
Abstract
The electronic and topological properties of MoS2 monolayers with n-p codoping effect are investigated by using first-principles calculations. Two types of the doped Nb atoms play the roles of the p-type and n-type dopants, respectively. The n-p codoping is found inducing a large valley polarization, associated with the strong magnetization induced by the Nb dopants. Interestingly, the system simultaneously owns a perfect Chern insulating band gap opened exactly at the Fermi level. The nontrivial band gap comes from the lifting of the degeneracy of the dxz and dyz orbitals of Nb2 atoms after the spin-orbit coupling is considered. Our work inspires exciting prospects to tune the novel properties of materials with n-p codoping effects.
Similar content being viewed by others
Introduction
Recently, transition-metal dichalcogenides (TMDs) have been proposed as excellent candidates for electronics, spintronics, and valleytronics materials by manipulating the charge, spin, and valley degrees of freedom in the system1,2,3,4. For example, the experimental realization of valley polarization could be through optical pumping5,6 in MoS2 monolayers (MLs) or externally applied magnetic fields7,8,9 in WSe2 and MoSe2 monolayers. The approach of optical pumping is, however, restricted by the limited carrier lifetimes in dynamical process. And the valley polarization achieved through an external magnetic field is generally quite small. An alternative way to control the valley degree of freedom in TMDs is through magnetic atom doping10,11 or the proximate effect from magnetic substrates12.
N-p codoping, with both n-type and p-type dopants in one material, has been proved to be an effective strategy to tune the electronic properties13,14,15. Ferromagnetic (FM) order was reported in graphene with Ni-B codoping13. And quantum anomalous Hall effect was predicted in graphene14 and Sb2Te315 through n-p codoping. In this work, we explore the electronic structures and valleytronics in the MoS2 monolayer with n-p codoping. Very large valley polarization at the MoS2 valence bands is obtained, attributed to the imbalance of K and K′ bands aroused by the magnetic Nb dopants. Chern insulating states are also found in the system. The coexistence of valley polarization and Chern insulating effects in the MoS2 ML with Nb n-p codoping demonstrates that this kind of system has potential applications in not only valleytronics, but also electronics and spintronics, which will greatly facilitate the device integration in practice.
Results and discussion
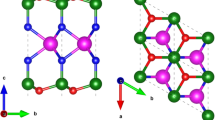
For the 3 × 3 supercell MoS2 ML with one Mo substituted by Nb atom (Nb1), the Nb tends to substitute the Mo atom instead of the S atom for both rich Mo and S cases, consistent with the tendency reported in previous researches16,17,18,19,20. The Nb1 substituted MoS2 ML is also proved to be dynamically stable (without imaginary frequency) through the density functional perturbation theory (DFPT)21. Based on this structure, four typical high-symmetry adsorption sites are considered for Nb2 atom, with the adsorption energy calculated as: \({E}_{{\rm{a}}}={E}_{{\rm{sample}}}+{E}_{{\rm{adatom}}}-{E}_{{\rm{total}}}\). The Esample, Eadatom, and Etotal are the total energies of the MoS2 ML with Nb1 doping, the single Nb atom, the MoS2 ML with Nb1-Nb2 codoping, respectively. As shown in Table 1, the most stable configuration is N (see Fig. 1(a)). For comparison, the obtained adsorption energy of the most stable configuration M for the pristine MoS2 ML with Nb2 adsorption is also given in Table 1. As the definition indicates, the adsorption energy reflects the interaction strength between the Nb2 atom and the two-dimensional sheet. For the pristine MoS2 ML, the adsorption energy is 2.485 eV, while if the sheet is the Nb1 doped MoS2 ML (Fig. 1(a)), the adsorption energy increases to 3.789 eV. Thus, we infer that the interaction strength of the Nb2 adatom in the Nb1-Nb2 codoping case is enhanced (by 1.3 eV). This behavior can be ascribed to the strong electrostatic attractive interaction between the n-type (Nb2) and p-type (Nb1) dopants. In the following, we primarily focus on the most stable N configuration for the Nb1-Nb2 codoping case.

(a) Top and side views of the 3 × 3 supercell of the MoS2 ML with Nb n-p codopants. The side view is for the N configuration. (b) Reciprocal momentum space structure, the red and black hexagons/arrows are the reciprocal lattices/vectors for the 1 × 1 and 3 × 3 supercells, respectively. The special k-points for the 1 × 1 supercell are marked with subscript p. (c) Evolution of the band structures for the pristine MoS2 ML (i) and the Nb1 doped (ii), Nb2 doped (iii), and Nb1-Nb2 codoped (iv) MoS2 MLs.
The band structure and the densities of states (DOSs) for the MoS2 ML with Nb1-Nb2 codoping are given in Fig. 2(a) and the upper panel of Fig. 2(b), respectively. The spin-orbit coupling (SOC) is not yet considered. The Nb1 DOSs distribute over a wide range of energy, similar to those of Mo atoms in the pristine MoS222, indicating strong bonds formed between Nb1 and its neighboring S atoms. The DOSs of the Nb2 are, however, mainly located within the band gap of the pristine MoS2. To further comprehend the roles of the two types of Nb atoms, the electronic structures of the pristine MoS2 ML with solely Nb1 or Nb2 are also calculated. For the MoS2 ML with one Mo atom substituted by one Nb atom in the 3 × 3 supercell, labeled as MoS2 (Nb1), the Fermi level (EF) is now located below the top of the valence bands of the pristine MoS2 (middle panel of Fig. 2(b)), implying the p-type acceptor character of Nb1 dopant. When one Nb atom is adsorbed on the top of one of the Mo atoms of the pristine MoS2 ML, marked as MoS2 (Nb2), the EF of the system is very close to the bottom of the conduction bands of the pristine MoS2 (lower panel of Fig. 2(b)). Thus, Nb2 adatom acts as the n-type donor. The d orbital of Nb2 atom splits into A (dz2), E1 (dxz, dyz), and E2 (dxy, dx2−y2) (Fig. 2(a,b)), due to the C3v symmetry owned.

(a) Band structure of the Nb1-Nb2 codoped MoS2 ML without SOC. The red and blue curves show the spin-up and spin-down components, respectively. The black curves in the upper, middle, and lower panels of (b) are the total DOSs of the Nb1-Nb2 codoped, Nb1 doped, and Nb2 doped MoS2 MLs, respectively. The red/blue curves are the partial DOSs of the Nb1/Nb2 atom. The gray areas in the middle and lower panels of (b) are the total DOSs of the pristine MoS2 ML. The Nb1 and Nb2 partial DOSs are quadrupled and doubled to obtain a clear view. The positive and negative values correspond to the spin-up and spin-down components, respectively. (c,d) are the normal and enlarged views of the band structures of the Nb1-Nb2 codoped MoS2 ML with SOC. The small red/blue circles in (a,c) indicate the spin-up/spin-down component located around Γ, folded from the Kp and K′p in the primitive cell.
The band evolution from the pristine MoS2 ML to the Nb1-Nb2 codoped MoS2 ML is summarized in Fig. 1(c). For the pristine MoS2 ML, the EF is within the band gap. For the MoS2 (Nb1) case, the EF moves downwards to the valence bands of the pristine MoS2, while for the MoS2 (Nb2) case, the EF moves upper to the conduction bands of the pristine MoS2, indicating Nb1 and Nb2 act as p-type and n-type dopants, respectively. For the MoS2 (Nb1, Nb2) case, the compensation effect of the n-type and p-type doping causes the EF located exactly at the E1 bands.
Besides the movements of the EF position, the magnetic behaviors of the Nb1 and Nb2 atoms in the systems are also different from each other. For example, the MoS2 (Nb1) is weakly spin polarized with a total magnetic moment of 0.73 μB (the middle panel in Fig. 2(b)), while the total magnetic moment is 5.00 μB for the MoS2 (Nb2). The strong magnetism of the MoS2 (Nb2) can be comprehended through strong atomic behavior of Nb2 atoms, consistent with the case of the 3d transition metal atom adsorption in MoS2 MLs in previous studies23,24. For the Nb n-p codoping case, the total magnetic moment of 4.00 μB, instead of 5.00 μB, is obtained for the most stable N configuration. The decrease of the magnetism in the n-p codoping case can be ascribed to the p-type Nb1 in the n-p codoping case gaining one unpaired electron from Nb2. The local magnetic moments of the Nb1 and Nb2 in the n-p codoped system are about 0.05 μB and 2.49 μB, respectively. Thus, the magnetism in the n-p codoped MoS2 ML is primarily induced by the Nb2 adatoms. To find whether the above FM structure is the magnetic ground state, the total energies of non-magnetic (NM) and antiferromagnetic (AFM) structures are also calculated. The total energy of the FM ordering per Nb1-Nb2 pair is found to be lower than that of the NM ordering by 355 meV and AFM ordering by 2 meV. Thus, the FM ordering is the most stable magnetic configuration for the Nb codoped system. The Curie temperature (Tc) estimated with the mean-field approximation25 is about 15.5 K.
The C3v symmetry owned by the system makes the E1 (dxz, dyz) and E2 (dxy, dx2−y2) bands both be degenerate at the Γ point with quadratic non-Dirac band dispersions before the SOC is included (Fig. 2(a)). Under this case, the system could be called as a spin-gapless semiconductor (SGS)26,27,28 for the bands at the EF are 100% spin polarized. When the SOC is included, the doubly degenerate energy points at the Γ point are lifted and band gaps are opened, as shown in Fig. 2(c,d). Additionally, the degeneracy of the tops of the valence bands of the pristine MoS2 (around −1.2 eV in Fig. 2(a)) is also lifted (Fig. 2(c)). E1 is exactly located at the EF without the SOC (Fig. 2(a)) and a global band gap of 15.0 meV is opened by the SOC in the system (Fig. 2(d)). We also check the results by using the HSE06 functional29. The calculations show that the main feature keeps undistributed with the HSE06 functional, i.e., the E1 bands are still located around the EF and a global band gap is also opened. The opened band gap is 36.4 meV, larger than the result obtained from the metaGGA method, due to the stronger exchange-correlation interactions predicted for the Nb dxz and dyz orbitals in the HSE06 functional. The real value of the band gap may be determined by future experimental measurements. Hence, the n-p codoped MoS2 ML is an insulator. As shown in Fig. 1(c), if there is no Nb1 in the system, the E1 bands are totally occupied by electrons. It is, thus, the Nb n-p codoping that gives rise to the E1 bands located exactly at the EF.
Due to the strong magnetism of the Nb2 adatoms, legible spin polarization of about 0.2 eV is induced at the tops of the valence bands of MoS2 (around −1.2 eV in Fig. 2(a)). This magnetism together with the broken space-inversion symmetry in the system may lead to valley polarization effect30,31. However, the use of the 3 × 3 supercell in the calculations causes band folding. To explore the valley polarization of the n-p codoped MoS2 ML, the bands shown in Fig. 2(a,c) are unfolded onto the k-points in the Brillouin zone (BZ) of the 1 × 1 primitive cell (Fig. 1(b)) by using a k-projection method32,33,34. Figure 3(a,b) show the unfolded bands for the Nb n-p codoped MoS2 ML without SOC. The obvious spin polarization in the valence bands of the MoS2 is also outstanding. The bands at Kp and K′p for both the spin-up and spin-down components are degenerate, whose sketches are displayed in the (i) case of Fig. 3(d). Thus, no valley polarization appears in the bands.

Unfolded band structures for the Nb n-p codoped MoS2 ML. (a,b) are for the spin-up and spin-down bands without SOC, respectively. (c) is for the SOC calculation. The small red/blue arrow indicates the spin-up/spin-down bands. (d) Schematic diagrams of the bands around the Kp and K′p points without (i) and with (ii) SOC in the primitive cell. The red and blue curves represent the bands with different spin components.
When the SOC is included, a valley polarization of 125 meV can be observed, defined as \(\Delta {V}_{K{K}^{{\rm{{\prime} }}}}={E}_{{K{\rm{{\prime} }}}_{p}}^{V}-{E}_{{K}_{p}}^{V}\). When the HSE06 functional is employed, an almost same value of the valley polarization is obtained, due to the small exchange-correlation interaction for the Mo d orbitals mainly contributing to the valley bands in the system. Only in the unfolded bands (Fig. 3(c)), the valley polarization can be distinguished. How the tops of the valence bands of the MoS2 in Fig. 2(c) correspond to the Kp or K′p points in the MoS2 primitive cell is also illustrated. Figure 3(d) shows the schematic diagrams of the valley polarization formed in the Nb n-p codoped MoS2 ML. Reference12 has found that the magnitude of the valley polarization is limited by the smaller spin splitting arising from the SOC or the exchange field. Therefore, the valley polarization obtained (125 meV) should be limited by the smaller SOC strength other than the spin polarization (200 meV). The valley polarization may be enhanced further with the increase of the SOC, by such as substituting the atoms with heavier elements. The obtained valley polarization is actually an intrinsic attribute owned by the system and no magnetic field or optical pumping is necessary to realize the effect. This valley polarization makes the Nb n-p codoped MoS2 ML can be called as a ferrovalley material. If the spin orientations of the Nb atoms are reversed, the band features at K and K′ are exchanged, similar to the case in ref. 35. Thus, the ferrovalley property still exists in the system, with the same valley polarization strength, but an opposite sign. Since the total energies of the two systems are the same, the two ferrovalley states can be regarded as energy degenerate states.
We now identify the possible topological behaviors of the band gap opened at the EF (Fig. 2(d)). The bands are fitted by using maximally localized Wannier functions (MLWFs) method36,37,38 (Fig. 4(a)). The Berry curvatures can then be calculated. Two peaks of the Berry curvatures appear around the Γ point (Fig. 4(b)), ascribed to the parabolic band dispersions of E1 (Fig. 2(a)) without SOC39. The calculated Chern number of 1 gives a direct evidence for the existence of Chern insulating state in the system. Figure 4(c) displays the band structure of a one-dimensional Nb n-p codoped MoS2 nanoribbon, calculated by using the tight-binding model constructed from the MLWFs. The two edge states (red curves in Fig. 4(c)) connecting the conduction and valence bands exist inside the SOC induced band gap, representing one chiral dissipationless conducting channel existing on each side of the nanoribbon sample. Besides, the edge states should be 100% spin-polarized and half-metallic since the two-fold E1 bands in Fig. 2(a) are spin up40,41, availing the applications of the Nb n-p codoped MoS2 ML system in spintronic devices. The quantized Hall conductivity of e2/h at EF (Fig. 4(d)) matches well with the nontrivial band gap contributed by E1 orbitals, while the Hall conductivity of −e2/h at −0.8 eV appears due to the E2 orbitals. The forming mechanism of these topological states can be ascribed to the SOC-induced band splitting of the degenerate quadratic non-Dirac bands at the Γ point39,40,41. Thus, the large ferrovalley effect and the Chern insulating state achieved in the Nb n-p codoped MoS2 monolayer are contributed from the bands at the K (K′) and Γ points in the momentum space, respectively.

Wannier-function fitted bands around the EF (a), Berry curvatures (b), edge states (c), and Hall conductivity (d) for the Nb1-Nb2 codoped MoS2 ML.
The electronic and topological properties of the Nb codoped systems with different Nb doping concentrations are also studied. If the Nb doping concentration is increased, such as one pair of Nb1-Nb2 doped in one 2 × 2 supercell, the band structure is found being changed much and becomes metallic, ascribed to the strong interaction between the different Nb pairs. If the Nb doping concentration is lowered, for example, one pair of Nb1-Nb2 doped in one 4 × 4 supercell, the valley polarization and the nontrivial bands can still be clearly observed, although global band gaps may not be opened. Therefore, the Nb doping concentration should be carefully examined to give perfect Chern insulting states and large valley polarization in the MoS2 MLs.
In experiments, the n-p codoping has been realized in some materials, such as dilute magnetic semiconductors of ZnMnAlO and ZnCoAlO42. Based on these techniques, the n-p codoped MoS2 ML may be fabricated by following two steps. First, synthesize the p-type doped MoS2 ML with Nb1 atoms substituting the Mo atoms via e-beam evaporation plus chemical vapor deposition (CVD), as implemented in ref. 16 or chemical vapor transport (CVT) in refs. 17,18. Second, deposit some Nb atoms onto the surface of the prepared p-type MoS2 ML. Due to the electrostatic attraction between the n-type and p-type dopants, these deposited Nb atoms (Nb2) tend to be located on the top of the Nb1 atoms, as the obtained adsorption energies indicate. In this way, the Nb n-p codoped system might be achieved in experiments.
Conclusion
The electronic states of the Nb n-p codoped MoS2 monolayer are studied with ab initio calculations. Two Nb atoms (Nb1 and Nb2) serve as p-type and n-type dopants, respectively. Large valley polarization is predicted, caused by the induced magnetism together with the broken space-inversion symmetry and the SOC interaction in the MoS2 system. The Nb n-p codoped MoS2 monolayer is also a Chern insulator, whose edge can conduct the pure spin-up current without energy dissipation. The coexistence of the large valley polarization and Chern insulting states provides encouraging routes in applying the codoped two-dimensional materials in the fabrication of valleytronic, microelectronic, and spintronic devices.
Methods
Since experimental studies have shown that the Mo atoms in MoS2 thin films can be substituted by Nb atoms as efficient acceptors16,17,18, we build the MoS2 ML with n-p codoping by first substituting one of the Mo atoms in the 3 × 3 supercell with an Nb atom, marked as Nb1 in Fig. 1(a). Another Nb atom (Nb2) is then considered to adsorb at the surface of the MoS2 ML. Totally four typical adsorption sites are explored for the Nb2 atom: H (Hollow), M (Mo-top), N (Nb1-top), and S (S-top) (Fig. 1(a))43. Some other adsorption sites, far away from the Nb1 atom, are also considered, which are, however, found being unfavorable in energy, compared with the above corresponding sites. The calculations of the electronic structures are performed by using first-principles methods44. The exchange-correlation interaction is described with the metaGGA with SCAN form45. The energy cutoff is set as 450 eV. Monkhorst-Pack k-point meshes with 5 × 5 × 1 are adopted for structural relaxation and electronic structure calculations. The experimental lattice constant of 3.16 Å46 is used and the vacuum space along the z direction is set about 20 Å. The test calculations show that the Nb dopants can expand slightly the lattice structure. Since the energy bands are found insensitive to the lattice constant, the experimental lattice is employed in the subsequent calculations. The convergences of the total energy and Hellmann-Feynman forces are set to be 10−6 eV and 0.01 eV/Å, respectively.
References
Manzeli, S., Ovchinnikov, D., Pasquier, D., Yazyev, O. V. & Kis, A. 2D transition metal dichalcogenides. Nat. Rev. Mater. 2, 17033 (2017).
Wei, Z. et al. Various structures of 2D transition-metal dichalcogenides and their applications. Small Methods 2, 1800094 (2018).
Han, G. H., Duong, D. L., Keum, D. H., Yun, S. J. & Lee, Y. H. van der Waals metallic transition metal dichalcogenides. Chem. Rev. 118, 6297–6336 (2018).
Wei, X. Y. et al. Chern insulators without band inversion in MoS2 monolayers with 3d adatoms. Phys. Rev. B 95, 075419 (2017).
Mak, K. F., McGill, K. L., Park, J. & McEuen, P. L. The valley Hall effect in MoS2 transistors. Science 344, 1489 (2014).
Zeng, H., Dai, J., Yao, W., Xiao, D. & Cui, X. Valley polarization in MoS2 monolayers by optical pumping. Nat. Nanotechnol. 7, 490 (2012).
Aivazian, G. et al. Magnetic control of valley pseudospin in monolayer WSe2. Nat. Phys. 11, 148 (2015).
MacNeill, D. et al. Breaking of valley degeneracy by magnetic field in monolayer MoSe2. Phys. Rev. Lett. 114, 037401 (2015).
Srivastava, A. et al. Valley Zeeman effect in elementary optical excitations of monolayer WSe2. Nat. Phys. 11, 141–147 (2015).
Chen, X., Zhong, L., Li, X. & Qi, J. Valley splitting in the transition-metal dichalcogenide monolayer via atom adsorption. Nanoscale 9, 2188 (2017).
Singh, N. & Schwingenschlögl, U. A Route to permanent valley polarization in monolayer MoS2. Adv. Mater. 29, 1600970 (2017).
Li, N. B., Zhang, J. Y., Xue, Y., Zhou, T. & Yang, Z. Q. Large valley polarization in monolayer MoTe2 on a magnetic substrate. Phys. Chem. Chem. Phys. 20, 3805 (2018).
Qi, S., Chen, H., Xu, X. & Zhang, Z. Diluted ferromagnetic graphene by compensated n–p codoping. Carbon 61, 609–615 (2013).
Deng, X. et al. Realization of quantum anomalous Hall effect in graphene from n-p codoping-induced stable atomic adsorption. Phys. Rev. B 95, 121410(R) (2017).
Qi, S. et al. High-Temperature Quantum anomalous Hall effect in n-p codoped topological insulators. Phys. Rev. Lett. 117, 056804 (2016).
Laskar, M. R. et al. p-type doping of MoS2 thin films using Nb. Appl. Phys. Lett. 104, 092104 (2014).
Suh, J. et al. Reconfiguring crystal and electronic structures of MoS2 by substitutional doping. Nat. Commun. 9, 199 (2018).
Suh, J. et al. Doping against the native propensity of MoS2: degenerate hole doping by cation substitution. Nano Lett. 14, 6976–6982 (2014).
Dolui, K., Rungger, I., Das Pemmaraju, C. & Sanvito, S. Possible doping strategies for MoS2 monolayers: an ab initio study. Phys. Rev. B 88, 075420 (2013).
Onofrio, N., Guzman, D. & Strachan, A. Novel doping alternatives for single-layer transition metal dichalcogenides. J. Appl. Phys. 122, 185102 (2017).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Feng, W. X. et al. Intrinsic spin Hall effect in monolayers of group-VI dichalcogenides: a first-principles study. Phys. Rev. B 86, 165108 (2012).
Wang, Y. et al. First-principles study of transition-metal atoms adsorption on MoS2 monolayer. Physica E 63, 276–282 (2014).
Li, X. D., Fang, Y. M., Wu, S. Q. & Zhu, Z. Z. Adsorption of alkali, alkaline-earth, simple and 3d transition metal, and nonmetal atoms on monolayer MoS2. AIP Adv. 5, 057143 (2015).
Shi, H. L., Pan, H., Zhang, Y.-W. & Yakobson, B. I. Strong ferromagnetism in hydrogenated monolayer MoS2 tuned by strain. Phys. Rev. B 88, 205305 (2013).
Wang, X. L. Proposal for a new class of materials: Spin Gapless Semiconductors. Phys. Rev. Lett. 100, 156404 (2008).
Wang, X. T., Li, T. Z., Cheng, Z. X., Wang, X.-L. & Chen, H. Recent advances in Dirac spin-gapless semiconductors. App. Phys. Rev. 5, 041103 (2018).
Wang, X. T., Cheng, Z. X., Wang, J. L., Wang, X.-L. & Liu, G. D. Recent advances in the Heusler based spin-gapless semiconductors. J. Mater. Chem. C 4, 7176 (2016).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. J. Chem. Phys. 118, 8207 (2003).
Tong, W.-Y., Gong, S.-J., Wan, X. & Duan, C.-G. Concepts of ferrovalley material and anomalous valley Hall effect. Nat. Commun. 7, 13612 (2016).
Zhou, T., Zhang, J. Y., Jiang, H., Žutić, I. & Yang, Z. Q. Giant spin-valley polarization and multiple Hall effect in functionalized bismuth monolayers. npj Quantum Mater. 3, 39 (2018).
Chen, M. X. & Weinert, M. Layer k-projection and unfolding electronic bands at interfaces. Phys. Rev. B 98, 245421 (2018).
Chen, M. X., Chen, W., Zhang, Z. & Weinert, M. Effects of magnetic dopants in (Li0.8M0.2OH) FeSe (M = Fe, Mn, Co): density functional theory study using a band unfolding technique. Phys. Rev. B 96, 245111 (2017).
Chen, M. X. & Weinert, M. Revealing the substrate origin of the linear dispersion of Silicene/Ag(111). Nano Lett. 14, 5189–5193 (2014).
Zhai, B. et al. Spin-dependent Dirac electrons and valley polarization in the ferromagnetic stanene/CrI3 van der Waals heterostructure. Phys. Rev. B 100, 195307 (2019).
Marzari, N. & Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 56, 12847 (1997).
Souza, I., Marzari, N. & Vanderbilt, D. Maximally localized Wannier functions for entangled energy bands. Phys. Rev. B 65, 035109 (2001).
Mostofi, A. A. et al. wannier90: A tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 178, 685 (2008).
Liang, Q.-F., Yu, R., Zhou, J. & Hu, X. Topological states of non-Dirac electrons on a triangular lattice. Phys. Rev. B 93, 035135 (2016).
Xue, Y., Zhang, J. Y., Zhao, B., Wei, X. Y. & Yang, Z. Q. Non-Dirac Chern insulators with large band gaps and spin-polarized edge states. Nanoscale 10, 8569 (2018).
Xue, Y. et al. Novel Chern insulators with half-metallic edge states. NPG Asia Mater. 10, 467 (2018).
Xu, X. H. et al. Carrier-induced ferromagnetism in n-type ZnMnAlO and ZnCoAlO thin films at room temperature. New J. Phys. 8, 135 (2006).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Sun, J., Ruzsinszky, A. & Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
Böker, T. et al. Band structure of MoS2, MoSe2, and a-MoTe2: angle-resolved photoelectron spectroscopy and ab initio calculations. Phys. Rev. B 64, 235305 (2001).
Acknowledgements
This work was supported by National Natural Science Foundation (NNSF) of China under Grant Nos. 11874117, 11574051, 11604134, 11904250, and 61971143 and Natural Science Foundation of Shanghai under Grant No. 19ZR1402600.
Author information
Authors and Affiliations
Contributions
Xinyuan Wei carried out the first-principles calculations with the assistance of Jiayong Zhang and Bao Zhao. Xinyuan Wei, Zhongqin Yang, and Jiayong Zhang wrote the manuscript. All authors analyzed the results. Zhongqin Yang guided the whole project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, X., Zhang, J., Zhao, B. et al. Coexistence of valley polarization and Chern insulating states in MoS2 monolayers with n-p codoping. Sci Rep 10, 9851 (2020). https://doi.org/10.1038/s41598-020-66912-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66912-y
This article is cited by
-
Transport properties in a monolayer MoS2 with time-periodic potential
Indian Journal of Physics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
