning (Baseline 1), thirty minutes after CS constitution or one hour later for the SHAM group (Baseline 2) and every hour during a three-hour period (Fig. 1). Animal were sacrificed by intravenous injection of a lethal dose of sodium pentobarbital (≥ 100 mg/kg) before proceeding to sternotomy and heart explantation.

Study protocol. BL1 = baseline 1; BL2 = Baseline 2; H + 1 = 60 min after BL2; H + 2 = 120 min after BL2; H + 3 = 180 min after BL2.
Myocardial necrosis quantification
A sternotomy was performed at the end of experimentation. An aortic supra-coronary clamp was placed and Evans blue dye injected at the aortic root to colour the perfused cardiac tissues. The heart was then extracted and sectioned perpendicular to the main axis, to obtain 1 cm-thick sections. The sections were weighed, bathed for 10 min in a 2,3,5-triphenyl tetrazolium chloride (TTC) solution to discriminate infarcted and the area-at-risk (AAR). Both sides of each section were photographed and analysed using NIH Image J software to obtain a planimetric quantification of the infarcted area, expressed as a percentage of the left ventricle AAR and total ventricular mass; values for all sections from each heart were averaged.
Myocardial histopathology
One section per animal was used for histologic and protein analyses. Biopsies of healthy and infarcted myocardium were processed for the following analyses:
Optical histology-specimens were fixed in 10% formalin, cut and stained with haematoxylin–eosin (H/E) or Masson’s trichrome (M/T), after which they were examined for pathological changes in the infarcted myocardium and alcohol-injured LAD region.
Electron Microscopy-specimens were snap frozen directly in optimum cutting temperature (OCT) compound; after being frozen at − 80 °C, samples were cut into sections measuring 20 nm and analysed for tissue oedema, mitochondrial morphology and disruption of the intercellular tight-junctions in order to evaluate the effects of ventricular wall stress.
Statistical analysis
Results are expressed as mean ± SEM. A student’s t-test was used to compare two mean values. An ANOVA was performed for comparison of multiple means and post-hoc test analysis was performed using a Newman–Keuls test. A p-value < 0.05 was required to reject the null hypothesis.
Results
Closed-chest ischemic cardiogenic shock model
Twenty-four animals underwent CS induction (SHOCK), according to our protocol, and seven expired prior to completion of the protocol due to refractory rhythmic complications, thus the procedure had a 71% survival rate. Among the remaining 17 SHOCK animals, 5 required a very low dose (mean dose ≤ 0.01 µg kg−1 min−1) continuous epinephrine infusion as a rescue therapy after reversal of ventricular fibrillation or tachycardia to maintain systolic arterial pressure ≥ 55 mmHg, after the induction of CS. The mean intracoronary alcohol volume necessary to obtain CS was 6.1 ± 0.8 mL. The mean time between beginning of the alcoholisation procedure and shock instauration was 84 ± 8 min. There were no significant differences in electrolytes concentration between the SHOCK and SHAM groups (Na+ = 140 ± 2 mmol L−1; Cl− = 105 ± 4 mmol L−1; K+ = 2.7 ± 0.3 mmol L−1 and Ca2+ = 2.17 ± 0.12 mmol L−1). Ethanol concentrations were undetectable in blood obtained from animals in which CS was induced.
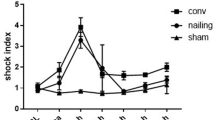
The haemodynamic profile of CS is summarized in Table 1. It was characterized by a significant decrease in mean arterial pressure (− 33%), cardiac output (− 29%), end-systolic left ventricular pressure (− 26%) and dP/dtmax (− 28%). A greater decrease in the dP/dtmin was observed (− 55%). Carotid blood flow (− 22%) and left ventricular fractional shortening (− 28%) also decreased, while left ventricle tele-diastolic pressure increased (+ 38%). Due to the progressive complexity of our hemodynamic monitoring, Millar-derived parameters were not available for all animals (SHOCK n = 5, SHAM n = 6). The Millar-derived parameter ESPVR was significantly reduced (− 51%). A significant decrease in SvO2 was also observed. Lactate and cardiac troponin I levels increased from 1.4 ± 0.2 to 4.9 ± 0.7 mmol/L (p = 0.001) and from 0.05 ± 0.02 to 14.74 ± 2.59 µg/L (p = 0.001), respectively. These variations remained stable in SHOCK animals during the follow-up period while SHAM animals (n = 8) did not exhibit any variation at any timepoint (Tables 2, 3 and Fig. 2).

Changes in haemodynamic parameters between baseline 1 (BL1) and baseline 2 (BL2) in SHAM (open circles) and SHOCK (closed circles) groups are presented as mean ± SEM. Representative data from n = 17 CS and n = 8 SHAM animals is presented. A paired Sudent's t test was used to compare two mean values. *p<0.05 versus BL1. MAP mean arterial pressure, CI cardiac index, SVI Stroke volume index, LVESP Left ventricular end-systolic pressure, dP/dtmax maximal positive left ventricular pressure derivative, ESPVR End-systolic pressure-volume relationship, MI myocardial infarction.
Echocardiography parameters
All SHOCK animals uniformly showed apico-antero-septal akinesis associated with a decrease in the fractional area change (Table 1).
Morphometric and histological analysis
SHOCK animals (n = 5) uniformly showed transmural perfusion defects in the apico-antero-septal wall; the necrotic volume and overall ischemic region (AAR) represented respectively 24.0 ± 1.9% and 43.9 ± 3.8% of LV volume, yielding an ‘infarct to AAR ratio’ of 0.55 ± 0.03 (Fig. 3). Pathological observations (in two animals) with H/E staining demonstrated loss of cross striations, contraction bands, some oedema with haemorrhage and neutrophil infiltrate, which are all signs of lesions associated with myocardial cellular degeneration (Fig. 3). Analysis by electron microscopy showed clear differences between SHAM (n = 1) and SHOCK (n = 1) animals. In samples from the SHAM animal the mitochondria were very dense and not swollen, and the inner membrane folding (Cristae) was well preserved (Fig. 4a, b). On the contrary, in samples from the SHOCK animal the mitochondria were swollen and broken with the inner membrane folding (Cristae) completely altered (Fig. 4c, d).

Macroscopic and microscopic aspect of healthy and infarcted myocardium from two representative animals, one CS and one SHAM. Top of figure: macroscopic aspect of healthy (a) and infarcted (d) myocardium after Evans blue dye and 2,3,5-triphenyl tetrazolium chloride coloration. The dotted blue line delimits the ischemic territory. Bottom of figure: microscopic aspect of healthy (b, c) and infarcted myocardium (e, f). (b, e) scale: 50 μm; (c, f) scale: 20 μm. Red arrows: red blood cells. Blue arrows: leucocytes. Green arrows: sarcomeres. Dark red arrows: contraction bands. C: collagen. M: myocardial cells.

Electron microscopy of healthy (left panel: a, b) and infarcted (right panel: c, d) myocardium from two representative animals, one CS and one SHAM. ‘a’ and ‘c’ scale: 2 μm; ‘b’ and ‘d’ scale: 200 nm. In the healthy non-ischemic zone, mitochondria are aligned along the normally structured muscle fibres (a), very dense, not swollen and the inner membrane folding (Cristae) are well preserved (b). In the ischemic zone, the mitochondrial network is disorganized (c), mitochondria are swollen, the inner membrane folding (Cristae) is completely altered (d).
Discussion
We described here, for the first time, a reproductible CS model obtained in sheep by percutaneous intracoronary ethanol injection that demonstrates a significant decrease in mean arterial pressure (− 33%), cardiac output (− 29%), dP/dtmax (− 28%), dP/dtmin (− 55%), carotid blood flow (− 22%), left ventricular fractional shortening (− 28%), left ventricle end-systolic pressure–volume relationship (− 51%) and remained stable over a three-hours period. Microscopic analysis revealed signs of myocardial cellular degeneration, e.g. loss of cross striations, contraction bands, some oedema with haemorrhage and neutrophil infiltrate; electron microscopy analysis revealed swollen mitochondria with the inner membrane folding completely altered in infarcted tissue.
Large animal model
The use of large animal models in cardiac physiology offers some advantages, including clinical relevancy, the possibility to test delivery devices based on catheters and to study the dose–effect relationships13. Several characteristics make sheep a valuable species; unlike canine models and similar to humans, they do not sufficiently develop a collateral circulation network beneath the epicardium: as a result, it is easier to induce myocardial infarction in a reproducible manner, including the related issues of myocardial hibernation, myocardial stunning14, and LV aneurysm15. Moreover, unlike bovine models and similar to humans, sheep allow studies on angiogenesis16. Even if swine have a cardiac morphology very similar to that of humans17, they grow up relatively rapidly, which makes it harder to conduct long-term studies; furthermore, it is challenging to set up devices for thoracotomy and cardiopulmonary bypass in this species18.
Closed chest model
The pioneer sheep models of antero-apical MI19 and postinfarction heart failure20 had a disadvantage in that they required a thoracotomy due to the variability of coronary arterial anatomy. Contrary to them, all closed-chest models have the advantage to profoundly limit the huge trauma of thoracotomy, thus respecting cardiac and whole-body physiology and favouring recovery. Percutaneous methods also show reliability and reproducibility, and are able to induce myocardial infarction with a pathophysiology mimicking that of myocardial ischemia in human clinical setting. However, they have inherent limitations. (1) In percutaneous models employing a microsphere embolization technique, the precise quantity of particles injected is required14, the volume and the anatomical location of myocardial necrosis cannot be selectively produced, thus rendering the extent of myocardial damage poorly predictable21; moreover, in some dog models, left ventricular function is acutely and severely depressed, leading to a high mortality, while survivors experience an almost complete recovery, partially due to the rich collateral circulation of the canine heart22. (2) The exploration of well-defined ischemia and related cardioprotective therapies are barely explored by models established with coronary thrombosis because of the variability of time needed for occlusive thrombus formation, and of the difficult control of duration and severity of ischemia23. (3) Models employing angioplasty balloon inflation and deflation are typically used to explore the myocardial reperfusion phenomenon, but they are burdened with high incidence of left ventricular fibrillation24. (4) When a metallic coil embolization technique is utilized, it is almost impossible to determine in advance the site of the arterial obstruction, which results in considerable variability of the ischemic areas sizes among different animal species25. (5) Gelatine sponge embolization procedure as well is complicated by high rate of ventricular fibrillation, ventricular tachycardia and mortality2. In our view, this totally closed-chest sheep CS model addresses and overcomes many of these limitations and importantly, it has a high survival rate of (71%). Although few authors specifically reported this criterion26, this high survival rate appears to be another strength of this model: we provide, as additional data, a table summarizing the main features of known cardiogenic shock animal models (Annexe 1).
Hemodynamic profile of cardiogenic shock model
As expected, our closed-chest CS model is characterized by approximately a 30% decrease of systemic arterial pressure and cardiac output as well as inotropic parameters. As previously reported, myocardial infarction appears to affect more severely lusitropic properties27. Although our CS model was associated with a significant decrease in SvO2, it should be pointed out that the value remains relatively high in comparison to a previous reported model12. This difference may be explained by two reasons: (1) the fact that all our animals were maintained under general anaesthesia, including myorelaxant agents, which could be responsible for a profound decrease in oxygen consumption; (2) the high level of arterial oxygenation during the entirety of the experiment. However, a profound end-organ hypoperfusion state was confirmed by a significant increase in lactate value. More severe CS has been previously reported7,8,12, but most of these authors implanted a mechanical circulatory support device before the induction of CS or immediately after7, 8. In our model, heart rate was slightly but significantly lower after CS induction. Such a slight decrease is unlikely to be responsible for the drop in cardiac output, or the low systolic volume and consequently affected left ventricular ejection fraction. Most previously reported models of CS noted a compensatory tachycardia8,28, rather than bradycardia. However, our findings may be easily explained by the fact that at the beginning of the experiment, all animals received analgesia based on sufentanyl associated to a mixture of xylocaine, amiodarone and sulphate magnesium to prevent arrhythmic irregularities. These drugs are known to induce bradycardia and could have influenced the hemodynamic profile of our model29,30,31. These bradycardizing effects could have been accentuated by ischemic dysfunction of the sinus node. Finally, as we were unable to detect any alcohol plasma concentrations in animals, an inhibition of baroreflex bradycardia through GABA receptors by ethanol injection32 is highly unlikely. It should be noted that an absence of compensatory tachycardia has been widely reported by others developing animal model of CS7,33,34. Lastly, five animals received epinephrine. Because the mean dose was ≤ 0.01 µg kg−1 min−1, it is highly unlikely that a such dose could significantly modify the hemodynamic profile of cardiogenic shock in our model.
Coronary alcoholisation
Intracoronary alcohol injection was firstly used to treat arrhythmias in patients suffering from chronic ischemic heart disease or myocarditis35, and to relieve the ventricular pressure gradient in symptomatic patients suffering from obstructive hypertrophic cardiomyopathy36. Inspired by these clinical settings, intracoronary alcohol injection has become a supplemental approach in animal models of ischemic myocardial dysfunction: under video-fluoroscopic guidance, an ethyl alcohol solution (at variable percentage) is injected in specific coronary vessels according to predetermined experimental goals. This method has proven to be simple and reproducible37,38 and, compared to other techniques, it enables the TIMI flow grade evaluation immediately after the injection and without removing the balloon device13.
Alcohol concentration and Infarct Size
Previous findings have shown that solutions with slight ethyl alcohol concentration are not adequate to induce tissue damage39 and that the severity of myocardial damage tends to be greater with solutions that have a higher ethyl alcohol concentration (50 to 100%)35,37. The use of pure alcohol for coronary embolization in our experimental model allowed us to obtain homogeneous infarct sizes of 24 ± 4% of ventricular mass. These results differ from measures reported by a recent review paper27 in which infarct size measured by TTC staining is described as ranging from 30 to 60% of the total LV volume. We are not able to fully explain these discrepancies: studies cited by Linsdey27 vary in several ways (animal size, infarct induction, time elapsed from infarct induction until sacrifice) whereas studies describing alcohol concentration do not express infarct size as a percentage of total ventricular mass35,37.
AAR/infarct size ratio
It has previously been demonstrated that infarct size strongly depends on the extension of the occluded coronary bed, rather than the anatomic occlusion site, regardless of the oxygen demand-supply balance40. Accordingly, although a variable number of regions of the LAD territory were injected with alcohol, we were able to obtain a homogeneous percentage of AAR and “infarct size to AAR ratio” (infarct to AAR volume ratio 0.55 ± 0.03).
Histological effects of coronary alcoholisation
Differential effects of intracoronary alcohol injection on coronary vessels or myocardial tissue have been previously described37. As regards intraluminal response, a persistent and extensive thrombi constitution is prompted by high concentrations of alcohol, distally to the injection site35 due to sub-endothelium exposure. Progressively, thrombus is organized and calcified and a chronic total occlusion ensues18. As regards myocardial structure, myocardial necrosis occurs from 100% ethanol injection into the canine coronary circulation, either arterial or venous: it is focal or patchy and non-confluent, and involves more than half the thickness of the left ventricular wall35. Direct intra-myocardial alcohol injection induces myocardial necrosis without intraluminal thrombosi41.
If severe ischemia lasts more than 20- to 30-min, cardiomyocytes in the subendocardial area undergo irreversible changes ranging from sarcolemmal disruption to striking perturbations in mitochondrial architecture; typical mitochondrial ultrastructural alterations are represented by amorphous matrix densities and severe swelling. All cardiomyocytes showing these ultrastructural alterations cannot be rescued and they will constitute the infarct environment after death42. According to studies on reperfused infarction in sheep, it would seem that the mid-myocardium is more vulnerable to ischemic injury whereas the sub-endocardium is relatively resistant43.
Accordingly, we found thrombi in ischemic tissue and coagulative necrosis at an early stage. Optical microscopy revealed that the full myocardial thickness was involved in the ischemic process, with muscular fibres appearing to be hyper-contracted, while nuclei were still present because animals were sacrificed before their disappearance (18–24 h). Electron microscopic analysis revealed that the mitochondrial network was disorganized, mitochondria swollen, and the inner membrane folding completely altered; indicating that the mitochondria were broken and unable to provide any energetic supply for cell survival44.
Correlation “AAR/infarct size ratio”: haemodynamic
The fall in coronary blood flow is immediately followed by myocardial ischemia, which implies myocardial anaerobic metabolism, fast ATP depletion, and metabolic waste amassing; the myocardial mechanism is severely altered and results in harsh systolic and diastolic dysfunction, and the ischemic muscle stretches instead of shortens during systole45. As ischemia progresses (less than 15 min), the myocardium turns into stunning and is not forthwith responsive to reperfusion46. Whenever injury continues, myocytes death leads to infarction42,47. Several clinical and experimental observations48,49 suggest that CS complicating myocardial infarction is accompanied by extensive myocardial injury and that at least 40% of left ventricular volume should be ischemic to induce CS. Nevertheless, in the clinical scenario, a severe degree of myocardial insult is not always necessary to cause CS45.
Although in our experimental model we observed homogeneous infarct sizes lower than those meant to induce CS, the overall ischemic region, including the AAR, represents 43.9 ± 3.8% of LV volume. This is consistent with the concept that myocardial dysfunction can develop even in the presence of viable tissue: the presumed mechanism underlying this phenomenon is myocardial stunning, i.e. a condition of ischemic injury without myocardial necrosis or cell death46. Moreover, previous studies showed that hemodynamic variables become abnormal when the area at risk is larger than 25%50 or the infarct size is between 12 and 13%28 (Annexe 1). Of course, the alternative evolution towards a complete myocardial recovery or a myocardial necrosis cannot be determined because stunning may persist for hours to weeks47. Accordingly, we speculate that by prolonging the post-induction period and delaying the sacrifice, more myocardial tissue in the AAR would have become necrotic.
Study limitations
No model is devoid of flaws.
- 1.
Myocardial infarction is defined as myocardial necrosis consequent to myocardial ischemia51. In our model, a chemical injury rather than ischemia was responsible of the heart damage. Although the mechanism is not the same, the histological appearance, both on optical and electron microscopy, and the left ventricular kinetic pattern of the affected area are quite similar to classical infarct by myocardial ischemia.
- 2.
Very few specimens have been used for microscopy investigation: one SHAM and two SHOCK for each histological approach (optical and electron): nevertheless, all findings found in SHOCK animals are pathognomonic of infarcted cardiac tissue and not present in controls.
- 3.
The use of an ischemia–reperfusion model is usually suggested in order to explore potential strategies prompting a reduction of acute myocardial infarct size25: our model exposes myocardial tissue to a rapid sequence of ischemia–reperfusion phases when the balloon catheter is progressively pulled from the distal to the final proximal site of ethyl alcohol injection. Nevertheless, the intracoronary thrombosis remains irreversible.
- 4.
We used healthy animals, whereas human patients with acute ischemic heart failure belong a population exhibiting several risk factors, such as diabetes, hypertension, and atherosclerosis11; nevertheless, almost all animal models are confronted with this limitation.
- 5.
Lastly, the observation time was limited to three consecutive hours. This study period may appear to be short but it is similar to those proposed by other author8. The main reason of our choice to limit the study period is practical. The preparation of the experimental setting and CS induction took approximately 4–5 h. Consequently, an observation time of only three consecutive hours was compatible with operatory room organization.
Possible utility of our model for future research
As previously outlined, few large animal models of consistent and stable refractory cardiogenic shock are available for research purpose to-date, and the previously established models do not completely mimic the clinical scenario of CS induced by selective coronary occlusion. In our experimental model, we do not use coronary hypoxemia or hypercarbia thus minimizing the hibernating myocardial condition, and we are able to obtain homogeneous trans-mural myocardial damage without jeopardizing cellular death in a precise coronary artery territory. Furthermore, permanent coronary occlusion mimics the clinical situation of patients enduring acute myocardial without timely or successful reperfusion because of contraindications or logistic issues52. In such a specific context, our experimental set-up could lead to a better understanding of mechanical interaction between short-term mechanical circulatory support or LVAD and an acutely failing ventricle, thus allowing a precise tuning of flow and pressure generated by an assist device. The potential usefulness or harmfulness of vasoactive and inotropic drugs (calcium inhibitors, alpha-2-antagonists, sympathomimetics or other like levosimendan) in patients assisted with those devices might also be explored in this model. Moreover, the impact of ventricular support on early remodelling of non-ischemic territory could be more fully investigated, as well as the time-course evolution of the AAR in cases where it is not possible to reperfuse: while LV mechanical unloading prior to coronary revascularization may theoretically reduce infarct size by reducing the AAR10, no experimental investigation has been undertaken in the case of permanent coronary occlusion.
Conclusions
We described and exhaustively characterized a unique, reproductible, homogenous and stable sheep model of CS obtained by percutaneous intracoronary ethanol administration. This technique grants trans-mural infarct formation with a clinical profile close to the human one. We think that our model may be appropriate to better understand the mechanical interaction between short-term mechanical circulatory support and/or LVAD and an acutely failing ventricle, the impact of ventricular support on early remodelling of non-ischemic territory, as well as the time-course evolution of AAR in the case of impossible reperfusion.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Harjola, V. P. et al. Clinical picture and risk prediction of short-term mortality in cardiogenic shock. Eur. J. Heart Fail. 17, 501–509. https://doi.org/10.1002/ejhf.260 (2015).
Reffelmann, T. et al. A novel minimal-invasive model of chronic myocardial infarction in swine. Coron. Artery Dis. 15, 7–12 (2004).
Weil, B. R., Konecny, F., Suzuki, G., Iyer, V. & Canty, J. M. Jr. Comparative hemodynamic effects of contemporary percutaneous mechanical circulatory support devices in a porcine model of acute myocardial infarction. JACC Cardiovasc. Interv. 9, 2292–2303. https://doi.org/10.1016/j.jcin.2016.08.037 (2016).
Meyns, B., Stolinski, J., Leunens, V., Verbeken, E. & Flameng, W. Left ventricular support by catheter-mounted axial flow pump reduces infarct size. J. Am. Coll. Cardiol. 41, 1087–1095 (2003).
Ostadal, P. et al. Novel porcine model of acute severe cardiogenic shock developed by upper-body hypoxia. Physiol. Res. 65, 711–715 (2016).
Simonsen, C., Magnusdottir, S. O., Andreasen, J. J., Rohde, M. C. & Kjaergaard, B. ECMO improves survival following cardiogenic shock due to carbon monoxide poisoning: an experimental porcine model. Scand. J. Trauma Resusc. Emerg. Med. 26, 103. https://doi.org/10.1186/s13049-018-0570-6 (2018).
Moller-Helgestad, O. K. et al. Impella CP or VA-ECMO in profound cardiogenic shock: left ventricular unloading and organ perfusion in a large animal model. EuroIntervention 14, e1585–e1592. https://doi.org/10.4244/EIJ-D-18-00684 (2019).
Ostadal, P. et al. Electrocardiogram-synchronized pulsatile extracorporeal life support preserves left ventricular function and coronary flow in a porcine model of cardiogenic shock. PLoS ONE 13, e0196321. https://doi.org/10.1371/journal.pone.0196321 (2018).
Vanhuyse, F. et al. Moderate hypothermia improves cardiac and vascular function in a pig model of ischemic cardiogenic shock treated with veno-arterial ECMO. Shock 47, 236–241. https://doi.org/10.1097/SHK.0000000000000712 (2017).
Kapur, N. K. et al. Mechanically unloading the left ventricle before coronary reperfusion reduces left ventricular wall stress and myocardial infarct size. Circulation 128, 328–336. https://doi.org/10.1161/CIRCULATIONAHA.112.000029 (2013).
Crisostomo, V. et al. Development of a closed chest model of chronic myocardial infarction in Swine: magnetic resonance imaging and pathological evaluation. ISRN Cardiol. 2013, 781762. https://doi.org/10.1155/2013/781762 (2013).
Stenberg, T. A. et al. The acute phase of experimental cardiogenic shock is counteracted by microcirculatory and mitochondrial adaptations. PLoS ONE 9, e105213. https://doi.org/10.1371/journal.pone.0105213 (2014).
Isorni, M. A. et al. Comparative analysis of methods to induce myocardial infarction in a closed-chest rabbit model. Biomed. Res. Int. 2015, 893051. https://doi.org/10.1155/2015/893051 (2015).
Ikram, H. et al. An ovine model of acute myocardial infarction and chronic left ventricular dysfunction. Angiology 48, 679–688. https://doi.org/10.1177/000331979704800803 (1997).
Maxwell, M. P., Hearse, D. J. & Yellon, D. M. Species variation in the coronary collateral circulation during regional myocardial ischaemia: a critical determinant of the rate of evolution and extent of myocardial infarction. Cardiovasc. Res. 21, 737–746. https://doi.org/10.1093/cvr/21.10.737 (1987).
Bertho, E. & Gagnon, G. A comparative study in three dimension of the blood supply of the normal interventricular septum in human, canine, bovine, procine, ovine and equine. Dis. Chest 46, 251–262 (1964).
Terp, K. et al. The hemodynamic impact of diffuse myocardial ischemic lesions: an animal experimental model based on intracoronary microembolization. Heart Vessels 13, 132–141 (1998).
Kim, W. et al. A porcine model of ischemic heart failure produced by intracoronary injection of ethyl alcohol. Heart Vessels 26, 342–348. https://doi.org/10.1007/s00380-010-0022-3 (2011).
Markovitz, L. J. et al. Large animal model of left ventricular aneurysm. Ann. Thorac. Surg. 48, 838–845 (1989).
Millner, R. W., Mann, J. M., Pearson, I. & Pepper, J. R. Experimental model of left ventricular failure. Ann. Thorac. Surg. 52, 78–83 (1991).
Heusch, G., Schulz, R., Baumgart, D., Haude, M. & Erbel, R. Coronary microembolization. Prog. Cardiovasc. Dis. 44, 217–230. https://doi.org/10.1053/pcad.2001.26968 (2001).
Smiseth, O. A., Lindal, S., Mjos, O. D., Vik-Mo, H. & Jorgensen, L. Progression of myocardial damage following coronary microembolization in dogs. Acta Pathol. Microbiol. Immunol. Scand. A 91, 115–124 (1983).
Bush, L. R. & Shebuski, R. J. In vivo models of arterial thrombosis and thrombolysis. FASEB J. 4, 3087–3098. https://doi.org/10.1096/fasebj.4.13.2210155 (1990).
Garner, D., Ginzton, L. E., Jagels, G. & Laks, M. M. A new technique for producing myocardial infarction using coronary artery balloon occlusion. Cardiovasc. Res. 22, 42–46. https://doi.org/10.1093/cvr/22.1.42 (1988).
Mitsos, S., Katsanos, K., Dougeni, E., Koletsis, E. N. & Dougenis, D. A critical appraisal of open- and closed-chest models of experimental myocardial ischemia. Lab. Anim. 38, 167–177. https://doi.org/10.1038/laban0509-167 (2009).
Gotberg, M. et al. Mild hypothermia reduces acute mortality and improves hemodynamic outcome in a cardiogenic shock pig model. Resuscitation 81, 1190–1196. https://doi.org/10.1016/j.resuscitation.2010.04.033 (2010).
Lindsey, M. L. et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Heart Circ. Physiol. 314, H812–H838. https://doi.org/10.1152/ajpheart.00335.2017 (2018).
Wouters, P. F. et al. Left ventricular assistance using a catheter-mounted coaxial flow pump (Hemopump) in a canine model of regional myocardial ischaemia. Eur. Heart J. 14, 567–575. https://doi.org/10.1093/eurheartj/14.4.567 (1993).
Al Rasheed, N. M., Al Sayed, M. I., Al Zuhair, H. H., Al Obaid, A. R. & Fatani, A. J. Effects of two newly synthesized analogues of lidocaine on rat arterial blood pressure and heart rate. Pharmacol. Res. 43, 313–319. https://doi.org/10.1006/phrs.2000.0783 (2001).
Freye, E., Schmidhammer, H. & Latasch, L. 14-methoxymetopon, a potent opioid, induces no respiratory depression, less sedation, and less bradycardia than sufentanil in the dog. Anesth. Analg. 90, 1359–1364. https://doi.org/10.1097/00000539-200006000-00018 (2000).
Michelin, M. T., Cheucle-Beaughard, M. & Duchene-Marullaz, P. Comparative effects of amiodarone, bepridil and perhexiline on coronary venous flow and several cardiovascular parameters. Arch. Int. Pharmacodyn. Ther. 245, 236–248 (1980).
Varga, K. & Kunos, G. Ethanol inhibition of baroreflex bradycardia: role of brainstem GABA receptors. Br. J. Pharmacol. 101, 773–775. https://doi.org/10.1111/j.1476-5381.1990.tb14155.x (1990).
Feola, M., Haiderer, O. & Kennedy, J. H. Experimental graded “pump failure” of the left ventricle. J. Surg. Res. 11, 325–341. https://doi.org/10.1016/0022-4804(71)90110-7 (1971).
Udesen, N. L. J. et al. Impact of concomitant vasoactive treatment and mechanical left ventricular unloading in a porcine model of profound cardiogenic shock. Crit. Care. 24, 95. https://doi.org/10.1186/s13054-020-2816-8 (2020).
Inoue, H., Waller, B. F. & Zipes, D. P. Intracoronary ethyl alcohol or phenol injection ablates aconitine-induced ventricular tachycardia in dogs. J. Am. Coll. Cardiol. 10, 1342–1349 (1987).
Maron, B. J. Role of alcohol septal ablation in treatment of obstructive hypertrophic cardiomyopathy. Lancet 355, 425–426. https://doi.org/10.1016/S0140-6736(00)82005-X (2000).
Haines, D. E., Whayne, J. G. & DiMarco, J. P. Intracoronary ethanol ablation in swine: effects of ethanol concentration on lesion formation and response to programmed ventricular stimulation. J. Cardiovasc. Electrophysiol. 5, 422–431 (1994).
Joudinaud, T. M. et al. An experimental method for the percutaneous induction of a posterolateral infarct and functional ischemic mitral regurgitation. J. Heart Valve Dis. 14, 460–466 (2005).
Ellman, B. A., Green, C. E., Eigenbrodt, E., Garriott, J. C. & Curry, T. S. Renal infarction with absolute ethanol. Invest. Radiol. 15, 318–322 (1980).
Reimer, K. A. & Jennings, R. B. The, “wavefront phenomenon” of myocardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and collateral flow. Lab. Invest. 40, 633–644 (1979).
Weismuller, P. et al. Chemical ablation by subendocardial injection of ethanol via catheter–preliminary results in the pig heart. Eur. Heart J. 12, 1234–1239. https://doi.org/10.1093/eurheartj/12.11.1234 (1991).
Jennings, R. B., Murry, C. E., Steenbergen, C. Jr. & Reimer, K. A. Development of cell injury in sustained acute ischemia. Circulation 82, 2–12 (1990).
Sakamoto, H. et al. Effect of reperfusion on left ventricular regional remodeling strains after myocardial infarction. Ann. Thorac. Surg. 84, 1528–1536. https://doi.org/10.1016/j.athoracsur.2007.05.060 (2007).
Anversa, P. et al. Ischemic cardiomyopathy: myocyte cell loss, myocyte cellular hypertrophy, and myocyte cellular hyperplasia. Ann. N. Y. Acad. Sci. 752, 47–64. https://doi.org/10.1111/j.1749-6632.1995.tb17405.x (1995).
Frangogiannis, N. G. Pathophysiology of myocardial infarction. Compr. Physiol. 5, 1841–1875. https://doi.org/10.1002/cphy.c150006 (2015).
Bolli, R. Why myocardial stunning is clinically important. Basic Res. Cardiol. 93, 169–172 (1998).
Aymong, E. D., Ramanathan, K. & Buller, C. E. Pathophysiology of cardiogenic shock complicating acute myocardial infarction. Med. Clin. N. Am. 91, 701–712. https://doi.org/10.1016/j.mcna.2007.03.006 (2007).
Boor, P. J. & Reynolds, E. S. Myocardial infarct size: clinicopathologic agreement and discordance. Hum. Pathol. 8, 685–695 (1977).
Page, D. L., Caulfield, J. B., Kastor, J. A., DeSanctis, R. W. & Sanders, C. A. Myocardial changes associated with cardiogenic shock. N. Engl. J. Med. 285, 133–137. https://doi.org/10.1056/NEJM197107152850301 (1971).
Kaul, S. et al. The importance of defining left ventricular area at risk in vivo during acute myocardial infarction: an experimental evaluation with myocardial contrast two-dimensional echocardiography. Circulation 75, 1249–1260 (1987).
Alpert, J. S. & Thygesen, K. A new global definition of myocardial infarction for the 21st century. Pol. Arch. Med. Wewn 117, 485–486 (2007).
Gharacholou, S. M. et al. Implications and reasons for the lack of use of reperfusion therapy in patients with ST-segment elevation myocardial infarction: findings from the CRUSADE initiative. Am. Heart J. 159, 757–763. https://doi.org/10.1016/j.ahj.2010.02.009 (2010).
Acknowledgements
The authors owe their sincere gratitude to David Gonthier and Virginie Loyer for the outstanding quality of their technical assistance during the experimental work in the operative setting. This work was financed by the French Society of Anaesthesiology and Critical Care (SFAR) through 2017 Research Grants, by INSERM (French National Institute of Health and Medical Research) Unit n°1034 through supplemental funding, and the Association des Anesthésistes-Réanimteurs COeur-THorax et VAisseaux (ARCOTHOVA).
Author information
Authors and Affiliations
Contributions
M.R., J.I. and A.O. conceived the study. M.R., J.I., P.P., T.C. and A.O. designed the study protocol. M.R., J.I., Y.E., C.S. and A.O. acquired and analysed data. M.R., J.I., Y.E., M.P., C.S., P.P. and A.O. interpreted data. M.R., J.I. and A.O. wrote the draft and with O.B., M.H., A.B. and T.C. revised it. All authors reviewed the manuscript and approved it in its final version.
Corresponding author
Ethics declarations
Competing interests
The authors of this paper, except Professor Ouattara, do not have any conflict of interests to declare regarding any direct financial relation with the commercial identities mentioned in the paper. Professor Ouattara has received honoraria as consultant to Abiomed Inc and ORION Pharma.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rienzo, M., Imbault, J., El Boustani, Y. et al. A total closed chest sheep model of cardiogenic shock by percutaneous intracoronary ethanol injection. Sci Rep 10, 12417 (2020). https://doi.org/10.1038/s41598-020-68571-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-68571-5
This article is cited by
-
The impact of ECPELLA on haemodynamics and global oxygen delivery: a comprehensive simulation of biventricular failure
Intensive Care Medicine Experimental (2024)
-
Extending the ‘host response’ paradigm from sepsis to cardiogenic shock: evidence, limitations and opportunities
Critical Care (2023)
-
An innovative ovine model of severe cardiopulmonary failure supported by veno-arterial extracorporeal membrane oxygenation
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
