
Abstract
Little is known about the epigenetic changes associated with delayed cerebral ischemia (DCI) pathogenesis after subarachnoid hemorrhage (SAH). Here, we investigated genome-wide DNA methylation profiles specifically associated with DCI, which is a major contributor to poor clinical outcomes. An epigenome-wide association study (EWAS) and quantitative real-time PCR (qRT-PCR) were conducted in 40 SAH patients (DCI, n = 13; non-DCI, n = 27). A replication study using bisulfite modification and methylation-specific PCR was further performed in 36 patients (DCI, n = 12; non-DCI, n = 24). The relative degree of methylation was described as the median and 25th–75th percentile. No significant differences in clinical characteristics between DCI and non-DCI groups were observed. Among the top 10 differentially methylated genes analyzed via EWAS, two aberrantly methylated CpG sites of cg00441765 (INSR gene) and cg11464053 (CDHR5 gene) were associated with decreased mRNA expression (2−ΔCt). They include INSR [0.00020 (0.00012–0.00030) in DCI vs. 0.00050 (0.00030–0.00068) in non-DCI] and CDHR5 [0.114 (0.053–0.143) in DCI vs. 0.170 (0.110–0.212) in non-DCI]. Compared with non-DCI cases, patients with DCI exhibited an increased degree of methylation in the replication study: INSR, 0.855 (0.779–0.913) in DCI vs. 0.582 (0.565–0.689) in non-DCI; CDHR5, 0.786 (0.708–0.904) in DCI vs. 0.632 (0.610–0.679) in non-DCI. Hypermethylation of two novel genes, INSR and CDHR5 may serve as a biomarker for early detection of DCI following SAH.
Similar content being viewed by others

Introduction
Delayed cerebral ischemia (DCI) is a major contributor to poor neurologic outcomes in patients diagnosed with subarachnoid hemorrhage (SAH)1. However, it is also a preventable and treatable complication. Well-known risk factors for DCI include female gender, cigarette smoking, hyperglycemia, high Hunt-Hess (HH) grade and thick hemorrhage at admission1,2,3. The majority of genetic studies investigating DCI have focused on gene expression or linkage analyses of candidate genes4. Candidate genes include those associated with inflammation, endothelial dysfunction, fibrinolysis, and brain metabolism. A previous meta-analysis5 showed that ApoE ε4 carriers had a higher risk of DCI than non- ε4 carriers. Kim et al.6 reported that DCI was more frequently observed in patients expressing haptoglobin (Hp) 2–2 than Hp 1–1 phenotype. In particular, SAH patients with Hp 2–1, and non-DCI patients showed higher α1intensities than DCI patients. A genome-wide association study (GWAS)7 revealed that SNP rs999662 encompassing solute carrier family 12 member 3 (SLC12A3) was significantly associated with high transcranial Doppler (TCD) velocities, such as angiographic vasospasm, which leads to DCI7.
Genetic background accounts for approximately 37.9% of stroke pathogenesis8,9,10. However, the frequency of susceptible genetic variants in stroke patients varies between 5 and 10%9, which indicates cryptic genetic changes beyond DNA sequences8,11. One of these cryptic variations involving novel epigenetic pathways is DNA methylation, histone modifications or non-coding RNA11. A positive correlation between higher methylation status of the inositol 1-,4-,5-trisphosphate receptor (ITPR3) and DCI development was observed in patients with SAH11. In their study, DCI patients manifested a lower expression of ITPR3 mRNA concomitant with increased expression of DNA methyltransferase1 and a decrease in ten-eleven translocation methylcytosine dioxygenase 1 (TET1). Nevertheless, little is known about the epigenetic changes associated with DCI pathogenesis following SAH. Herein, we report an epigenome-wide association study (EWAS) comparing DNA methylation and DCI development to determine its role as a biomarker in DCI pathogenesis following SAH.
Results
Clinical characteristics of enrolled patients
Patient’s baseline characteristics are presented in Table 1. Variables such as female gender, HTN, DM, hyperlipidemia and smoking did not differ significantly between the DCI and non-DCI groups throughout the study period, although SAH patients in the discovery phase tended to be younger than those in the replication phase. In the discovery phase, higher HH grade was more frequently observed in DCI compared with non-DCI, but not statistical significant (p = 0.311). Most patients (80.0%) underwent coil embolization and 2 of 13 DCI patients (15.4%) underwent chemical angioplasty entailing intraarterial infusion of vasodilators during cerebral angiography to reverse cerebral vasospasm. In the replication phase, DCI patients appear to exhibit higher HH grade and Fisher grade, but was not statistically significant (p = 0.095 and p = 0.421, respectively). Most aneurysms were located in the anterior cerebral circulation.
Altered DNA methylation and mRNA expression in the discovery phase
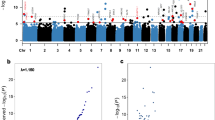
We evaluated significant epigenome-wide associations with DCI in 40 SAH patients. A total of 35 CpG sites passed the cutoff (Fig. 1). Among the differentially methylated CpG sites (DMCpGs) identified, cg00441765 and cg11464053 were the top two CpG sites showing hypermethylation in DCI patients compared with non-DCI patients (Table 2). Hierarchical clustering of CpG sites based on differences in DNA methylation in patients with DCI is presented in Fig. 2A. Interestingly, cg00441765 and cg11464053 were located in the second intron of insulin receptor (INSR) gene and in the exon 13 of the cadherin-related family member 5 (CDHR5).

Manhattan plot of genome-wide DNA methylation analysis.

(A) Hierarchical clustering of differences in DNA methylation between delayed cerebral ischemia (DCI) and non-DCI patients. (B) Transcription analysis of the two differentially methylated gene candidates using qRT-PCR. The results showed a lower expression of INSR and CDHR5 in DCI patients compared with non-DCI patients.
We evaluated the mRNA expression of the top 10 genes in 40 SAH patients via quantitative real-time PCR (qRT-PCR) analysis (Table 2). After exlcusion of the two genes, MIR640 (microRNA) and LOC389641 (long non-coding RNA), we further evaluated the relative mRNA expression (2−ΔCt) of 8 genes. DCI patients showed decreased transcription of INSR than in non-DCI patients (0.00020 [0.00012–0.00030] vs. 0.00050 [0.00030–0.00068]; p = 0.006). In addition, DCI patients showed lower CDHR5 expression than the non-DCI patients (0.114 [0.053–0.143] vs. 0.170 [0.110–0.212]; p = 0.010) (Figs. 2B). Associations between the degree of methylation of the remaining six genes and their corresponding mRNA expression were not correlated significantly with other genes according to DCI development (Supplemental Table S2).
MSP of INSR and CDHR5 and mRNA expression in the replication phase
To evaluate the methylation status of selected INSR and CDHR5 in DCI and non-DCI groups, the MSP primer sets were designed against the identical region annotated in Infinium MethylationEPIC assay (Fig. 3A) (Supplemental Table S1). The methylation level of two genes was measured using MSP in 36 patients with SAH in the replication phase (Figs. 3B,C)11. DCI patients exhibited a higher degree of methylation than non-DCI patients: INSR, 0.855 (0.779–0.913) in DCI vs. 0.582 (0.565–0.689) in non-DCI; p = 0.002; CDHR5, 0.786 (0.708–0.904) in DCI vs. 0.632 (0.610–0.679) in non-DCI; p = 0.017, respectively (Figs. 3A,B). Additionally, qRT-PCR was conducted to analyze the mRNA expression of the corresponding genes in 36 SAH patients. Patients with DCI had a lower level of INSR mRNA compared with those without DCI [0.00021 (0.00017–0.00024) vs. 0.00044 (0.00033–0.00065); p < 0.001]. DCI patients also expressed a lower level of CDHR5 than non-DCI patients [0.121 (0.090–0.134) vs. 0.185 (0.151–0.229); p < 0.001] (Supplemental Fig. S3).

(A) Schematic representation of methylation-specific PCR (MSP) analysis associated with intron 13/15 above INSR, or exon 2/21 of the CDHR5 genes. (B) Representative subsets of MSP of the INSR and CDHR5 regions using methylated or unmethylated specific primer sets in 36 subarachnoid hemorrhage patients in the replication phase. (C) DCI patients show higher methylation level than non-DCI patients. Nucleotide sequences of INSR and CDHR5, and MSP primers are listed in supplemental data.
Discussion
In this study, we identified two novel hypermethylated CpG sites of INSR and CDHR5 in DCI patients following SAH. Our findings suggested that initial brain injury due to sudden surge in intracranial pressure (ICP) might affect DNA methylation patterns concomitant with decreased mRNA expression, resulting in DCI.
The INSR gene is distributed in the brain extensively, especially in choroid plexus, olfactory bulb and areas of the striatum and cerebral cortex. INSR signaling is associated with brain metabolism, maintaining neuronal function, synaptogenesis and mitochondrial activity12. Hancock et al.13 reported nuclear translocation of the insulin receptor from the cell surface leading to the formation of RNA polymerase II complex on chromatin. In particular, most INSRs bound to the promoters, suggesting regulation of genes linked to insulin function. Approximately, 75% of the SAH patients exhibited hyperglycemia at admission due to activated hypothalamic–pituitary–adrenal axis and increased inflammatory response14,15. Kruyt et al.15 reported that hyperglycemia is associated with DCI and poor clinical outcome in SAH patients. Hyperglycemia under ischemia–reperfusion injury can increase reactive oxygen species formation and anaerobic glycolysis associated with aggravation of infarct volume, disruption of homeostasis and subsequent brain injury15. However, the correlation between intensive insulin treatment and neurologic outcomes has yet to be firmly established15. To verify the correlation between DNA methylation of INSR (cg00441765) in the whole blood and brain, we used the UNAGE-CpG tool16. Spearman rho value of variable CpGs in the brain and blood was 0.64 (p = 0.0021, Supplemental Fig. S1), suggesting correlation of differentially expressed genes between the two tissues. Nevertheless, SAH severities at admission and intensive glucose monitoring via microdialysis to prevent hypoglycemia can skew the results. Therefore, studies investigating the effect of INSR dysfunction on DCI pathogenesis in vivo and in vitro are necessary, focusing on the advantages for patients with inhibited INSR methylation.
CDHR5 is usually expressed in the kidney, liver and gastrointestinal tract. CDHR5 is found mainly at the brush border of renal proximal tubules and intestinal epithelium17,18. CDHR5 has been investigated as a biomarker for solid tumors such as colon cancer or renal cell carcinoma. Blasius et al.17 reported that CDHR5 was expressed in approximately 75% of the renal cell carcinoma. Particularly, CDHR5 expression was associated with a significantly longer survival time. In SAH, renal dysfunction has been reported in approximately 7% of the patients19. Zacharia et al.20 reported that SAH patients who were at risk for renal failure showed two-fold poor outcomes. Renal proximal tubule regulated blood pressure and plasma volume via sodium absorption in the glomerular filtrate21. Vrsajkov et al.22 reported that hyponatremia was associated with DCI and poor functional outcomes. In our study, DCI patients exhibited decreased transcription of CDHR5 compared with non-DCI patients. In addition, hyponatremia was observed frequently in DCI patients. Nevertheless, various factors including SAH-induced sympathetic activation, contrast use, antibiotic therapy, surgical intervention, and aggressive management of fluid volume can increase the risk of renal failure23. Therefore, additional studies are needed to investigate the epigenetic effect of CDHR5 on DCI, focusing on electrolyte imbalance.
In stroke research, epigenetic analysis has been performed in patients with ischemic stroke and atherosclerotic risk. Atherosclerotic aorta showed a higher degree of global DNA methylation of cytosine at CpG sites than in healthy human aorta24. Using EWAS, Rask-Andersen et al.25 reported myocardial infarction-specific methylation pattern in 211 CpG-sites of individuals living in northern Sweden. However, no significant stroke-associated methylation pattern was observed in 27 stroke patients8. Baccarelli et al.26 showed an association between hypomethylation and stroke occurrence or mortality. No significant difference in global methylation patterns was observed between stroke subtypes among patients with large-artery atherosclerosis, small-artery disease and cardio-aortic embolism27. Nevertheless, differences in biological age between stroke patients and healthy controls can affect the interpretation. Soriano-Tárraga et al.28 reported that ischemic stroke patients were biologically older than the healthy controls by an average age of 2.5 years. Due to variations in DNA methylation pattern in aging, a further epigenetic study investigating the role of age on stroke is required.
This study has some limitations. First, smoking status and sex were not included as covariates in the analysis due to the small sample size. Second, the expression of the two genes (INSR and CDHR5) was low, although the difference reached statistical significance. In addition, one non-DCI sample showed similar methylation pattern compared with the DCI sample. As a marker to distinguish DCI from non-DCI cases, the reliability of this CpG site should be further validated using in vivo and in vitro studies. Third, the difference in beta value is relatively less than expected. We have further analyzed the differences in beta value of all CpG sites via histogram (Supplemental Fig. S2). The results indicated that two CpG sites were significantly hypermethylated in DCI patients compared with non-DCI patients, although the difference in beta value was not dramatically altered. Fourth, we used bioinformatics tools to explore the molecular mechanisms of DNA methylation in DCI pathogenesis (Supplemental Table S3, Fig. S4). However, the proportion of the enrolled genes in the biological process and molecular pathway was small. Therefore, additional investigations involving a large cohort of SAH patients are further needed11.
Conclusions
To the best of our knowledge, this is the first genome-wide epigenetic study analyzing the quantitative expression of the corresponding susceptibility genes to DCI following SAH. Two novel hypermethylation sites of INSR and CDHR5 represent valuable biomarkers for the early detection of DCI. Studies investigating the precise mechanism underlying the methylation of candidate genes and DCI pathogenesis are further required.
Methods and materials
Study population
The derivation cohort was obtained from the stroke database of the Chuncheon Sacred Heart Hospital. The cohort is a prospective and observational project in the regional center of the district of Chuncheon city, the capital city of Gangwon Province in Korea11,29,30,31. In this database, we enrolled SAH patients with the following conditions: (1) adult SAH patients aged above 18 years; and (2) SAH due to ruptured aneurysm associated with saccular appearance. We excluded patients with the following conditions: (1) fusiform, dissection, traumatic and infectious aneurysms; (2) concomitant cerebrovascular diseases such as arteriovenous malformation or dural arteriovenous fistula; and (3) angiogram-negative SAH32,33. This study was performed in two phases: discovery and replication. In the discovery phase, the susceptible epigenetic marker was identified in 40 patients with SAH from September 2016 to October 2017 using EWAS. In the replication phase, an independent cohort of 36 patients between September 2017 and July 2019 were subjected to methylation-specific PCR.
We investigated the epigenetic patterns of blood representing potential markers for the prediction of DCI after SAH. DCI was defined by new neurologic deficits including motor weakness, sensory changes, dysphasia and decreased level of consciousness with concomitant cerebral vasospasm6,11. Clinical demographics regarding gender, age, hypertension (HTN), diabetes mellitus (DM), hyperlipidemia, smoking, and aneurysms detected radiologically were reviewed. This study was approved by the Institutional Review Board (No. 2016-3, 2017-9 and 2018-6) and informed consent was obtained from the patients or their relatives.
Extraction and quantification of genomic DNA
Genomic DNA (gDNA) was extracted from buffy coat, which was collected by centrifuging whole blood samples at 3,000×g for 4 min at room temperature. The buffy coat fractions were stored at − 80 ℃ until ready for further processing. The gDNA was extracted from buffy coat with FlexiGene DNA kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The concentration and purity of gDNA were determined by measuring the absorbance ratio (A260/A280) using UV Eppendorf BioSpectrometer (Eppendorf, Hamburg, Germany)11.
Epigenome-wide association study
In the discovery phase, we evaluated genome-scale DNA methylation profiles. Genomic DNA was analyzed using the Infinium MethylationEPIC (EPIC) assay with sodium bisulfite using the EZ-96 DNA methylation kit obtained from Zymo Research (CA, USA) according to manufacturer’s instructions. DNA methylation was quantified using the Infinium MethylationEPIC BeadChip (Illumina, CA, USA). Potentially existing raw quality probes in the raw data were filtered by the minfi package (version 1.24.0)34. First, samples with non-significant mean detection p-values (> 0.05) were excluded. The functional normalization algorithm (preprocessFunnorm) for Illumina methylation microarrays implemented in the minifi package was used to eliminate undesirable variation by regressing out variability with the control probes present in our methylation microarray. Finally, the differentially methylated CpG sites (DMCpGs) were identified with a q-value cutoff of 0.00005. To report methylation levels, the beta (β) value, which is an estimate of methylation level based on the ratio of intensity between methylated and unmethylated alleles, was calculated for each CpG site.
Quantitative real-time PCR
Total RNA was extracted from fresh whole blood using QIAamp RNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA quality and quantity were determined using the UV Eppendorf BioSpectrometer (Eppendorf, Hamburg, Germany). Isolated RNA (2 µg) was used for cDNA synthesis using Maxime RT PreMix Kit (iNtRON Biotechnology, Korea). The gene expression level was measured by quantitative real-time PCR (qRT-PCR) using the Power SYBR Green PCR master Mix (Thermo Fisher Scientific, MA, USA). PCR was performed for 45 cycles under the following conditions: 94 °C for 15 s, 55 °C for 30 s, and 70 °C for 30 s in the Rotor-Gene Q (Qiagen, Hilden, Germany). The qRT-PCR primers used in this study are listed in the supplemental data. Actin was used as an internal reference gene.
Methylation-specific PCR
In the replication phase, methylation-specific PCR (MSP) was further performed after selecting the two candidate genes. Approximately, 2 µg of DNA was subjected to bisulfite modification using the EpiTect Bisulfite Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions11. The modified DNA was subjected to MSP11,35. MSP primer sets were designed using the MethPrimer (https://www.urogene.org/methprimer/index1.html). The primer sequences, product sizes and target regions of PCR amplification are described in the supplemental data. Forty-five cycles of PCR using both unmethylated and methylated primers were performed under the following conditions: 95 °C for 15 s, 55 °C for 15 s and 72 °C for 30 s using 1.25 U Taq (iTaq, Intron Biotechnology, Seoul, Korea) in a final volume of 20 µl. PCR products were analyzed by electrophoresis on 3% agarose gel and visualized on UV.
Statistical analysis
Differentially methylated CpG sites (DMCpGs) were identified with an adjusted p-value cutoff of 0.0000536. To compare the methylation profiles between the DCI and non-DCI groups, the scatter plots for probe methylation values and the box plots for genomic features of each group were drawn by in-house R scripts. The relative methylation intensities are expressed as medians with interquartile range6. Relative methylation intensity was calculated by dividing the degree of methylation by the combined intensity of methylation and unmethylation11. The degree of relative methylation and mRNA expression were compared using the Mann–Whitney U test. p-value < 0.05 was considered statistically significant. The analysis was performed using MedCalc software (Medcalc, Mariakerke, Belgium).
Ethical approval
Sample collection and study design were performed according to the principles of the Declaration of Helsinki and were approved by Coordinating Ethnics Committee of the Chuncheon Sacred Heart Hospital.
Data availability
Data are available from the corresponding author (JPJ) upon ethical approval from the IRB of the participating hospital.
References
Duan, W. et al. Risk factors and clinical impact of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: Analysis from the china national stroke registry. Neuroepidemiology 50, 128–136 (2018).
Nahed, B. V. et al. Genetics of intracranial aneurysms. Neurosurgery 60, 213–225 (2007).
Geraghty, J. R. & Testai, F. D. Delayed cerebral ischemia after subarachnoid hemorrhage: Beyond vasospasm and towards a multifactorial pathophysiology. Curr. Atheroscler. Rep. 19, 50 (2017).
Ducruet, A. F. et al. Genetic determinants of cerebral vasospasm, delayed cerebral ischemia, and outcome after aneurysmal subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 30, 676–688 (2010).
Hu, X. et al. Role of apolipoprotein e genotypes in aneurysmal subarachnoid hemorrhage: Susceptibility, complications, and prognosis. World Neurosurg. 118, e666–e676 (2018).
Kim, B. J., Kim, Y., Kim, S. E. & Jeon, J. P. Study of correlation between hp alpha1 expression of haptoglobin 2–1 and clinical course in aneurysmal subarachnoid hemorrhage. World Neurosurg. 117, e221–e227 (2018).
Kim, H. et al. Cerebral vasospasm after sub-arachnoid hemorrhage as a clinical predictor and phenotype for genetic association study. Int. J. Stroke 8, 620–625 (2013).
Krupinski, J. et al. DNA methylation in stroke. Update of latest advances. Comput. Struct. Biotechnol. J. 16, 1–5 (2018).
Bevan, S. et al. Genetic heritability of ischemic stroke and the contribution of previously reported candidate gene and genomewide associations. Stroke 43, 3161–3167 (2012).
Domingues-Montanari, S., Mendioroz, M., del Rio-Espinola, A., Fernandez-Cadenas, I. & Montaner, J. Genetics of stroke: A review of recent advances. Expert Rev. Mol. Diagn. 8, 495–513 (2008).
Kim, B. J. et al. Correlation between altered DNA methylation of intergenic regions of itpr3 and development of delayed cerebral ischemia in subarachnoid hemorrhage patients. World Neurosurg. https://doi.org/10.1016/j.wneu.2019.06.113 (2019).
Kleinridders, A., Ferris, H. A., Cai, W. & Kahn, C. R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 63, 2232–2243 (2014).
Hancock, M. L. et al. Insulin receptor associates with promoters genome-wide and regulates gene expression. Cell 177, 722–736 (2019).
Kruyt, N. D., Biessels, G. J., Devries, J. H. & Roos, Y. B. Hyperglycemia in acute ischemic stroke: Pathophysiology and clinical management. Nat. Rev. Neurol. 6, 145–155 (2010).
Kruyt, N. D. et al. Hyperglycemia in aneurysmal subarachnoid hemorrhage: A potentially modifiable risk factor for poor outcome. J. Cereb. Blood Flow Metab. 30, 1577–1587 (2010).
Spindola, L. M. et al. Detecting multiple differentially methylated cpg sites and regions related to dimensional psychopathology in youths. Clin. Epigenet. 11, 146 (2019).
Blasius, F. M. et al. Loss of cadherin related family member 5 (cdhr5) expression in clear cell renal cell carcinoma is a prognostic marker of disease progression. Oncotarget 8, 75076–75086 (2017).
Crawley, S. W. et al. Intestinal brush border assembly driven by protocadherin-based intermicrovillar adhesion. Cell 157, 433–446 (2014).
Solenski, N. J. et al. Medical complications of aneurysmal subarachnoid hemorrhage: A report of the multicenter, cooperative aneurysm study. Participants of the multicenter cooperative aneurysm study. Crit. Care Med. 23, 1007–1017 (1995).
Zacharia, B. E. et al. Renal dysfunction as an independent predictor of outcome after aneurysmal subarachnoid hemorrhage: A single-center cohort study. Stroke 40, 2375–2381 (2009).
Horita, S. et al. The role of renal proximal tubule transport in the regulation of blood pressure. Kidney Res. Clin. Pract. 36, 12–21 (2017).
Vrsajkov, V., Javanovic, G., Stanisavljevic, S., Uvelin, A. & Vrsajkov, J. P. Clinical and predictive significance of hyponatremia after aneurysmal subarachnoid hemorrhage. Balkan Med. J. 29, 243–246 (2012).
Chen, S. et al. The harmful effects of subarachnoid hemorrhage on extracerebral organs. Biomed. Res. Int. 2014, 858496 (2014).
Zaina, S. et al. DNA methylation map of human atherosclerosis. Circ. Cardiovasc. Genet. 7, 692–700 (2014).
Rask-Andersen, M. et al. Epigenome-wide association study reveals differential DNA methylation in individuals with a history of myocardial infarction. Hum. Mol. Genet. 25, 4739–4748 (2016).
Baccarelli, A. et al. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology. 21, 819–828 (2010).
Soriano-Tarraga, C. et al. Global DNA methylation of ischemic stroke subtypes. PLoS ONE 9, e96543 (2014).
Soriano-Tarraga, C. et al. Ischemic stroke patients are biologically older than their chronological age. Aging (Albany, N.Y.) 8, 2655–2666 (2016).
Hong, E. P. et al. Genomic variations in susceptibility to intracranial aneurysm in the Korean population. J. Clin. Med. 8, 275 (2019).
Kim, C. H., Jeon, J. P., Kim, S. E., Choi, H. J. & Cho, Y. J. Endovascular treatment with intravenous thrombolysis versus endovascular treatment alone for acute anterior circulation stroke: A meta-analysis of observational studies. J. Korean Neurosurg. Soc. 61, 467–473 (2018).
Cho, Y. D. et al. Protected versus unprotected carotid artery stenting: Meta-analysis of the current literature. J. Korean Neurosurg. Soc. 61, 458–466 (2018).
Hong, E. P. et al. A novel association between lysyl oxidase gene polymorphism and intracranial aneurysm in Koreans. Yonsei Med. J. 58, 1006–1011 (2017).
Hong, E. P., Kim, B. J., Kim, C., Choi, H. J. & Jeon, J. P. Association of sox17 gene polymorphisms and intracranial aneurysm: A case-control study and meta-analysis. World Neurosurg. 110, e823–e829 (2018).
Aryee, M. J. et al. Minfi: A flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Herman, J. G., Graff, J. R., Myohanen, S., Nelkin, B. D. & Baylin, S. B. Methylation-specific pcr: A novel pcr assay for methylation status of cpg islands. Proc. Natl. Acad. Sci. U.S.A. 93, 9821–9826 (1996).
Pera, J. et al. Gene expression profiling of blood in ruptured intracranial aneurysms: In search of biomarkers. J. Cereb. Blood Flow Metab. 33, 1025–1031 (2013).
Acknowledgements
This study received funding from the National IT Industry Promotion Agency (R-20190315-004075), the Korea Technology and Information Promotion Agency for SMEs (S-2693922), and BioGreen21 (PJ01313901) grant from the Rural Development Administration.
Author information
Authors and Affiliations
Contributions
J.P.J. designed and obtained funding for the study. B.J.K., Y.K. and D.H.Y. were responsible for the molecular experiments. J.J.P., J.K.R., H.C.K. and K.K. interpreted the data and performed statistical analyses. J.P.J. provided input for the final version of the manuscript. All authors reviewed the manuscript and provided editorial feedback.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, B.J., Kim, Y., Youn, D.H. et al. Genome-wide blood DNA methylation analysis in patients with delayed cerebral ischemia after subarachnoid hemorrhage. Sci Rep 10, 11419 (2020). https://doi.org/10.1038/s41598-020-68325-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-68325-3
This article is cited by
-
Differential DNA methylation associated with delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: a systematic review
Neurosurgical Review (2024)
-
Ischemic preconditioning modulates the DNA methylation process of the rat heart to provide tolerance to withstand ischemia reperfusion injury and its associated mitochondrial dysfunction
3 Biotech (2024)
-
Blood DNA Methylation Analysis Reveals a Distinctive Epigenetic Signature of Vasospasm in Aneurysmal Subarachnoid Hemorrhage
Translational Stroke Research (2024)
-
Methylated MFGE8 Promotes Early Brain Injury After Subarachnoid Hemorrhage and Inhibiting Autophagy of Nerve Cell
Translational Stroke Research (2023)
-
Fine-mapping of intracranial aneurysm susceptibility based on a genome-wide association study
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
