
Abstract
Grade 4 glioma or GBM has poor prognosis and is the most aggressive grade of glioma. Accurate diagnosis and classification of tumor grade is a critical determinant for development of treatment pathway. Extensive genomic sequencing of gliomas, different cell types, brain tissue regions and advances in bioinformatics algorithms, have presented an opportunity to identify molecular markers that can complement existing histology and imaging methods used to diagnose and classify gliomas. ‘Cancer stem cell theory’ purports that a minor population of stem cells among the heterogeneous population of different cell types in the tumor, drive tumor growth and resistance to therapies. However, characterization of stem cell states in GBM and ability of stem cell state signature genes to serve as diagnostic or prognostic molecular markers are unknown. In this work, two different network construction algorithms, Weighted correlation network analysis (WGCNA) and Multiscale Clustering of Geometric Network (MEGENA), were applied on publicly available glioma, control brain and stem cell gene expression RNA-seq datasets, to identify gene network regulatory modules associated with GBM. Both gene network algorithms identified consensus or equivalent modules, HuAgeGBsplit_18 (WGCNA) and c1_HuAgeGBsplit_32/193 (MEGENA), significantly associated with GBM. Characterization of HuAgeGBsplit_18 (WGCNA) and c1_HuAgeGBsplit_32/193 (MEGENA) modules showed significant enrichment of rodent quiescent stem cell marker genes (GSE70696_QNPbyTAP). A logistic regression model built with eight of these quiescent stem cell marker genes (GSE70696_QNPbyTAP) was sufficient to distinguish between control and GBM samples. This study demonstrates that GBM associated gene regulatory modules are characterized by diagnostic quiescent stem cell marker genes, which may potentially be used clinically as diagnostic markers and therapeutic targets in GBM.
Similar content being viewed by others

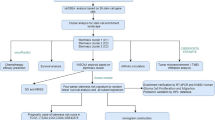

Introduction
GBM is a tumor that occurs in brain and can spread to spinal cord1. Prognosis of GBM is poor and treatment options are limited, with most patients not surviving the disease2. For diagnosis and treatment of GBM, identification of glioma grade is key to devise tumor specific treatment pathways3. Gliomas are classified as grade 2, grade 3 and grade 4 gliomas, with increasing aggressiveness and decreasing survival rates4,5. Gliomas of different grades do not respond uniformly to treatment as they are distinctly different from each other6. To properly test efficacy of drugs and biologics in glioma clinical trials, it is important to understand how their effectiveness changes in tumor grades7. Research and market for GBM treatment is predicted to rapidly grow from $465 million in 2016 to $1 billion by 20258.
Glioma grade is a key determinant of metastasis and tumor relapse in patients. Presently, tumor imaging, limited molecular profiling and histological analysis of tumor biopsy are techniques used to grade gliomas in clinical settings7. However, application of a wider range of molecular analysis can complement these existing techniques and improve glioma classification. Cancer therapy driven by molecular classification holds promise to improve patient specific tumor grading for application of precision medicine for improved treatment outcome. It is speculated that gliomas originate from mutation in proliferative stem cells in the brain and number of mutations grow during the course of tumor development. Therefore, it maybe possible to grade gliomas at the molecular level based on the stem cell signature genes9,10. Moreover, stem cell signature genes could potentially serve as targets for gene therapy and drugs11,12.
‘Stem cell theory of cancer’ states that resistance of cancer to chemotherapy and radiotherapy, is due to resident tumor stem cells13. Chemotherapy and radiotherapy treatments work by specifically targeting proliferative stem cells, but non-proliferative stem cells that are in a resting state called quiescence, escape this treatment strategy14. Resting stem cells in cancer under suitable conditions can become proliferative, replenish tumor cells and mutate further to reestablish the tumor after chemotherapy and radiotherapy. Thus, it is essential to determine stem cell molecular markers underlying GBM associated gene networks to improve treatment outcome and patient survival.
An emerging field of drug development is ‘network medicine’, where a combination of drugs are picked to target multiple subnetworks or modules of a disease gene regulatory network15,16. ‘Omics’ or genomics data and biomedical literature text data are two major sources of gene and gene interaction information in human health and disease. Both omics data and text data resources are used to identify druggable gene networks and pathways underlying disease symptoms. Extensive research endeavors led to establish collections of RNA-sequencing (RNA-seq), chromatin immunoprecipitation sequencing (ChIP-seq), ES (Exome Sequencing), Whole Exome Sequencing (WES), high-throughput proteomics and other ‘omics’ patient data17. The Cancer Genome Atlas (TCGA), International Cancer Genome Consortium (ICGC), Chinese Glioma Genome Atlas (CGGA) and NCBI GEO are some examples of publicly available databases for collection and storage of patient omics data18,19,20,21. Additionally, NCBI GEO archives a multitude of genome-wide molecular transcriptome profiles, including stem cell transcriptome data from human and non-human animal species19. Presently, a RNA-seq meta-analysis was performed to identify stem cell enriched GBM modules and develop a diagnostic model based on stem cell genes, which is able to distinguish between control and GBM samples.
Here, lists of quiescent and proliferative state stem cell signature genes were identified underlying GBM specific gene regulatory modules. Two network analysis methods, WGCNA and MEGENA, were utilized to build a glioma gene regulatory network and GBM associated sub-networks or modules based on gene–gene co-expression patterns22,23,24. Both methods identified comparable GBM associated modules, which suggests that GBM modules identified in this study are biologically robust. GBM modules, HuAgeGBsplit_18 (WGCNA) and its equivalent c1_HuAgeGBsplit_32/193 (MEGENA), were enriched with quiescent stem cell signature genes (GSE70696_QNPbyTAP) and expression of these quiescent stem cell signature genes was sufficient to build a logistic regression diagnostic model capable of distinguishing between control and GBM samples. In summary, a set of quiescent stem cell markers enriched in GBM modules was identified with potential application in diagnostic GBM screening.
Methods
Glioma RNA-seq data sources
RNA-seq gene expression datasets were obtained from NCBI Sequence Read Archive (SRA) and NCBI Gene Expression Omnibus (GEO)19,25. RNA-seq gene expression datasets SRP027383 and SRP091303 for glioma belonging to grade 2, grade 3 and GBM (grade 4) was obtained from NCBI SRA18. SRP027383 dataset is also available on NCBI GEO under series number GSE48865. RNA-seq gene expression datasets were obtained from NCBI GEO, belonging to various parts of brain to use as controls: cortex (series numbers GSE64810, GSE53697), hippocampus (series number GSE67333) and optic chiasma (series number GSE100297)26,27,28,29.
Stem cell RNA-seq data sources
Five different proliferative and quiescent stem cell datasets were used in this study. Two human RNA-seq gene expression datasets were obtained from NCBI GEO for GBM grown in quiescent and proliferative culture conditions, with series numbers GSE93991 and GSE11457430,31. Both RNA-seq and microarray gene expression datasets for adult rodent brain stem cells in proliferative and quiescent states, were obtained from NCBI GEO with series numbers GSE68270, GSE70696 and GSE9977732,33,34.
Processing of RNA-seq reads
RNA-seq reads were mapped to genome and genes were annotated as previously described33,35. Briefly, sra raw read files were converted to fastq using sratoolkit.2.9.1 and quality was assessed with fastqc_v0.11.725,36. Alignment of RNA-seq raw reads on reference genome was done with tophat 2.1.1 aligner using gene annotation information with parameters: tophat2-b2 -very-fast -no-coverage-search -no-novel-juncs37. Following reference genome and annotation files were used: (1) for human: Homo_sapiens.GRCh38.dna.primary_assembly.fa and Homo_sapiens.GRCh38.92.gtf, (2) for mouse: Mus_musculus.GRCm38.dna.primary_assembly.fa and Mus_musculus.GRCm38.92.gtf, (3) for rat: rnor6_ensemble_seq_whole_genome.fa and Rattus_norvegicus.Rnor_6.0.95.gtf. Mapped output bam files were analyzed with HTSeq 0.10.0 to estimate gene abundance and obtain count reads overlapping a gene with parameters: htseq-count -r pos -t gene_name38.
Glioma gene network analysis with WGCNA and MEGENA
Gene counts from HTSeq 0.10.0 were normalized to TPM values and filtered to keep values > 1 in atleast 2 samples39. TPM values were further scaled to log2TPM + 1 and visualized with volcano plots, barplots and density plots in ggplot2_3.1.0 R package40. To compare samples before and after TPM normalization, prcomp function in stats4_3.5.0 R package and corrplot_0.84 R package were used to perform PCA and correlation analysis, respectively40,41. To remove effects of covariants other than glioma, such as gender, study or batch, age, tissue and surrogate variables (hidden variables) on gene expression, a surrogate variable adjustment and linear model (SVA + LM) method was applied to log2TPM + 1 values, following previously published methods using sva_3.30.1 and limma_3.38.3 R packages35,42,43,44,45. After SVA + LM adjustment the resultant normalized log2TPM + 1 gene expression was used to build a glioma gene network with subnetworks or modules using WGCNA_1.66 and MEGENA_1.3.7 R packages23,24,35,46.
In WGCNA, default parameters and a minimum module size of 100 was used to calculate a topology overlap matrix (TOM) based on gene expression correlations. Hierarchical clustering was then used to build a glioma gene network consisting of interconnected subnetworks or modules22,23. In MEGENA, default parameters and a minimum module size of 100 was used to calculate a planar filtered network (PFN) from gene expression correlations. Multiscale clustering method was applied to build a glioma gene network consisting of interconnected subnetworks or modules24. WGCNA computes scale-free or single scale networks, while MEGENA computes multi-scale networks to include different possible variations of gene–gene interactions. Therefore, in MEGENA a given gene or node can be part of multiple modules representing different possible interactions, while in WGCNA a given gene or node is assigned to only a single module. To determine module trait correlations, module eigengenes were computed with moduleEigengenes R function and correlations were calculated22. To compare WGCNA and MEGENA modules, previously published module preservation analysis and hypergeometric enrichment tests were used35,47. Briefly, hypergeometric test was implemented with userListEnichment R function widely used to compare WGCNA gene network modules with each other and with user supplied gene lists22,23,46. Both module preservation and userListEnrichment R functions are from WGCNA23,47. After module preservation analysis between WGCNA and MEGENA modules, significant overlap of genes between WGCNA and MEGENA modules was done with userListEnrichment R function for all WGCNA and MEGENA modules i.e. each WGCNA module was compared with each MEGENA module. WGCNA and MEGENA modules that significantly overlapped and were significantly associated with GBM were retained.
Stem cell differential gene expression analysis with limma, edgeR and simple comparison of means
Differentially expressed genes (DEGs) between proliferative and quiescent stem cell states were identified using R packages limma_3.38.3, edgeR_3.24.3 and simple comparison expression means18,45,48. To calculate genes enriched in proliferative stem cells, gene expression of samples annotated to proliferative stem cells were compared with gene expression of samples annotated to quiescent stem cells for each of the five datasets (GSE68270, GSE70696, GSE99777, GSE93991 and GSE114574). Similarly, to calculate genes enriched in quiescent stem cells, gene expression of samples annotated to quiescent stem cells were compared with gene expression of samples annotated to proliferative stem cells for each of the five datasets (GSE68270, GSE70696, GSE99777, GSE93991 and GSE114574). In simple comparison of means method, mean expression of genes were simply compared between proliferative and quiescent stem cell states to determine DEGs. In limma and edgeR model design included variables stem cell state, study or batch, gender, age and tissue, and stem cell states were contrasted to determine DEGs, while all other variables were held constant in the model. In limma and edgeR methods Benjamini and Hochberf (BH) corrections for multiple testing is included as a large number of genes were included in analysis49. DEGs with BH corrected adjP-values < 0.05 and fold change > 1.25 were considered significant DEGs. To visualize gene expression values of DEGs volcano plots, barplots and density plots were made using ggplot2_3.1.1 R package40. Consensus DEGs were obtained by overlapping DEG lists produced by limma, edgeR and simple comparison of means with a significance of overlap p-value < 0.05 as calculated with GeneOverlap_1.18.0 and visualized by VennDiagram_1.6.20 R packages50,51. Consensus DEGs that belonged to atleast two of the three DEG lists produced by limma, edgeR and simple comparison of means were designated significantly enriched genes or DEGs in proliferative and quiescent stem cell states—simply referred to as (1) proliferative stem cell marker genes and (2) quiescent stem cell marker genes.
Following is a detailed description of all DEG analysis contrasts, sample size for each dataset and abbreviations used to represent proliferative and quiescent stem cell marker genes: (A) Adult proliferative neural progenitor cells (PNPCs) vs adult quiescent neural stem cells (QNSCs) DEG analysis to identify genes enriched in PNPCs relative to QNSCs in mouse dataset with series number GSE68270 and sample size of 4 each, abbreviated as GSE68270_PNPCbyQNSC32 (B) Adult quiescent neural stem cells (QNSCs) vs adult proliferative neural progenitor cells (PNPCs) DEG analysis to identify genes enriched in QNSCs relative to PNPCs in mouse dataset with series number GSE68270 and sample size of 4 each, abbreviated as GSE68270_QNSCbyPNPC32 (C) Adult hippocampal stem cells in proliferative condition or transient amplifying progenitor cells (TAPs) vs adult hippocampal stem cells in quiescent condition or quiescent progenitor cells (QNPs) DEG analysis to identify genes enriched in TAPs relative to QNPs in rat dataset with series number GSE70696 and sample size of 2 each, abbreviated as GSE70696_TAPbyQNP33 (D) Adult hippocampal stem cells in quiescent condition or quiescent progenitor cells (QNPs) vs adult hippocampal stem cells in proliferative condition or transient amplifying progenitor cells (TAPs) DEG analysis to identify genes enriched in QNPs relative to TAPs in rat dataset with series number GSE70696 and sample size of 2 each, abbreviated as GSE70696_QNPbyTAP33 (E) Adult proliferative sub-ventricular zone stem cells (PSVZSCs) vs adult quiescent sub-ventricular zone stem cells (QSVZSCs) DEG analysis to identify genes enriched in PSVZSCs relative to QSVZSCs in mouse microarray dataset with series number GSE99777 and sample size of 3 each, abbreviated as GSE99777_PSVZSCbyQSVZSC34 (F) Adult quiescent sub-ventricular zone stem cells (QSVZSCs) vs adult proliferative sub-ventricular zone stem cells (PSVZSCs) DEG analysis to identify genes enriched in QSVZSCs relative to PSVZSCs in mouse microarray dataset with series number GSE99777 and sample size of 3 each, abbreviated as GSE99777_QSVZSCbyPSVZSC34 (G) GBM cells cultured in proliferative condition or proliferative GBM cells (PGBCs) vs GBM cells cultured in quiescent condition or quiescent GBM cells (QGBCs) DEG analysis to identify genes enriched in PGBCs relative to QGBCs in human dataset with series number GSE93991 and sample size of 9 and 6, respectively, abbreviated as GSE93991_PGBCbyQGBC30 (H) GBM cells cultured in quiescent condition or quiescent GBM cells (QGBCs) vs GBM cells cultured in proliferative condition or proliferative GBM cells (PGBCs) DEG analysis to identify genes enriched in QGBCs relative to PGBCs in human dataset with series number GSE93991 and sample size of 6 and 9, respectively, abbreviated as GSE93991_QGBCbyPGBC30 (I) GBM organoids cultured in proliferative condition or proliferative GBM organoids (PGBOs) vs GBM organoids cultured in quiescent condition or quiescent GBM cells (QGBOs) DEG analysis to identify genes enriched in PGBOs relative to QGBOs in human dataset with series number GSE114574 and sample size 6, abbreviated as GSE114574_PGBObyQGBO31 and (J) GBM organoids cultured in quiescent condition or quiescent GBM organoids (QGBOs) vs GBM organoids cultured in proliferative condition or proliferative GBM organoids (PGBOs) DEG analysis to identify genes enriched in QGBOs relative to PGBOs in human dataset with series number GSE93991 and sample size of 6, abbreviated as GSE114574_QGBObyPGBO31.
Enrichment of proliferative and quiescent stem cell marker genes in glioma modules
Enrichment of proliferative and quiescent stem cell marker genes identified by differential gene expression analysis above, in WGCNA and MEGENA modules was determined using useListEnrichment R function22,23,46. Additionally, a supplementary table containing a set of 336 genes potentially involved in transition of GBM from stem-like state to differentiation identified by SWIM tool were downloaded directly from the published paper52,53. Enrichment of SWIM GBM list in WGCNA and MEGENA modules was also determined using useListEnrichment R function22,23,46. Briefly, hypergeometric test was implemented with userListEnichment R function from WGCNA R package23,46. Hypergeometric test was implemented with userListEnrichment R function to determine: (A) Significant overlap of a WGCNA module set vs a set of proliferative stem cell marker genes. This was done for all WGCNA modules and all proliferative stem cell marker gene sets (GSE68270_PNPCbyQNSC, GSE70696_TAPbyQNP, GSE99777_PSVZSCbyQSVZSC, GSE93991_PGBCbyQGBC and GSE114574_PGBObyQGBO). (B) Significant overlap of a WGCNA module set vs a set of quiescent stem cell marker genes. This was done for all WGCNA modules and all quiescent stem cell marker gene sets (GSE68270_QNSCbyPNPC, GSE70696_QNPbyTAP, GSE99777_QSVZSCbyPSVZSC, GSE93991_QGBCbyPGBC and GSE114574_QGBObyPGBO). (C) Significant overlap of a WGCNA module set vs a set of SWIM 336 GBM gene list. This was done for all WGCNA modules and the SWIM 336 GBM gene list. (D) Significant overlap of a MEGENA module set vs a set of proliferative stem cell marker genes. This was done for all WGCNA modules and all proliferative stem cell marker gene sets (GSE68270_PNPCbyQNSC, GSE70696_TAPbyQNP, GSE99777_PSVZSCbyQSVZSC, GSE93991_PGBCbyQGBC and GSE114574_PGBObyQGBO). (E) Significant overlap of a MEGENA module set vs a set of quiescent stem cell marker genes. This was done for all WGCNA modules and all quiescent stem cell marker gene sets (GSE68270_QNSCbyPNPC, GSE70696_QNPbyTAP, GSE99777_QSVZSCbyPSVZSC, GSE93991_QGBCbyPGBC and GSE114574_QGBObyPGBO). And (F) Significant overlap of a MEGENA module set vs a set of SWIM 336 GBM gene list. This was done for all MEGENA modules and the SWIM 336 GBM gene list. The results from (A) to (F) analysis were filtered to retain WGCNA and MEGENA GBM modules significantly enriched with proliferative and quiescent stem cell marker genes.
Gene ontology (GO) analysis of GBM modules significantly enriched with proliferative and quiescent stem cell marker genes
EnrichR_1.0 R package was used to determine biological process gene ontology (GO) functional characterization of the Glioblastoma associated module gene members and p-value < 0.05 was kept as significant54.
GBM logistic regression diagnostic model with stem cell genes and Hosmer–Lemeshow goodness of fit (GOF) test
To distinguish between control and GBM samples, a logistic regression model was built with stem cell marker genes most significantly enriched in GBM modules and most upregulated in stem cell DEG list. Logistic regression was performed with glm R function from stats4_3.5.0 R package with selected genes and gene–gene interaction terms: glm(formula = DiseaseGrade_4 ~ gene1 + gene2 + …gene1*gene2*…, family = binomial(link = logit), data = data, na.action = na.exclude). Hosmer–Lemeshow GOF test was used to evaluate how well logistic regression model fits data using R package ResourceSelection v0.3–555,56. In other words, Hosmer–Lemeshow GOF test was used to evaluate how well probability values predicted by logistic regression model (expected probabilities) matched observed probabilities. For Hosmer–Lemeshow GOF test, H0 (null hypothesis) is that observed and expected probabilities do not differ significantly (good fit), while Ha (alternate hypothesis) is that observed and expected probabilities differ significantly (poor fit). A p-value < 0.05 and a large difference between observed and expected probabilities, indicates poor model fit on data so model can be rejected (reject Ho, accept Ha). On the other hand, p-value > 0.05 and a small difference between observed and expected probabilities, indicates there is no evidence of poor fit so model can be accepted as a good fit (accept Ho).
Consensus significantly enriched genes or DEGs in stem cell proliferation and quiescence
To determine proliferative and quiescent stem cell marker genes common between all five stem cell datasets (GSE68270, GSE70696, GSE99777, GSE93991 and GSE114574), the following consensus significantly enriched genes or DEGs in proliferative and quiescent states were overlapped, respectively, using online tool https://www.molbiotools.com/listcompare.html : (A) For proliferation comparison of (GSE68270_PNPCbyQNSC + GSE70696_TAPbyQNP + GSE99777_PSVZSCbyQSVZSC + GSE93991_PGBCbyQGBC + GSE114574_PGBObyQGBO) and (B) For quiescence comparison of (GSE68270_QNSCbyPNPC + GSE70696_QNPbyTAP + GSE99777_QSVZSCbyPSVZSC + GSE93991_QGBCbyPGBC + GSE114574_QGBObyPGBO).
To determine proliferative and quiescent stem cell marker genes common to both normal stem cells and GBM in culture, following consensus significantly enriched genes or DEG lists in proliferative and quiescent states were overlapped, respectively, using online tool https://www.molbiotools.com/listcompare.html (A) comparison of normal stem cells (GSE68270_PNPCbyQNSC + GSE70696_TAPbyQNP + GSE99777_PSVZSCbyQSVZSC) and GBM cell cultures (GSE93991_PGBCbyQGBC + GSE114574_PGBObyQGBO) DEG lists in proliferative conditions (B) Comparison of normal stem cells (GSE68270_QNSCbyPNPC + GSE70696_QNPbyTAP + GSE99777_QSVZSCbyPSVZSC) and GBM cell cultures (GSE93991_QGBCbyPGBC + GSE114574_QGBObyPGBO) DEGs in quiescent conditions.
Code availability
The computational pipeline used in this work is open-sourced and available both on github https://github.com/smukher2/GithubScientificReportsGlioblastomaStemApril2020 and protocol exchange research square https://dx.doi.org/10.21203/rs.3.pex-977/v1.
Results
SVA + LM approach reduces batch effects in RNA-seq datasets
To identify GBM specific transcriptome features, a meta-analysis was performed with glioma and control brain human RNA-seq samples. SVA + LM normalization reduced variability due to batch effects as indicated by greater overlap of expression data in density plots after SVA + LM normalization (Fig. 1A). Box-Whiskers plots showed a slightly skewed mean expression in GSE67333 dataset that was fixed by SVA + LM normalization (Fig. 1B). PCA plots showed that SVA + LM normalization reduces dispersion of samples from same study (Fig. 1C). Correlation plots to evaluate correlation of gene expression values among different studies, showed an increase in positive correlation after SVA + LM normalization (Fig. 1D). Thus, SVA + LM normalization may be used to achieve reduction in batch effects in global gene expression when RNA-seq studies are combined.

Effect of SVA + LM adjustment on Glioblastoma (SRP027383, SRP091303) and control brain (GSE67333, GSE64810, GSE100297, GSE53697) RNA-seq studies. (A) Density plot representation of gene expression per study before and after SVA + LM adjustment (B) Box and whisker plot representation of gene expression per study before and after SVA + LM adjustment. (C) Principal component (PC) projections of datasets before and after SVA + LM adjustment. (D) Pearson correlation plot of the studies before and after SVA + LM adjustment.
Network analysis reveals GBM associated modules within glioma network
Glioma gene co-expression networks were constructed with WGCNA and MEGENA to uncover underlying molecular mechanisms. WGCNA identified 39 modules with largest module HuAgeGBsplit_01 comprising of 7,969 genes and smallest module HuAgeGBsplit_38 comprising of 113 genes (Table 1). MEGENA identified 235 modules with largest module c1_HuAgeGBsplit_13 comprising of 1,483 genes and smallest modules c1_HuAgeGBsplit_1081/181/1895 comprising of 100 genes each (Table 2).
To identify GBM associated modules, a module eigengene was used to represent overall expression pattern of each module produced by WGCNA and MEGENA. Spearman correlations were calculated for GBM and other clinical traits, such as batch, age, and gender. Eight modules in WGCNA, with module size ranging from 673 genes in HuAgeGBsplit_08 to 121 genes in Hu_AgeGBsplit_36, were found to be significantly associated with GBM (Fig. 2A,B). All WGCNA modules, including GBM associated modules, were preserved with MEGENA modules (Fig. 2C). Comparison of GBM specific WGCNA modules with MEGENA modules showed a significant overlap of all WGCNA GBM modules with MEGENA modules (Fig. 2D). All 20 MEGENA modules that significantly overlapped with GBM WGCNA modules, with module size ranging from 101 genes in c1_HuAgeGBsplit_605 to 708 genes in c1_HuAgeGBsplit_24, were also significantly associated with GBM (Fig. 2E,F). Thus, WGCNA and MEGENA complemented each other and helped identify GBM specific modules in largely preserved glioma WGCNA and MEGENA gene networks.

Identification of WGCNA and MEGENA Glioblastoma modules. (A) Bar-plot showing number of genes in Glioblastoma associated WGCNA modules. (B) Spearman correlation coefficient and p-values for significant tumor Grade 4 or Glioblastoma associated WGCNA modules (C) Preservation analysis between all WGCNA modules and all MEGENA modules using WGCNA module labels. (D) Table showing MEGENA modules that significantly overlapped with WGCNA Glioblastoma associated modules. (E) Bar-plot showing number of genes in Glioblastoma associated MEGENA modules that significantly overlap with Glioblastoma associated WGCNA modules. (F) Spearman correlation coefficient and p-values for significant tumor Grade 4 or Glioblastoma associated MEGENA modules that also significantly overlap with tumor Grade 4 or Glioblastoma associated WGCNA modules.
Differential gene expression analysis reveals proliferative and quiescent stem cell marker genes
To identify genes specific to quiescent and proliferative states of stem cells, differential gene expression analysis was performed on different stem cell datasets as described under methods. In mouse adult hippocampal stem cell dataset, 5,306 (3,959 human gene symbols) and 4,072 (3,188 human gene symbols) DEGs with a fold change of 1.25 (p-value < 0.05) were identified in proliferation (GSE68270_PNPCbyQNSC) and quiescence (GSE68270_PNPCbyQNSC), respectively (Fig. 3A,B). In rat adult hippocampal stem cell dataset, 5,122 (4,362 human gene symbols) and 3,290 (2,792 human gene symbols) DEGs with a fold change of 1.25 (p-value < 0.05) were identified in proliferation (GSE70696_TAPbyQNP) and quiescence (GSE70696_QNPbyTAP), respectively (Fig. 3C,D). In mouse adult subventricular zone stem cell dataset, 5,216 (4,235 human gene symbols) and 5,733 (4,658 human gene symbols) DEGs with a fold change of 1.25 (p-value < 0.05) were identified in proliferation (GSE99777_PSVZSCbyQSVZSC) and quiescence (GSE99777_QSVZSCbyPSVZSC), respectively (Fig. 3E,F). In human GBM cell culture dataset, 7,857 and 3,363 (human gene symbols) DEGs with a fold change of 1.25 (p-value < 0.05) were identified in proliferation (GSE93991_PGBCbyQGBC) and quiescence (GSE93991_QGBCbyPGBC), respectively (Fig. 3G,H). In human GBM organoid culture dataset, 1928 and 3,315 (human gene symbols) DEGs with a fold change of 1.25 (p-value < 0.05) were identified in proliferation (GSE114574_PGBObyQGBO) and quiescence (GSE114574_QGBObyPGBO), respectively (Fig. 3I,J). Stem cell marker genes common between all normal stem cells and GBM culture datasets (GSE68270, GSE70696, GSE99777, GSE93991 and GSE114574), consisted of 7 proliferation genes or DEGs (ACYP1, AKAP12, LRP11, MYSM1, SLC20A1, TERT, TSPAN13) and 5 quiescence genes or DEGs (ETHE1, FZD9, NINJ1, P2RX4, PTP4A3) (Tables 3, 4). Overlapping stem cell marker genes obtained from normal stem cell datasets (GSE68270, GSE70696 and GSE99777), with GBM culture datasets (GSE93991 and GSE114574), showed an overlap of 4,176 proliferation genes (45.87% of GBM proliferation DEGs) and only 1598 quiescence genes (25.97% of GBM quiescence DEGs (Supplementary Tables S1A,B).

(A–J) Venn Diagram showing number of overlapping genes and heat-map showing significance of overlap for different differential gene expression calculation methods. Upregulated differentially expressed genes (DEGs) for comparisons between GSE68270_PNPCbyQNSC (A), GSE68270_QNSCbyPNPC (B), GSE70696_TAPbyQNP (C), GSE70696_QNbyTAP (D), GSE99777_PSVZSCbyQSVZSC (E), GSE99777_QSVZSCbyPSVZSC (F), GSE93991_PGBCbyQGBC (G), GSE93991_QGBCbyPGBC (H), GSE114574_PGBObyQGBO (I) and GSE114574_QGBObyPGBO (J) as determined by Limma, edgeR and simple comparison of means methods are shown.
Proliferative and quiescent stem cell marker genes underly GBM modules
Hypergeometric test showed that HuAgeGBsplit_18 WGCNA GBM module and its equivalent c1_HuAgeGBsplit_193/32 MEGENA GBM modules, were the only GBM modules significantly enriched with proliferative and quiescent stem cell marker genes (Fig. 4A,B). Comparison of genes in c1_HuAgeGBsplit_193 and c1_HuAgeGBsplit_32 MEGENA modules, revealed that all genes in c1_HuAgeGBsplit_193 were also present in c1_HuAgeGBsplit_32 (Fig. 4A). Though proliferative and quiescent stem cell markers from different stem cell datasets (GSE68270, GSE70696, GSE99777, GSE93991 and GSE114574) were enriched in GBM modules (HuAgeGBsplit_18 WGCNA and c1_HuAgeGBsplit_193/32 MEGENA), quiescent adult hippocampal rat stem cell marker genes (GSE70696_QNPbyTAP) were most significantly enriched (Fig. 4B). However, no significant enrichment of SWIM 336 Glioblastoma gene list was found in HuAgeGBsplit_18 WGCNA module (p-value 0.411, overlap of 9 genes) and its equivalent MEGENA modules, c1_HuAgeGBsplit_32/193 MEGENA modules (p-value 1, overlap of 6 genes in c1_HuAgeGBsplit_32 and p-value 1, overlap of 2 genes in c1_HuAgeGBsplit_193).

Stem cell genes enriched WGCNA and MEGENA Glioblastoma modules. (A) Number of genes overlapping between Glioblastoma modules, HuAgeGBsplit_18 WGCNA and its equivalent c1_HuAgeGBsplit_32, and HuAgeGBsplit_193 MEGENA modules. HuAgeGBsplit_193 is a subset of HuAgeGBsplit_32 MEGENA module. (B) Table showing most significant overlap of rat quiescent hippocampal stem cell genes (QNPbyTAP) in Glioblastoma associated HuAgeGBsplit_18 WGCNA and c1_HuAgeGBaplit_32/193 MEGENA modules. (C, D, E) Gene Ontology (GO) Biological Process analysis of quiescent stem cell (QNPbyTAP) signature enriched HuAgeGBsplit_18 (C), c1_HuAgeGBsplit_32 (D) and c1_HuAgeGBsplit_193 (E).
Gene ontology (GO) annotation of GBM modules enriched with quiescent stem cell marker genes
To determine biological processes underlying GBM modules (HuAgeGBsplit_18 WGCNA and c1_HuAgeGBsplit_32/193 MEGENA modules) enriched with quiescent stem cell marker genes (GSE70696_QNPbyTAP), biological process gene ontology (GO) analysis was done. Lipid metabolic processes, “ether lipid metabolic process (GO:0046485)” and “cellular lipid biosynthetic process (GO:0097384)”, signaling pathways, “regulation of glucocorticoid receptor signaling pathway (GO:2000322)” and “negative regulation of response to cytokine stimulus (GO:0060761)”, and biomolecule modification processes, "protein O-linked fucosylation (GO:0036066)", "DNA dealkylation (GO:0035510)" and "rRNA base methylation (GO:0070475)", were top hits in HuAgeGBsplit_18 (Fig. 4C). Lipid metabolic processes, “phosphatidylserine metabolic process (GO:0006658)”, signaling pathways, “regulation of canonical Wnt signaling pathway (GO:0060828)” and “regulation of glucocorticoid receptor signaling pathway (GO:2000322)”, and biomolecule modification processes, “rRNA base methylation (GO:0070475)” and “positive regulation of mRNA splicing, via spliceosome (GO:0048026)”, were top hits in c1_HuAgeGBsplit_32/193 MEGENA modules (Fig. 4D,E). Most biological process GO categories showed comparable profiles for WGCNA and MEGENA GBM modules (HuAgeGBsplit_18 WGCNA and c1_HuAgeGBsplit_32/193 MEGENA modules) enriched with quiescent stem cell marker genes (GSE70696_QNPbyTAP). This suggests that networks identified by WGCNA and MEGENA network analysis are consistent and biologically robust.
Logistic regression model built with quiescent stem cell marker genes in GBM modules
A logistic regression model was built with select quiescent stem cell marker genes (GSE70696_QNPbyTAP) to diagnostic between control and GBM samples. From a total of 110 genes from GSE70696_QNPbyTAP enriched in GBM WGCNA module HuAgeGBsplit_18, genes that were atleast 40-fold upregulated in QNP relative to TAP were selected (CD151, CEND1, DCHS1, SMPD1, TPP1, GATD1, RNH1 and SMCR8) for logistic regression. Effects plots showed that probability of GBM relative to control increases with increased expression of CEND1, DCHS1, TPP1, GATD1, RNH1 and SMCR8 (Fig. 5B,C,E–H) and decreases with increased expression of CD151 and SMPD1 (Fig. 5A,D). Logistic regression model with these 8 genes without gene–gene interaction term was not significant (Chi-square p-value = 0.9847), while with gene–gene interactions the model was a significant (Chi-square p-value = 0.00799) predictor for GBM (Fig. 5I,J).

Logistic regression model for prediction of Glioblastoma using QNPbyTAP genes that are atleast 20-fold-upregulated in QNP relative to TAP and are significantly enriched in in Glioblastoma HuAgeGBsplit_18 WGCNA module. Effects plot showing probability of Glioblastoma (DiseaseGrade_4) with change in expression of CD151 (A), CEND1 (B), DCHS1 (C), SMPD1 (D), TPP1 (E), GATD1 (F), RNH1 (G) and SMCR8 (H). Chi-square test and Hosmer and Lemeshow goodness of fit (GOF) test for logistic regression model without gene–gene interaction (I) and with gene–gene interaction (J).
Hosmer–Lemeshow GOF test on without gene–gene interaction logistic regression model showed a large difference between observed and expected probabilities, and there was significant evidence of poor fit (p-value = 0.006, less than 0.05) (Fig. 5I, Table 5). Therefore, in logistic regression model without gene–gene interaction, Ho is rejected and Ha is accepted, and the model is rejected for being a poor fit for the data. Hosmer–Lemeshow GOF test on with gene–gene interaction logistic regression model showed a small difference between observed and expected probabilities, and there was no significant evidence of poor fit (p-value = 1, greater than 0.05) (Fig. 5J, Table 6). Therefore, in logistic regression model with gene–gene interaction, Ho is accepted and the model is accepted as a good fit for the data.
Discussion
Tumors are commonly treated by surgical removal, chemotherapy, radiotherapy and immunotherapy57. Safe surgical removal of glioma is challenging due to its critical location in brain. Additionally, high grade glioma or GBM is highly resistant to chemotherapy and radiotherapy. Immunotherapy treatments are FDA approved for certain blood cancers, but for solid tumors such as GBM immunotherapy ineffective due to incomplete infiltration of immunotherapeutic agent and immune suppression by tumor microenvironment58,59. Diagnosis is the first step in development of an effective treatment plan for any disease, including glioma. GBM is the most aggressive form of glioma that advances quickly giving healthcare providers limited time for diagnosis and treatment60. Therefore, to gain deeper understanding of glioma, especially GBM, with hope to develop early accurate diagnosis tools and novel therapies, molecular profiling and network medicine have emerged as research forerunners.
Molecular profile or gene based classification and diagnosis help physicians plan treatment and predict clinical outcome. For example, IDH1 gene mutation is highly correlated with glioma survival and is therefore used for glioma classification61. IDH1 mutation is lowest in grade 1 glioma that correlates with slow tumor growth and good survival4. On the other hand, IDH1 mutation is highest in grade 4 glioma or GBM that correlates with fast tumor growth and poor survival4. Improvement in technology, reduced cost of high-throughput sequencing, extensive collaborations and data sharing have made a plethora of glioma molecular profiling datasets such as RNA-seq available to research community18,20. With the explosion of molecular profiling genomics big data, research focus has now shifted from big data mining to big data analysis to prioritize a set of genes with diagnostic value that would eliminate need to profile all 23 K protein coding genes from glioma samples. However, prioritization of a subset of genes for glioma diagnosis and classification has been challenging due to high cellular heterogeneity across and within tumor samples of glioma62. Stem cell-like cells in glioma are thought to be responsible for tumor initiation, progression and recurrence63. Chemotherapy and radiotherapy kill proliferative stem cells, but are unable to kill quiescent stem cells in the tumor. Quiescent stem cells left in the tumor at end of treatment enter a proliferative state and reconstitute tumor, which leads to tumor recurrence64. Therefore, here the goal was to identify distinct sets of proliferative and quiescent stem cell marker genes in GBM that can be used for diagnosis and can serve as potential drug targets.
A meta-analysis was performed on publicly available high-throughput gene expression datasets from human glioma samples, control human brains, normal stem cells and GBM cells in quiescent and proliferative states18,26,27,28,29,30,31,32,33,34. DEGs specific to stem cell states were identified from normal stem cell and Glioblastoma cell culture datasets in quiescent and proliferative states. Interestingly, only 45.87% and 25.97% of genes from GBM cell cultures in proliferative and quiescent states were common with normal stem cells in proliferative and quiescent states, respectively (Supplementary Table S1 A,B). This suggests that cancer stem cells, especially those in quiescent state are distinctly different from normal stem cells.
Network analysis facilitates grouping of genes with highly correlated gene expression patterns into modules. It is assumed that modules corelate with distinct biological and cellular states, such as diseases and cell types, respectively. Presently, network analysis identified 9 WGCNA modules and 20 MEGENA modules that were highly correlated with GBM (Fig. 2B,F). One of these WGCNA modules, HuAgeGBsplit_18 WGCNA module (equivalent c1_HuAgeGBsplit_32/193 MEGENA modules) was also significantly enriched with adult hippocampal rodent quiescent stem cell genes (GSE70696_QNPbyTAP) (Fig. 4B). Interestingly, though this quiescent stem cell marker enriched HuAgeGBsplit_18 WGCNA module (equivalent c1_HuAgeGBsplit_32/193 MEGENA modules) had a significant correlation with GBM (p-value 0.007) it had a small correlation value of 0.14 with GBM (Fig. 2B). Possible reasons for this small but significant correlation are discussed here: (A) The result is consistent between WGCNA and MEGENA, two completely different network analysis algorithms. This supports that the results are biologically robust and not a computational artifact that would alter based on alterations in default algorithm settings. (B) SVA + LM normalization was used in this study to retain effects of glioma on gene expression and remove effects of all other covariants. It is possible that effects of covariants such as batch effects are not completely removed by this normalization, which is confounding glioma gene expression effects. (C) It is possible there are other covariants that significantly effect gene expression, such as patients’ comorbidities. However, as this information was not available, it could not be included in SVA + LM normalization and therefore glioma effects could not be effectively retained. (D) Controls used in this study comprise of RNA-seq datasets from different parts of the brain derived from humans other than the patients themselves. Ideally control tissue should be derived from the same patient who has the glioma, but presently such patient matched controls were not available for analysis. Lack of patient matched controls is a common challenge in the field of glioma and human disease research.
Quiescent stem cells exist in non-proliferative G0 cell cycle phase, but retain ability to reversibly enter cell cycle in response to stimuli. Depletion of surrounding proliferative stem cells and differentiated cells stimulate quiescent stem cells, which are multipotent and have self-renewal potential, to enter cell cycle and replenish the tissue65. Quiescent stem cell properties of rodent hippocampal stem cells have been extensively experimentally characterized33,66. Though proliferative stem cell marker genes of GBM origin (PGBCbyQGBC) were significantly enriched in HuAgeGBsplit_18 WGCNA GBM module (equivalent c1_HuAgeGBsplit_32/193 MEGENA GBM modules) with p-value 1.62E-07, it was normal quiescent stem cell marker genes from rodent hippocampal stem cells (GSE70696_QNPbyTAP) that were most significantly enriched with p-value 4.39E-20 (Fig. 4B). Normal stem cells transition from quiescent state to proliferative state and further to differentiated state. Recently, a set of 336 genes were identified in GBM with SWIM network analysis method, which are potentially involved in transition from stem-like state to differentiated state52,53. Interestingly, no significant enrichment (p-value 0.411, overlap of 9 genes) of SWIM 336 Glioblastoma gene list was found in HuAgeGBsplit_18 GBM WGCNA module (equivalent c1_HuAgeGBsplit_32/193 MEGENA GBM modules). This supports that quiescent stem cell marker genes (GSE70696_QNPbyTAP) enriched in HuAgeGBsplit_18 WGCNA GBM module (equivalent c1_HuAgeGBsplit_32/193 MEGENA GBM modules) represent an undifferentiated quiescent stem cell state distinct from differentiating stem cells.
Gene Ontology (GO) analysis of GSE70696_QNPbyTAP enriched HuAgeGBsplit_18 WGCNA GBM module (equivalent c1_HuAgeGBsplit_32/193 MEGENA GBM modules) revealed enrichment of biomolecule synthesis GO terms, such as lipid metabolism, DNA modification, protein post-translational modification, ribosome RNA processing, cell cycle GO terms (G2/M transition, mitotic cell cycle) and signaling pathways such as Wnt signaling (Fig. 4C–E). This is consistent with cell cycle and ribosome biogenesis GO terms, previously reported in the rodent hippocampal stem cell dataset from which GSE70696_QNPbyTAP signature genes were identified33. As quiescent stem cells can replenish tumor after proliferative stem cells are killed by chemotherapy and radiotherapy, a combinatorial therapy that targets both proliferative and quiescent stem cells could be more effective in GBM treatment. Quiescent stem cell marker genes (GSE70696_QNPbyTAP) enriched in HuAgeGBsplit_18 WGCNA GBM module (equivalent c1_HuAgeGBsplit_32/193 MEGENA GBM modules) reported here, could serve as potential GBM quiescent stem cell drug targets. Small molecule DYRK1B inhibitors were recently shown to target quiescent stem cells and potentiate treatment benefits of chemotherapy67. This supports development of treatment strategies to target both proliferative and quiescent stem cells in GBM.
Gene expression of eight genes (CD151, CEND1, DCHS1, SMPD1, TPP1, GATD1, RNH1 and SMCR8) from GSE70696_QNPbyTAP enriched in HuAgeGBsplit_18 WGCNA GBM module (equivalent c1_HuAgeGBsplit_32/193 MEGENA GBM modules), were sufficient to build a logistic regression diagnostic model that could distinguish between GBM and control samples (Fig. 5J). Four of the eight genes (CD151, CEND1, SMPD1 and RNH1) used in the model have previously been reported to be important in GBM, other types of cancer or in development68,69,70,71. CD151 is a member of tetraspanins scaffolding protein family that is involved in cell–cell adhesion, integrin interaction, cell signaling, cancer progression and metastasis72. In GBM, CD151 associates with α3β1 integrin to potentiate EGFR signaling, drive cancer cell motility and tumor aggressiveness71. CEND1 or BM88 protein acts as a cell-cycle inhibitor to negatively regulated proliferation and promote differentiation in spinal cord development70. In cell membrane lipid bilayer, apoptosis and cellular growth is regulated by balance between sphingosine-1-phosphate and ceramide molecules73. SMPD1 gene that regulates ‘ceramide sphingosine-1-phosphate rheostat’ drives tumor growth and immune escape in non-small cell lung cancer69. RNH1 repairs DNA in response to DNA damage and is a known diagnostic and prognostic marker of glioma68. Taken together, this provides support for biological relevance of the eight genes (CD151, CEND1, DCHS1, SMPD1, TPP1, GATD1, RNH1 and SMCR8) used here to build a logistic regression diagnostic model for GBM. A molecular screening kit could be developed with these eight genes for faster and accurate screening of GBM. However, as results of present study were obtained by using computational meta-analysis alone, further experimental validation is required. Overall, this study deconvolutes highly heterogeneous glioma molecular profiles and provides a new perspective to diagnose and develop therapeutic strategies using a small number of quiescent stem cell markers in GBM.
Data availability
Glioblastoma RNA-seq datasets, SRP027383 and SRP091303, were obtained from NCBI SRA. Other RNA-seq datasets for control brains (GSE67333, GSE64810, GSE100297 and GSE53697) and stem cells (GSE68270, GSE70696, GSE99777, GSE93991 and GSE114574), were obtained from NCBI GEO. The author is most grateful to NCBI SRA and NCBI GEO for storing and making these datasets open access. The author also appreciates the organizations and investigators who generously submitted these datasets for sharing to NCBI GEO and NCBI SRA.
References
Timmons, J. J. et al. Literature review of spinal cord glioblastoma. Am. J. Clin. Oncol. 41, 1281–1287. https://doi.org/10.1097/COC.0000000000000434 (2018).
Delgado-Lopez, P. D. & Corrales-Garcia, E. M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 18, 1062–1071. https://doi.org/10.1007/s12094-016-1497-x (2016).
Davis, M. E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 20, S2-8. https://doi.org/10.1188/16.CJON.S1.2-8 (2016).
Louis, D. N. et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 131, 803–820. https://doi.org/10.1007/s00401-016-1545-1 (2016).
Louis, D. N. et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109. https://doi.org/10.1007/s00401-007-0243-4 (2007).
Noroxe, D. S., Poulsen, H. S. & Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 1, e000144. https://doi.org/10.1136/esmoopen-2016-000144 (2016).
Shergalis, A., Bankhead, A. 3rd., Luesakul, U., Muangsin, N. & Neamati, N. Current challenges and opportunities in treating glioblastoma. Pharmacol. Rev. 70, 412–445. https://doi.org/10.1124/pr.117.014944 (2018).
Alphandery, E. Glioblastoma treatments: An account of recent industrial developments. Front. Pharmacol. 9, 879. https://doi.org/10.3389/fphar.2018.00879 (2018).
Kaiser, J. The cancer stem cell gamble. Science 347, 226–229. https://doi.org/10.1126/science.347.6219.226 (2015).
Almiron Bonnin, D. A. et al. Secretion-mediated STAT3 activation promotes self-renewal of glioma stem-like cells during hypoxia. Oncogene 37, 1107–1118. https://doi.org/10.1038/onc.2017.404 (2018).
Yoshida, G. J. & Saya, H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 107, 5–11. https://doi.org/10.1111/cas.12817 (2016).
Safa, A. R., Saadatzadeh, M. R., Cohen-Gadol, A. A., Pollok, K. E. & Bijangi-Vishehsaraei, K. Emerging targets for glioblastoma stem cell therapy. J. Biomed. Res. 30, 19–31. https://doi.org/10.7555/JBR.30.20150100 (2016).
Wang, K., Wu, X., Wang, J. & Huang, J. Cancer stem cell theory: therapeutic implications for nanomedicine. Int. J. Nanomed. 8, 899–908. https://doi.org/10.2147/IJN.S38641 (2013).
Chen, W., Dong, J., Haiech, J., Kilhoffer, M. C. & Zeniou, M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016, 1740936. https://doi.org/10.1155/2016/1740936 (2016).
Barabasi, A. L., Gulbahce, N. & Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 12, 56–68. https://doi.org/10.1038/nrg2918 (2011).
Conte, F. et al. A paradigm shift in medicine: A comprehensive review of network-based approaches. Biochim. Biophys. Acta Gene Regul. Mech. https://doi.org/10.1016/j.bbagrm.2019.194416 (2019).
Scelfo, C. et al. Towards precision medicine: The application of omics technologies in asthma management. F1000Research 7, 423. https://doi.org/10.12688/f1000research.14309.2 (2018).
Zhao, Z. et al. Comprehensive RNA-seq transcriptomic profiling in the malignant progression of gliomas. Sci. Data 4, 170024. https://doi.org/10.1038/sdata.2017.24 (2017).
Edgar, R., Domrachev, M. & Lash, A. E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. https://doi.org/10.1093/nar/30.1.207 (2002).
Tomczak, K., Czerwinska, P. & Wiznerowicz, M. The cancer genome atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 19, A68-77. https://doi.org/10.5114/wo.2014.47136 (2015).
International Cancer Genome Consortium et al. International network of cancer genome projects. Nature 464, 993–998. https://doi.org/10.1038/nature08987 (2010).
Langfelder, P. & Horvath, S. Fast R Functions for robust correlations and hierarchical clustering. J. Stat. Softw. 46, i11 (2012).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Song, W. M. & Zhang, B. Multiscale embedded gene co-expression network analysis. PLoS Comput. Biol. 11, e1004574. https://doi.org/10.1371/journal.pcbi.1004574 (2015).
Leinonen, R., Sugawara, H., Shumway, M. & International Nucleotide Sequence Database, C. The sequence read archive. Nucleic Acids Res. 39, D19-21. https://doi.org/10.1093/nar/gkq1019 (2011).
Labadorf, A. et al. RNA sequence analysis of human huntington disease brain reveals an extensive increase in inflammatory and developmental gene expression. PLoS ONE 10, e0143563. https://doi.org/10.1371/journal.pone.0143563 (2015).
Scheckel, C. et al. Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. eLife https://doi.org/10.7554/eLife.10421 (2016).
Magistri, M., Velmeshev, D., Makhmutova, M. & Faghihi, M. A. Transcriptomics profiling of Alzheimer’s disease reveal neurovascular defects, altered amyloid-beta homeostasis, and deregulated expression of long noncoding RNAs. J. Alzheimer’s Dis. 48, 647–665. https://doi.org/10.3233/JAD-150398 (2015).
Itoh, N. et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. U.S.A. 115, E302–E309. https://doi.org/10.1073/pnas.1716032115 (2018).
Aulestia, F. J. et al. Quiescence status of glioblastoma stem-like cells involves remodelling of Ca(2+) signalling and mitochondrial shape. Sci/ Rep/ 8, 9731. https://doi.org/10.1038/s41598-018-28157-8 (2018).
Tejero, R. et al. Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine 42, 252–269. https://doi.org/10.1016/j.ebiom.2019.03.064 (2019).
Walker, T. L. et al. Lysophosphatidic acid receptor is a functional marker of adult hippocampal precursor cells. Stem Cell Rep. 6, 552–565. https://doi.org/10.1016/j.stemcr.2016.03.002 (2016).
Mukherjee, S., Brulet, R., Zhang, L. & Hsieh, J. REST regulation of gene networks in adult neural stem cells. Nat. Commun. 7, 13360. https://doi.org/10.1038/ncomms13360 (2016).
Morizur, L. et al. Distinct molecular signatures of quiescent and activated adult neural stem cells reveal specific interactions with their microenvironment. Stem Cell Rep. 11, 565–577. https://doi.org/10.1016/j.stemcr.2018.06.005 (2018).
Mukherjee, S., Klaus, C., Pricop-Jeckstadt, M., Miller, J. A. & Struebing, F. L. A Microglial signature directing human aging and neurodegeneration-related gene networks. Front. Neurosci. 13, 2. https://doi.org/10.3389/fnins.2019.00002 (2019).
Brown, J., Pirrung, M. & McCue, L. A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics https://doi.org/10.1093/bioinformatics/btx373 (2017).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. https://doi.org/10.1093/bioinformatics/btp120 (2009).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—A python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. https://doi.org/10.1093/bioinformatics/btu638 (2015).
Conesa, A. et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 17, 13. https://doi.org/10.1186/s13059-016-0881-8 (2016).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, New York, 2016).
41Wei, T. & Simko, V. R package "corrplot": Visualization of a Correlation Matrix (Version 0.84). https://github.com/taiyun/corrplot (2017). Accessed 17 Aug 2019.
Leek, J. T. & Storey, J. D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3, 1724–1735. https://doi.org/10.1371/journal.pgen.0030161 (2007).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Leek, J. T. svaseq: Removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. https://doi.org/10.1093/nar/gku864 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Miller, J. A., Horvath, S. & Geschwind, D. H. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc. Natl. Acad. Sci. U.S.A. 107, 12698–12703. https://doi.org/10.1073/pnas.0914257107 (2010).
Langfelder, P., Luo, R., Oldham, M. C. & Horvath, S. Is my network module preserved and reproducible?. PLoS Comput. Biol. 7, e1001057. https://doi.org/10.1371/journal.pcbi.1001057 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Benjamini, Y., Drai, D., Elmer, G., Kafkafi, N. & Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 125, 279–284. https://doi.org/10.1016/s0166-4328(01)00297-2 (2001).
Shen, L. & Sinai, M. GeneOverlap: Test and visualize gene overlaps. R package version 1.22.0, https://shenlab-sinai.github.io/shenlab-sinai/ (2019). Accessed 11 Aug 2019.
Chen, H. & Boutros, P. C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 12, 35. https://doi.org/10.1186/1471-2105-12-35 (2011).
Fiscon, G., Conte, F. & Paci, P. SWIM tool application to expression data of glioblastoma stem-like cell lines, corresponding primary tumors and conventional glioma cell lines. BMC Bioinform. 19, 436. https://doi.org/10.1186/s12859-018-2421-x (2018).
Fiscon, G., Conte, F., Licursi, V., Nasi, S. & Paci, P. Computational identification of specific genes for glioblastoma stem-like cells identity. Sci. Rep. 8, 7769. https://doi.org/10.1038/s41598-018-26081-5 (2018).
Chen, E. Y. et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128. https://doi.org/10.1186/1471-2105-14-128 (2013).
Lele, S. R. & Keim, J. L. Weighted distributions and estimation of resource selection probability functions. Ecology 87, 3021–3028. https://doi.org/10.1890/0012-9658(2006)87[3021:wdaeor]2.0.co;2 (2006).
Hosmer, D. W., Hosmer, T., Le Cessie, S. & Lemeshow, S. A comparison of goodness-of-fit tests for the logistic regression model. Stat. Med. 16, 965–980. https://doi.org/10.1002/(sici)1097-0258(19970515)16:9<965::aid-sim509>3.0.co;2-o (1997).
Paolillo, M., Boselli, C. & Schinelli, S. Glioblastoma under Siege: An overview of current therapeutic strategies. Brain Sci. https://doi.org/10.3390/brainsci8010015 (2018).
Mukherjee, S. Genomics-guided immunotherapy for precision medicine in cancer. Cancer Biotherapy Radiopharm. https://doi.org/10.1089/cbr.2018.2758 (2019).
Nixon, N. A. et al. Current landscape of immunotherapy in the treatment of solid tumours, with future opportunities and challenges. Curr. Oncol. (Toronto, Ont.) 25, e373–e384. https://doi.org/10.3747/co.25.3840 (2018).
Urbanska, K., Sokolowska, J., Szmidt, M. & Sysa, P. Glioblastoma multiforme—An overview. Contemp. Oncol. 18, 307–312. https://doi.org/10.5114/wo.2014.40559 (2014).
Yan, W. et al. Molecular classification of gliomas based on whole genome gene expression: A systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro-oncology 14, 1432–1440. https://doi.org/10.1093/neuonc/nos263 (2012).
Inda, M. M., Bonavia, R. & Seoane, J. Glioblastoma multiforme: A look inside its heterogeneous nature. Cancers 6, 226–239. https://doi.org/10.3390/cancers6010226 (2014).
Cheray, M. et al. Glioblastoma (ed S. De Vleeschouwer) (2017).
Carrasco-Garcia, E. et al. Therapeutic strategies targeting glioblastoma stem cells. Recent Pat. Anti-Cancer Drug Discov. 8, 216–227 (2013).
Cho, I. J. et al. Mechanisms, hallmarks, and implications of stem cell quiescence. Stem Cell Rep. 12, 1190–1200. https://doi.org/10.1016/j.stemcr.2019.05.012 (2019).
Mira, H. et al. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem Cell 7, 78–89. https://doi.org/10.1016/j.stem.2010.04.016 (2010).
Becker, W. A wake-up call to quiescent cancer cells—Potential use of DYRK1B inhibitors in cancer therapy. FEBS J. 285, 1203–1211. https://doi.org/10.1111/febs.14347 (2018).
Kun, S., Duan, Q., Liu, G. & Lu, J. M. Prognostic value of DNA repair genes based on stratification of glioblastomas. Oncotarget 8, 58222–58230. https://doi.org/10.18632/oncotarget.17452 (2017).
Kachler, K. et al. Enhanced acid sphingomyelinase activity drives immune evasion and tumor growth in non-small cell lung carcinoma. Cancer Res. 77, 5963–5976. https://doi.org/10.1158/0008-5472.CAN-16-3313 (2017).
Politis, P. K. et al. BM88/CEND1 coordinates cell cycle exit and differentiation of neuronal precursors. Proc. Natl. Acad. Sci. U.S.A. 104, 17861–17866. https://doi.org/10.1073/pnas.0610973104 (2007).
Zhou, P. et al. CD151-alpha3beta1 integrin complexes are prognostic markers of glioblastoma and cooperate with EGFR to drive tumor cell motility and invasion. Oncotarget 6, 29675–29693. https://doi.org/10.18632/oncotarget.4896 (2015).
Sadej, R., Grudowska, A., Turczyk, L., Kordek, R. & Romanska, H. M. CD151 in cancer progression and metastasis: A complex scenario. Lab. Investig. 94, 41–51. https://doi.org/10.1038/labinvest.2013.136 (2014).
Tani, M., Ito, M. & Igarashi, Y. Ceramide/sphingosine/sphingosine 1-phosphate metabolism on the cell surface and in the extracellular space. Cell. Signal. 19, 229–237. https://doi.org/10.1016/j.cellsig.2006.07.001 (2007).
Acknowledgements
This work was conducted at the author’s home using personal resources at a) Sarat Ghosh Garden Road, Calcutta, West Bengal, India 700031 and b) Garland Avenue, Downtown Los Angeles, Los Angeles, U.S., 90017 on personal laptops (MacBook Pro and Windows OS) from 2017 to 2019. The author would like to express deepest gratitude to Springer Nature waivers team and Scientific Reports team for the generous discount on publication cost. Other open source and free resources utilized for this work are acknowledged below.
For full text publications access The author is most grateful to NIH/NLM (U.S. National Institute of Health’s National Library of Medicine) for open-access to full-text scientific publications on www.ncbi.nlm.nih.gov/pmc/ PubMed Central.
For computational pipelines Citations and descriptions of all computational pipelines have been given under methods sections. Additionally, the author would like to add special thanks to Dr. Steve Horvath, Professor at Department of Human Genetics and Biostatistics, University of California Los Angeles (UCLA) and Dr. Jeffrey T. Leek, Professor at Department of Biostatistics, Johns Hopkins for their publicly available computational pipelines on WGCNA/enrichment/Preservation analyses and SVA/Regression analyses, respectively. Dr. Won-Min Song and Dr. Bin Zhang from Department of Genetics and Genomic Sciences, Icahn Institute of Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai for their publicly available computational MEGENA pipeline on github. Dr. Steve Horvath, Dr. Jeffrey T. Leek, Dr. Won-Min Song and Dr. Bin Zhang’s computational tutorials were integral to the author’s learning and for execution of the work done in this paper.
For computational platform The research site or computation was performed at the author’s home in Calcutta, India and Los Angeles, U.S. on personal laptops (MacBook Pro and Windows OS) with high-speed internet connection and computational tools. RNA-seq datasets were mapped using NSF supported publicly available Cyverse Discovery Environment.
Author information
Authors and Affiliations
Contributions
Author contributions S.M. ideated and managed the work presented in this paper. S.M. also performed experimental design, wrote the manuscript, prepared the figures/tables and submitted the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mukherjee, S. Quiescent stem cell marker genes in glioma gene networks are sufficient to distinguish between normal and glioblastoma (GBM) samples. Sci Rep 10, 10937 (2020). https://doi.org/10.1038/s41598-020-67753-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67753-5
This article is cited by
-
Characterization of EGFR-reprogrammable temozolomide-resistant cells in a model of glioblastoma
Cell Death Discovery (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
