
Abstract
The Klotho (KL) gene is involved in phosphate homeostasis. Polymorphisms in this gene have been reported to be associated with the risk of cardiovascular disease. Here we used computational tools to predict the damage-associated single nucleotide polymorphisms (SNPs) in the human KL gene. We further investigated the association of SNPs in the KL gene and mortality in the Swedish multicenter prospective Osteoporotic Fractures in Men (MrOS) cohort. This study included 2921 men (aged 69–81 years) with mean 4.49 ± 1.03 years follow-up. 18 SNPs in the KL gene were genotyped using Sequenom. These SNPs were identified by in silico tools for the coding and noncoding genome to predict the damaging SNPs. After quality analyses, SNPs were analyzed for mortality risk using two steps approach on logistic regression model screening and then Cox regression model confirmation. Two non-synonymous SNPs rs9536314 and rs9527025 were found to be potentially damaging SNPs that affect KL protein stability and expression. However, these two SNPs were not statistically significantly associated with all-cause mortality (crude Hazard ratio [HR] 1.72, 95% confidence interval [CI] 0.96–3.07 in rs9536314; crude HR 1.82, 95% CI 0.998–3.33 in rs9527025) or cardiovascular mortality (crude HR 1.52, 95% CI 0.56–4.14 in rs9536314; crude HR 1.54, 95% CI 0.55–4.33 in rs9527025) in additive model using Cox regression analysis. In conclusion, these two potentially damaging SNPs (rs9536314 and rs9527025) in the KL gene were not associated with all-cause mortality or cardiovascular mortality in MrOs cohort. Larger scales studies and meta-analysis are needed to confirm the correlation between polymorphisms of the KL gene and mortality.
Similar content being viewed by others


Introduction
The Klotho (KL) gene, which composed of five exons, encodes transmembrane protein type I (1014 and 1012 amino acids in human) that encompasses an extracellular domain with two internal repeats (KL1 and KL2), a membrane-spanning segment, and a short intracellular domain1. The KL protein, which is expressed predominantly in the distal tubules of the kidney, parathyroid cells, choroid plexus, and pituitary glands1, exists in both secreted and membrane-bound forms2. It is involved in longevity1, cardiovascular (CV) health3, and calcium and phosphorous regulation in the kidney4. Altered human KL expression is related to osteopenia/osteoporosis5, suppression of Wnt signaling6, amelioration of vascular endothelial dysfunction7, and in mediating the role of fibroblast growth factor 23 (FGF23) in bone-kidney-parathyroid control of phosphate and calcium8.
The human KL gene, located on chromosome 13q12, spans approximately 50 kb in length and contains 5 exons and 4 introns, encoding a 5.2 kb mRNA transcript2. Polymorphisms in the KL gene have been found associated with risk of coronary artery disease, stroke, and hypertension in different populations. A functional variant named KL-VS, which harbors two amino acid changes (F352V; rs9536314 and C370S; rs9527025), was shown to alter the number of extracellular levels of klotho and the ratio of intracellular vs. extracellular levels in transiently transfected HeLa cells5. This polymorphism was also associated with mortality risk among elderly individuals5, increased risk of coronary artery disease in the middle-aged population9, and related to lower high-density lipoprotein levels3, hypertension3, and stroke3,10 risk in the homozygous KL-VS genotype. However, some reports do not support the increased risk of coronary artery disease with the KL-VS genotype (rs9536314 and rs9527025)11. Further, only a few human exceptional longevity studies have examined KL variation and have published inconsistent results5,12,13,14. Therefore, the established relationships between KL SNPs and mortality or CV disease have lacked consistency. These studies are relatively small and require replication.
Beyond the coding genome, the non-coding genome also has regulatory elements driving gene expression15, such as gene promoters, enhancers, or binding sites for proteins or regulatory RNA. The intronic rs577912 SNP was found to be associated with mortality risk in patients receiving hemodialysis16. Furthermore, the CC genotype of this SNP was associated with a 16–21% lower Klotho expression compared with the AA/AC genotype when introduced into lymphoblastoid cell lines16. Since the KL gene is highly correlated to mortality and CV disease, complete evaluation of both coding and non-coding SNPs of the KL gene is important. Determining which SNPs affect the clinical phenotype would make it possible to identify the molecular mechanisms of disease variation. We hypothesized that variants in the KL gene might have an impact on mortality risk. The effects were evaluated by a combination of different approaches, including bioinformatics and clinical study. The aims of our study included a test in the KL gene polymorphisms using a computational approach based on tools to differentiate the deleterious or disease-associated SNPs first and then evaluate the clinical impact of selected KL gene polymorphisms on mortality in a cohort of elderly Swedish men.
Methods
Study design
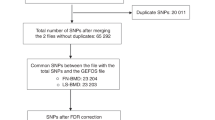
The present study investigated genotyped SNPs using computational tools to predict damage-associated SNPs in the coding and non-coding region of the human KL gene. The selected SNPs in the KL gene were then further investigated in the Swedish part of the Osteoporotic Fractures in Men Study (MrOS) cohort (Fig. 1).

Study design and flow chart.
Study population
The MrOS study was a prospective international study of osteoporosis and fracture risk in an elderly male cohort. It included participants in United Status, Sweden, and Hong Kong. We included Sweden MrOS participants aged 69–81 years. The participants (n = 3,014)were randomly selected from national population database, constituting three sub cohorts in Uppsala (n = 999), Göteborg (n = 1,010), and Malmö (n = 1,005). The MrOS Study in Sweden was approved by the ethics committees at Uppsala Universities (Ups 01–057), Göteborg (Gbg M 014–01), and Lund (LU 693-00). The study was performed in accordance with the declaration of Helsinki. Informed consent was obtained from all study participants.
Data collection, covariates definitions, and mortality assessment
Medical information, such as hypertension, diabetes, myocardial infarction, angina pectoris, stroke, and cancer, was recorded from a standardized questionnaire. Hypertension was defined by baseline self-reported blood pressure-lowering drugs or systolic blood pressure above 140 mmHg, which was measured once after 10 minutes rest in the spine position. Cardiovascular disease (CVD) comorbidity was defined as a history of angina pectoris, myocardial infarction, or stroke. Body mass index (BMI) was estimated as a person’s weight in kilograms, divided by height in meters squared.
Serum samples were collected and stored at −80 °C. Serum concentration of intact FGF23 was analyzed using ELISA (Kainos Laboratories International; Tokyo, Japan)17. The estimated glomerular filtration rate (eGFR) cystatin C (Cystatin C Immunoparticles, Dako A/S, Glostrup, Denmark) was calculated using the following formula: eGFR cystatin C = 79.901*(Cyst C [mg/L])−1.4389. This proxy for eGFR shows a strong correlation with the iohexol clearance rate (R2 = 0.956)18,19.
Mortality was retrieved from Statistics Sweden. Follow-up time was recorded between baseline visit (2001–2004) and the date of death or mortality data collection (March 1, 2008). The cause of death data was recorded by the International Classification of Diseases (ICD) codes based on death certificates from the Swedish Cause of Death Register. CV death was defined by ICD-10 codes I00 to I99 in this study.
Genotyping of the Klotho gene
DNA was extracted from whole blood using standard methods from 3014 participants. Tagging SNPs covering the KL gene and flanking 5’ and 3’ regions were selected using the HaploView 4.2 tagger algorithm20. Genotyping was performed using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) on a Sequenom MassARRAY system (Sequenom Inc., Newton, MA) with iPLEX assay. The primers were designed by MassARRAY Assay Design software (Version 3). Polymerase chain reaction (PCR) was performed according to the standard iPLEX methodology. Genotyping quality control was completed by excluding individual samples or SNPs with genotype call rates <95%, SNP assays with poor-quality spectra/cluster plots, and SNPs that significantly deviated from the Hardy-Weinberg equilibrium (p < 0.05) were excluded from the analysis. Genotype data on the SNPs were pairwise calculated regarding linkage disequilibrium and for identification of SNPs with the highest haplotype predictability. Successful genotyping was obtained from 23 SNPs, with an overall call rate of 98.9%. Allele frequencies for these SNPs were calculated and Hardy–Weinberg equilibrium (HWE) was tested for each SNP using chi-square goodness-of-fit statistics in the cohort for all SNP except two (rs1888057 and rs2283368) which were subsequently excluded from further analyses. Haploview 4.2 was accessed to generate haplotype blocks, linkage disequilibrium (LD) values and diagrams, as well as tagging SNPs using the tagger algorithm20. The data for chromosome location, minor genotype frequency, SNP function and regulation of the human KL gene, and wild or mutated residue of the non-synonymous SNPs (nsSNPs) were used according to the program requirements (Supplementary Tables 1–3).
Functional analysis Prediction by in silica tool
The function of nsSNPs was evaluated by 6 prediction tools, including SIFT (Sorting Intolerant From Tolerant)21, Polyphen-2 (Polymorphism Phenotyping v2)22, PROVEAN (Protein Variation Effect Analyzer)23, SNPs3D24, LS-SNP (Large-scale annotation of coding non-synonymous)25, and MutPred26 (Supplementary Table 4). In addition, computational tools for annotation of genetic variants on both protein-coding and noncoding variants were evaluated by Combined Annotation-Dependent Depletion (CADD)27, Deleterious annotation of genetic variants using neural networks (DANN)28, FATHMM (Functional Analysis through Hidden Markov Models)29, Funseq. 230, Genome-Wide Annotation of VAriants (GWAVA)31, PredictSNP232, PhD-SNP (Predictor of Human Deleterious Single Nucleotide Polymorphisms)33, and RegulomeDB34 (Supplementary Table 4). The prediction tools were selected by using different approaches in order to obtain a classification of the SNPs according to one or more features. Each program’s approach was detailed in the Supplementary Methods.
Statistical analysis
Patients’ baseline characteristics are reported as the mean ± SD and percentages for continuous and categorical variables, respectively. We performed a two-step procedure. Firstly, logistic regression is used to analyze all of the KL gene SNPs and mortality outcomes association as an initial filtering process. Secondly, Cox regression is fitted to those SNPs associated below P-value <0.05 threshold to obtain final results for these SNPs35. The association between genotypes for the SNPs and all-cause or CV mortality during follow-up was evaluated by odds ratios (OR) and 95% confidence interval (CI) with homozygotes major allele as reference using additive models. The models were tested: 1) unadjusted and 2) adjusted for age, body mass index, smoking, comorbidities (hypertension, diabetes, coronary artery disease, stroke, cancer), estimated glomerular filtration rate, phosphate, and FGF23 at baseline. In addition, dominant and recessive genetic models were also analyzed. Cox regression model with unadjusted Hazard Ratios (HR) and 95% CI was further analyzed to confirm the finding. Tests for the association and the estimates for the HR were computed with adjustment for age, BMI, and smoking. Since eGFR and FGF23 were strongly correlated with mortality, we further performed subgroup analysis on selected SNPs for mortality stratified by eGFR (≧60 ml/min/1.73 m2 and <60 ml/min/1.73 m2) and fibroblast growth factor 23 (≧60 pg/ml and <60 pg/ml). Kaplan-Meier curves with log-rank tests were used to examine 6 years of survival according to the KL variant. STATA 14 was used for calculations (Stata, College Station, TX, USA). A two-tailed P < 0.05 was considered statistically significant.
Results
Characteristics of the MrOS Sweden cohort
In MrOS Sweden, 2,921 participants were analyzed after excluding missing value of clinical variables (n = 1) and SNPs quality control (n = 92) (Fig. 1). The baseline characteristics of the MrOS cohort are shown in Table 1. Compared with survivors, participants who died had older age, more smoking habits, more comorbidities (diabetes, CAD, stroke, and cancer), higher FGF23 levels, and lower eGFR levels. The mean follow-up time of the total MrOS cohort was 4.49 ± 1.03 years, 364 deaths occurred and the mortality rate (95% CI) was 26.68 (23.77–29.95)/1,000 person-year in all-cause mortality and 10.93 (9.13–13.09)/1,000 person-year in CV mortality (Table 1).
Linkage disequilibrium of SNPs in the Klotho gene calculation
The SNP localizations in the KL gene and their degree of linkage (D’) are presented (Supplementary Fig. 1). There are three major haplotype blocks covering 48.6 kb of the gene, with overall linkage disequilibrium over the haplotype blocks exhibiting D’ values above 0.84 (mean max r2 in pairwise comparisons 0.983). SNPs rs1888057 and rs2283368 were excluded from further analysis due to deviation from HWE, and rs2249358 and rs541053 were excluded due to call rates less than the 95% predetermined cut-off. SNP rs22227122 was excluded due to a low minor allele frequency of 2.8%. In total, 18 SNPs were taken forward for further downstream analysis. These 18 SNPs included intron variant, transcript variant, missense, and synonymous variant based on Ensembl Variant Effect Predictor (Supplementary Fig. 2). SNPs chromosome position, minor allele frequency (MAF) in different databases, and estimated functional effects are presented in Supplementary Tables 1–3.
Analysis of SNPs using a combination of bioinformatics tools
Polymorphisms in the KL gene were analyzed using computational tools to predict coding and non-coding region SNPs. SNPs (rs9536314, rs9527025, rs9527026, rs564481) from the coding region (exon) were selected for further bioinformatic analysis using SIFT, PolyPhen-2, PROVEAN, SNPs3D, LS-SNP, and MutPred. SNP rs9536314 was predicted to be “damaging” in SIFT, “probably damaging” in PolyPhen-2, “deleterious” in PROVEAN, “deleterious” in SNPs3D and MutPred (Supplementary Table 5). SNP rs9527025 was predicted to be “damaging” in SNPs3D, LS-SNP, and MutPred (Supplementary Table 5).
Klotho SNP and risk of all-cause and cardiovascular mortality
The total genotype frequency, minor allele frequency, and genotype frequency of death and non-death were shown in Supplementary Tables 1, 6, 7. Using the univariate logistic regression model, there were 3 SNPs associated with increased risk of all-cause mortality in the additive model, including the TT genotype of rs9536282, GG genotype of rs9536314, and CC genotype of rs9527025 (Supplementary Table 8). However, none of the 18 SNPs were statistically significantly associated with the risk of CV mortality (Supplementary Table 8). In adjusted analyses controlling age, body mass index, smoking, comorbidities (hypertension, diabetes, coronary artery disease, stroke, cancer), estimated glomerular filtration rate, phosphate, and FGF23, rs9536282, rs9536314, and rs9527025 genotypes were not associated with all-cause mortality or CV mortality (Supplementary Table 9). Furthermore, in the analyses of dominant and recessive models, rs9527025 and rs9527026 were associated with all-cause mortality under the recessive model (Supplementary Tables 10 and 11). The Kaplan-Meier survival analysis according to genotypes of SNPs rs9536314 and rs9527025 are shown, respectively (Fig. 2). We performed Cox proportional hazards analysis to determine the relationship between rs9536314 and rs9527025 genotype and mortality. No statistical significance was found in the unadjusted and adjusted Cox model (Table 2 and Supplementary Table 12). Subgroup analysis of SNPs rs9536314 and rs9527025 using univariate Cox regression analysis in additive models demonstrated no significant risk difference of all-cause mortality or CV mortality stratified by eGFR or FGF23 (Fig. 3).

Kaplan-Meier curves of all-cause mortality cardiovascular mortality according to SNP rs9536314 and rs9527025.

Subgroup analysis of hazard ratio (95% confidence interval) on SNPs rs9536314 and rs9527025 for all-cause mortality (A) and cardiovascular mortality (B) using univariate Cox regression analysis with major homozygote as reference stratified by estimated glomerular filtration rate (≧60 ml/min/1.73 m2 and <60 ml/min/1.73 m2) or fibroblast growth factor 23 (≧60 pg/ml and <60 pg/ml).
Discussion
The SNPs situated in the KL gene were evaluated by computational programs and databases that use different methods to predict the damaging of SNPs. The nsSNP rs9536314 and rs9527025 are classified as the most damaging sites. In the clinical study, the risk association of all-cause mortality was found for the TT genotype of rs9536282, GG genotype of rs9536314, and CC genotype of rs9527025 by unadjusted logistic regression analysis using an additive model. However, no statistically significant association was found between polymorphisms in KL and mortality risk in the unadjusted Cox regression model or adjusted logistic regression model. The results were similar in the dominant and recessive models. Subgroup analysis of rs9536314 and rs9527025 stratified by eGFR or FGF23 also demonstrated negative results in the MrOS cohort.
In computational data, the nsSNPs are highly significantly responsible for amino acid residue substitutions subsequent in the functional diversity of proteins in humans. Functional variations can have neutral or deleterious effects on protein structure and function36. Damaging effects include altering gene regulation, destabilization of protein structure, affecting protein charge, hydrophobicity, geometry, dynamics, stability, translation and inter/intra protein interactions37,38,39. Therefore, it is assumed that nsSNPs will be linked to human disease. However, in order to keep important genetic variants in non-coding regions40, we also perform computational tools (CADD27, DANN28, FATHMM29, Funseq. 230, GWAVA31, PredictSNP232, PhD-SNP33, and RegulomeDB34) to discriminate non-coding pathogenic variants from benign variants for complete KL gene SNPs evaluation.
We demonstrated a non-significant trend of association between the nsSNPs rs9536314 and rs9527025 on all-cause mortality but not CV mortality. The SNPs of rs9536314 (F352V) and rs9527025 (C370S) in the exon have been reported to be related to CV diseases9,10. However, a meta-analysis of rs9536314 (F352V), rs9527025 (C370S) failed to indicate any statistically significant association with the additive genetic model, the dominant genetic model, or the recessive genetic model on CV disease41. In addition, the minor allele frequency difference of rs9536314 and rs9527025 between Sweden MrOs data and other databases (1000 Genomes EUR Population, HapMap CEU Population, or Genome Aggregation Database European Population) (Supplementary Table 1) may partly explain the outcomes difference. In our study, although the computational prediction of the “damaging” effect of the KL-VS genotype, no statistical significance was found in the study cohort. The contrary results may due to cohorts having different ages, gender, races, and comorbidities. The functional KL-VS variant results in amino acid substitutions, altering secretion, catalytic activities, and functionality of the Klotho protein5,42. However, a study has also reported that SNPs rs9536314 (F352V) and rs9527025 (C370S) substitutions alter amino acid-related shedding and trafficking but the effects are small and intragenic complementation may further minimize the effects in vivo43. As for another KL gene synonymous variant rs564481 (C1818T) located in the fourth exon, no all-cause or CV mortality association was demonstrated in our study, as compatible with previous studies that no CV disease correlation in this polymorphism10,41,44. Although rs564481 is a synonymous variant, it is likely to be functionally relevant, with reports associating it with CV risk factors (blood pressure, glucose metabolism, and lipid levels)45,46 and coronary artery disease47 in Asian populations. SNP rs577912 has previously been found to be related to higher mortality risk in patients receiving hemodialysis16 but negative findings in our clinical result. Interestingly, the computational tool FATHMM predicts rs577912 genotype could be a damaging intron non-coding region but no mortality outcome association was found in this study.
Study strengths and limitations
Strengths of this study include that the MrOS cohort represents a large sample size of community-dwelling men with nearly complete follow up of surviving cohort participants, and outcome measures are well-validated because of high-quality national registers to obtain information of all-cause and CV death. We perform 14 different computational tools as a bioinformatics approach as well as clinical study to complete the evaluation of KL gene polymorphism. Some limitations in this study still need to be addressed. Firstly, self-reported questionnaires were used at baseline visits, so we cannot exclude the possibility of smoking or disease prevalence underestimation. Secondly, the generalizability of the present study is limited to healthy community-dwelling Swedish males. Lastly, the minor allele frequency of rs9536314 homozygotes could be different in elderly and young subjects5. This could also affect our study result because of the enrolled elderly population in the MrOS cohort.
Conclusion
The two potentially damaging SNPs (rs9536314 and rs9527025) in the KL gene were not associated with all-cause mortality or CV mortality. Larger scales studies and meta-analysis are needed to confirm the correlation between polymorphisms of the KL gene and mortality.
References
Kuro-o, M. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 45–51 (1997).
Matsumura, Y. et al. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun 242, 626–630 (1998).
Arking, D. E., Atzmon, G., Arking, A., Barzilai, N. & Dietz, H. C. Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ Res 96, 412–418 (2005).
Torres, P. U. et al. Klotho: an antiaging protein involved in mineral and vitamin D metabolism. Kidney Int 71, 730–737 (2007).
Arking, D. E. et al. Association of human aging with a functional variant of klotho. Proc Natl Acad Sci USA 99, 856–861 (2002).
Liu, H. et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 317, 803–806 (2007).
Saito, Y. et al. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem Biophys Res Commun 276, 767–772 (2000).
Urakawa, I. et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774 (2006).
Arking, D. E. et al. KLOTHO allele status and the risk of early-onset occult coronary artery disease. Am J Hum Genet 72, 1154–1161 (2003).
Majumdar, V., Nagaraja, D. & Christopher, R. Association of the functional KL-VS variant of Klotho gene with early-onset ischemic stroke. Biochem Biophys Res Commun 403, 412–416 (2010).
Tavakkoly-Bazzaz, J. et al. Absence of kl-vs variant of klotho gene in Iranian cardiac patients (comparison to the world populations). Dis Markers 31, 211–214 (2011).
Novelli, V. et al. Lack of replication of genetic associations with human longevity. Biogerontology 9, 85–92 (2008).
Invidia, L. et al. The frequency of Klotho KL-VS polymorphism in a large Italian population, from young subjects to centenarians, suggests the presence of specific time windows for its effect. Biogerontology 11, 67–73 (2010).
Revelas, M. et al. Review and meta-analysis of genetic polymorphisms associated with exceptional human longevity. Mech Ageing Dev (2018).
Khurana, E. et al. Role of non-coding sequence variants in cancer. Nat Rev Genet 17, 93–108 (2016).
Friedman, D. J. et al. Klotho variants and chronic hemodialysis mortality. J Bone Miner Res 24, 1847–1855 (2009).
Yamazaki, Y. et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87, 4957–4960 (2002).
Flodin, M., Jonsson, A. S., Hansson, L. O., Danielsson, L. A. & Larsson, A. Evaluation of Gentian cystatin C reagent on Abbott Ci8200 and calculation of glomerular filtration rate expressed in mL/min/1.73 m(2) from the cystatin C values in mg/L. Scand J Clin Lab Invest 67, 560–567 (2007).
Siilin, H. et al. Prevalence of primary hyperparathyroidism and impact on bone mineral density in elderly men: MrOs Sweden. World J Surg 35, 1266–1272 (2011).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081 (2009).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7, Unit7 20 (2013).
Choi, Y. & Chan, A. P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Yue, P., Melamud, E. & Moult, J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics 7, 166 (2006).
Karchin, R. et al. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics 21, 2814–2820 (2005).
Li, B. et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744–2750 (2009).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315 (2014).
Quang, D., Chen, Y. & Xie, X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761–763 (2015).
Shihab, H. A. et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543 (2015).
Fu, Y. et al. FunSeq. 2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol 15, 480 (2014).
Ritchie, G. R., Dunham, I., Zeggini, E. & Flicek, P. Functional annotation of noncoding sequence variants. Nat Methods 11, 294–296 (2014).
Bendl, J. et al. PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PLoS Comput Biol 12, e1004962 (2016).
Capriotti, E. & Fariselli, P. PhD-SNPg: a webserver and lightweight tool for scoring single nucleotide variants. Nucleic Acids Res 45, W247–W252 (2017).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22, 1790–1797 (2012).
Staley, J. R. et al. A comparison of Cox and logistic regression for use in genome-wide association studies of cohort and case-cohort design. Eur J Hum Genet 25 (2017).
Capriotti, E. & Altman, R. B. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinformatics 12(Suppl 4), S3 (2011).
Petukh, M., Kucukkal, T. G. & Alexov, E. On human disease-causing amino acid variants: statistical study of sequence and structural patterns. Hum Mutat 36 (2015).
Chasman, D. & Adams, R. M. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J Mol Biol 307, 683–706 (2001).
Kucukkal, T. G., Petukh, M., Li, L. & Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr Opin Struct Biol 32, 18–24 (2015).
Hindorff, L. A. et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 106, 9362–9367 (2009).
Haiming Zhang, Y. S., Ma, F., Wang, L., Hou, Y. & Zhijun, Z. Association of Klotho single nucleotide polymorphisms with cardiovascular diseases: a systematic review and meta-analysis. Int J Clin Exp Med 10, 5721–5741 (2017).
Dubal, D. B. et al. Life extension factor klotho enhances cognition. Cell Rep 7, 1065–1076 (2014).
Tucker Zhou, T. B., King, G. D., Chen, C. & Abraham, C. R. Biochemical and functional characterization of the klotho-VS polymorphism implicated in aging and disease risk. J Biol Chem 288, 36302–36311 (2013).
Kim, H. K. & Jeong, B. H. Lack of functional KL-VS polymorphism of the KLOTHO gene in the Korean population. Genet Mol Biol 39, 370–373 (2016).
Rhee, E. J. et al. Relationship between polymorphisms G395A in promoter and C1818T in exon 4 of the KLOTHO gene with glucose metabolism and cardiovascular risk factors in Korean women. J Endocrinol Invest 29, 613–618 (2006).
Shimoyama, Y., Nishio, K., Hamajima, N. & Niwa, T. KLOTHO gene polymorphisms G-395A and C1818T are associated with lipid and glucose metabolism, bone mineral density and systolic blood pressure in Japanese healthy subjects. Clin Chim Acta 406, 134–138 (2009).
Rhee, E. J. et al. The differential effects of age on the association of KLOTHO gene polymorphisms with coronary artery disease. Metabolism 55, 1344–1351 (2006).
Acknowledgements
We thank the participants of MrOs US, Sweden. The MrOS Sweden study was supported by the Swedish Research Council, the Swedish Foundation for Strategic Research, The ALF/LUA research grant in Gothenburg, the Lundberg Foundation, the Torsten and Ragnar Söderberg’s Foundation.
Author information
Authors and Affiliations
Contributions
Ping-Hsun Wu and Per-Anton Westerberg made calculations and wrote the manuscript, Åsa Tivesten was a responsible collection of mortality data, Magnus K. Karlsson for handling the MrOS data in Malmoe, Claes Ohlsson and Dan Mellström for handling the MrOS data in Gothenburg, Östen Ljunggren for handling the MrOS data in Uppsala. Torbjörn Linde and Östen Ljunggren for planning the study. All authors have read, critically revised, and approved of the contents of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, PH., Westerberg, PA., Kindmark, A. et al. The association between Single Nucleotide Polymorphisms of Klotho Gene and Mortality in Elderly Men: The MrOS Sweden Study. Sci Rep 10, 10243 (2020). https://doi.org/10.1038/s41598-020-66517-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66517-5
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
