
Abstract
Wool fibre diameter (WFD) is one of the wool traits with higher economic impact. However, the main genes specifically regulating WFD remain unidentified. In this current work we have used Agilent Sheep Gene Expression Microarray and proteomic technology to investigate the gene expression patterns of body side skin, bearing more wool, in Aohan fine wool sheep, a Chinese indigenous breed, and compared them with that of small tail Han sheep, a sheep bread with coarse wool. Microarray analyses showed that most of the genes likely determining wool diameter could be classified into a few categories, including immune response, regulation of receptor binding and growth factor activity. Certain gene families might play a role in hair growth regulation. These include growth factors, immune cytokines, solute carrier families, cellular respiration and glucose transport amongst others. Proteomic analyses also identified scores of differentially expressed proteins.
Similar content being viewed by others


Introduction
Sheep fleece is a raw material extremely important for the textile industry. Since fibre diameter is one of the most economically significant attributes of sheep wool, the identification of genes regulating this characteristic would offer the opportunity to increase productivity and improve product quality and diversity. This could be achieved by developing breeding programs or through the production of transgenic lines with enhanced characteristics. Besides, drugs can also be develop in order to modify wool fiber properties through gene expression control1.
The single nucleotide polymorphisms (SNPs) and quantitative trait loci (QTLs), as well as the molecular mechanisms regulating wool and cashmere growth have been the subject of several studies1,2,3,4,5. In mammals, certain gene families, such as those encoding TNFs (tumor necrosis factors), TGFs (transforming growth factors), FGFs (fibroblast growth factors) or proteins involved in WNT signaling, have been previously linked to the development of hair follicle, as well as to hair morphogenesis and cycling6,7. Moreover, some molecular aspects of primary wool follicle initiation have been recently reported in Merino sheep8.
Microarray and other transcriptome analysis techniques have been successfully employed to analyze the properties of hair follicle stem cells in both mouse9,10 and human11,12. Different traits, such as resistance to parasites, mammary glands development and milk quality13,14,15, wool follicle development16, fleece resistance to rot17 or wool and skin pigmentation18 have also been studied on sheep and goat using microarrays19,20. MicroRNAs putatively involved in goat and sheep hair formation have been identified in the skin of these animals using this approach21,22,23. In addition to microarrays, RNA-sequencing (RNA-seq) has also been used to identify genes differentially expressed in cashmere goat skin during hair follicle initiation and cycling24. In fact, the comparative analysis of gene expression in primary and secondary follicles of a cashmere goat, using this technique, identified 51 differentially expressed genes25.
RNA-Seq was also recently used to find genes displaying differential expression in diverse sheep tissues26, including whole skin, and also between various sheep flocks producing wool with different diameters27. Besides, several studies have confirmed the effectiveness of cDNA microarray for establishing the expression profiling of different wool follicle growth stages in whole skin28,29,30,31,32.
Mean Wool Fibre diameter (MFD) is one of the main economic traits of wool. To explore the molecular mechanisms regulating MFD, the expression profiles of different stages of embryonic and adult sheep skin have been investigated through scanning of expressed sequence tags (ESTs) and cDNA microarray16,32,33. Mutations and epigenetic and post-translational modifications of any ligands or receptors in certain signalling pathways might also influence MFD1,34. However, the major pathways regulating MFD still remain unknown.
The Aohan fine wool sheep breed, developed in inner Mongolia, is an excellent sheep breed in terms of meat and wool production. This breed is also highly resistance to disease and to harsh environment and its wool has optimal traits: one animal yields up to 9 kg per year, with a wool length up to 10.5 cm, and a MFD lower than 22μm. Consequently, Aohan fine wool sheep could be regarded as a valuable resource for fine wool production. Only a few works have investigated the genetic characteristics of this particular sheep breed28,29,32. The aim of this present work was to explore and compare the gene expression profile of body side skin of Aohan fine wool sheep to its counterpart from small tail Han sheep, a breed producingcoarse wool, during wool follicle anagen phase. We performed both transcriptome and proteome analysis in order to pinpoint the genes and proteins potentially controlling wool diameter in wool sheep.
Results
Microarray analysis
All data obtained from the cDNA microarray have been submitted to NCBI’s Gene Expression Omnibus and are available through GEO Series accession number GSE85844 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE85844). 702 probes showed differential expression in the body side skin of the two sheep breeds in the anagen stage (time in August), including 280 probes up-regulated and 422 probes down-regulated in the comparisons of Aohan fine wool sheep versus small tail Han sheep (A/H), as shown in Table S1. Due to a lack of information, the majority of probes (560) were not assigned to specific genes/transcripts. The total number of annotated genes/transcripts was 135, of which 67 were up-regulated and 68 were down-regulated. In A/H, 2 genes, namely LOC443313 and interleukin-8 (IL8) were up-regulated by more than 10-fold, while 5 genes, namely intelectin 2 (ITLN2), 1-acylglycerol-3-phosphate O-acyltransferase 1 (AGPAT1), cytochrome P450 family 1 subfamily A member 1 (CYP1A1), fermitin family member 2 (FERMT2) and LOC101104557, were down-regulated by more than 10-fold.
Several gene families involved in the regulation of different aspects of hair follicle growth displayed differential expression in A/H (see Table S2). These include growth factors, solute carrier families, immune cytokines, cellular respiration and glucose transport among others.
Validation of microarray results by qPCR
In order to verify the previous results, we selected 8 differentially expressed (DE) genes, including IL8, cytochrome P450 family 1 subfamily A member 1 (CYP1A1), UBE2E1, SLC2A5, PNRC1, AMP18, VCAM1 and CD1D, to analyse their expression profiles by quantitative PCR (qPCR). The selection criterion for qPCR-validated transcripts is the extent of fold-change of differential expression. IL8 and CYP1A1 were included into the category of high fold-change (>10), while UBE2E1, SLC2A5 and PNRC1 were classified as medium fold-change (3–10), then AMP18, VCAM1 and CD1D were chosen as two representatives of low fold-change category (<3). As illustrated in Fig. 1, the qPCR results for all the genes analysed were in agreement with the microarray results. This clearly highlights the reliability of our microarray data.

q-PCR validation of the microarray data. P values (T-test) of the q-PCR data are 0.005 (IL8), 0.015 (CYP1A1), 0.006 (UBE2E1), 0.006 (SLC2A5), 0.004 (PNRC1), 0.003 (AMP18), 0.003 (VCAM1) and 0.005 (CD1D), respectively. Error bars show the standard errors of the mean estimates.
Hierarchical cluster and biological process Gene Ontology (GO) analyses and possible gene networks
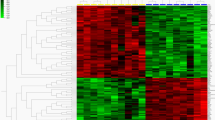
In order to further compare the two sheep breeds in terms of expression patterns of protein-coding genes, we carried out a cluster analysis using the Cluster 3.0 tool. This analysis was able to discriminate the body side skin from Aohan fine wool sheepfrom that of the small tail Han sheep (A/S) (Fig. 2).

Hierarchical cluster analysis of data between body side skin parts of the Aohan fine wool sheep and small tail Han sheep in anagen phase. Each column represents one sheep, and each horizontal line refers to a gene. Colour legend is at the top of the figure. Red indicates genes with a greater expression relative to the geometrical means, green indicates genes with a lower expression relative to the geometrical means. XJ1, XJ2 and XJ3 represent three repeats of body sideskin of Aohan fine wool sheep; RJ1, RJ2 and RJ3 represent three repeats of body sideskin of small tail Han sheep.Hierarchical cluster analysis of the data indicate that XJ1, XJ2 and XJ3repeats are classified in a tight cluster apparently different from anothercluster containing RJ1, RJ2 and RJ3.
An important number of the differentially expressed genes belonged to three specific signalling pathways: PI3K-AKT pathway, JAK-STAT pathway and FOXO pathway. Figure 3 shows the likely interplays between the differentially expressed genes of these three pathways. These interactions participate in apoptosis and cell cycle.

Biological pathways having more differentially expressed genes. (A) PI3K-AKT Pathway; (B) JAK-STAT Pathway; (C) FOXO Pathway. Gene name in red in the gene box indicates higher gene expression in A/S, green indicates lower gene expression, and black indicates no change of the gene expression.
Quantitative comparison and protein identification on 2-DE Gels
To pinpoint the differences between body side skins of the two sheep breeds at the protein level, we performed 2D gel electrophoresis with samples for each group in triplicate. Representatives of the results obtained are shown in Fig. 4. Ninety-nine protein spots showed significant differences in terms of expression levels (p < 0.05) between the AS and SS groups. Some of the spots showing significant differences could not be identified by MALDI-TOF/Mass Spectrometry analyses owing to incomplete polypeptide fragments, and some of them were too low in abundance to produce meaningful data. MALDI-TOF/MS analyses allowed the identification of a total of 51 proteins. A list of these proteins including accession numbers and protein/gene names is shown in Table S3. Correlation coefficient between transcriptome and proteome data is 0.1634. Not all the identified DE protein entries were differently expressed at the mRNA level. All MS data have been submitted to Peptide Atlas and are retrievable through Dataset Identifier PASS00797 (http://www.peptideatlas.org/PASS/PASS00797).

Representative image of 2-DE silver stained polyacrylamide gel.
Discussion
The best time point for identification of major genes determining wool fibre diameter
A reaction-diffusion mechanism controls the distribution, density and size of wool follicles35,36,37 and the size of wool follicle determines, in turn, the wool fibre diameter35,36. Wool fibre diameter, as well as follicle density are determinedduringthe initiation of wool follicle35,36, therefore primary follicles play more important roles than secondary follicles in determining wool fibre diameter. Since primary and secondary follicle development occur mainly at 50 and 80 days of gestational age, respectively35,36, further experiments should include the gene expression profile analysis of foetal sheep skin.
Analysis of highly differentially expressed genes (>10-fold)
Differential expression analyses performed between the two sheep breed (A/H) showed that 2 genes (LOC443313 and IL8) were up-regulated more than 10 folds in Aohan fine wool sheep.
According to a previous study comparing gene expression in relation to different wool fibre diameters, LOC443313 (type II small proline-rich protein, SPRR) was significantly down-regulated in the super-fine wool group versus the fine wool group27. The SPRR proteins constitute a new class of cornified envelope precursors encoded by geneslocated within the epidermal differentiation complex (EDC) region38. Two SPRR1, seven SPRR2, one SPRR3 and one SPRR4 genes are found in approximately 300 kb of the EDC39,40. These genes are expressed in the epidermis, hair follicles (HFs) and capillaries41,42. In our study, LOC443313 was up-regulated more than 10 folds in A/H. This apparent contradiction to previously published data27 needs to be further investigated.
IL8 was also significantly up-regulated in the A/D (August vs December)29 and S/G (body side skin vs groin skin) during the active growth phase of hair follicles32. Human IL8 was also found up-regulated in HF after 4-HC (a chemotherapeutic drug) treatment43. The expressionof IL8 gene was induced by the irritants in bulge-derived keratinocytes (BDKs)44. IL8 expression was significantly increased in psoriatic hair follicles45. Altogether, these results suggest that IL8 might be involved in wool growth regulation. However, whether IL8 plays a role specifically in diameter regulation needs to be determined.
Regarding the genes that were up-regulated, our analyses showed that in A/H, 5 genes were down-regulated by more than 10-fold: ITLN2, AGPAT1, CYP1A1, FERMT2 and LOC101104557.
Expression of sheep abomasal ITLN2 is up-regulated in both mRNA and protein levels after infection with Teladorsagia circumcincta46.
AGPAT1 promotes the attachment of fatty acyl moieties to the sn-2 position of the glycerol backbone in lysophosphatidic acid during triglyceride biosynthesis47. Since fat metabolism and wool synthesis are two processes closely related, down-regulation of AGPAT1 might occur in response to alterations in the barrier lipids of the skin26. Interestingly, AGPAT1 was found to be down-regulated in affected tissue of patients suffering from Lichen planopilaris (LPP), a cutaneous disorder that leads to the destruction of hair follicles48.
Rowe et al.49 confirmed that CYP1A1 was predominantly expressed in the sebaceous gland surrounding the hair shaft. CYP1A1 was also down-regulated more than 10 folds in body side skin vs groin skin at both August and December time points28 (unpublished data). The expression of CYP1A1 gene was increased scalp biopsies of subjects both unaffected and affected by LPP48. Interestingly, CYP1A1 has found to be down-regulated in the body side skin (hair and cashmere rich) of Cashmere goatssubjected to a short photoperiod compared with natural light conditions50. Why CYP1A1 expression was also down-regulated over 10 folds in A/H deserves further investigation.
Analysis of less highly differentially expressed genes (<10-fold)
The gene encoding one cell adhesion molecule, namely vascular cell adhesion molecule 1 (VCAM1) showed differential expression in A/S, suggesting that immune signal transduction had certain universality in the growth processes of wool follicle and wool diameter regulation.
Since different CD antigens are often specifically expressed in different immune cells, so a variety of CD antigens combinations have been used to identify the types of immune cells12. Mast cells, macrophages, dendritic cells are involved in immune privilege of hair follicle51,52, so the differential expression of CD1D antigen in A/S suggests that it might be associated with immune privilege mechanism.
Functional implication of differentially expressed proteins
The majority of the differentially expressed proteins found in our study were not mentioned in the previous discussion.
FGF18 protein was down-regulated in A/S. The higher expression of FGF18 in hair follicles is consistent with our studies. Interestingly, this protein is able to induce anagen hair growth when administered subcutaneously to mice in a uniform telogen state53.
Keratins are fibrous structural proteins that constitute the structural skeleton of epithelial cells. Keratins are further divided into cytokeratins and hair keratins. KRT1, KRT19 and KRT5 proteins were up-regulated in A/S, while KRT2.13 protein was down-regulated. KRT2.13, as a hair-like gene, was supposed to be inactive in hair follicles54. Dowling-Degos disease, characterized by abnormal hair follicles, is a result of at least three different loss-of-function mutations affecting the keratin 5 gene (KRT5)55.
Expression of FGFR3 was detected in precuticle cells surrounding the hair bulb56. However, in our study, we found that FGFR3 protein was down-regulated in wool bearing skin compared to the control group.
In the skin of foetal Merino sheep, BMP4 mRNA levels quickly raised at the time point of secondary follicle branching57. In our study, BMP4 protein was down-regulated in A/S. BMP4 was supposed to play inhibition roles in hair-follicle induction58. So BMP4 might play an important role in the process of wool growth.
Stratifin, a protein encoded by the SFN gene thatis required for hair follicle integrity and epidermal homeostasis59,60 was up-regulated in A/S. This would further support an essential role of this protein in wool follicle regulation.
Brief discussion on sampling specificity
In our study, we used two sheep breeds, Aohan fine wool sheep, with fine wool, and small tial Han sheep, with coarse wool, as two models for researching the molecular mechanisms regulating wool fibre diameter. So the main difference between these two sheep breeds is focused on wool fibre diameter. We speculated that the major difference in terms of gene expression profiles between these two breeds was derived from differences at the wool follicles. Certainly, due to the nature of the samples used in this study, we cannot completely exclude that some differences were due to skin tissue outside the wool follicle. But according to the results of a comparative study we conducted at the same time, the differences in gene expression profiles of skin tissues other than hair follicles is relatively small (comparison of gene expression profiles in the hairless region of skin between these two sheep breeds, data not shown). Besides, the fact thatmost of the differentially expressed genes obtained in this study were related to hair follicle development and wool growth regulation underscores our results.
In summary, the data presented here indicate that, at the anagen phase, the wool follicle in the bodyside skin of Aohan fine wool sheep shows a distinct expression pattern when compared to that of small tail Han sheep. Microarray analysis indicated that most of the genes putatively related to wool diameter regulation could be assigned into different categories, including regulation of receptor binding, growth factor activity and immune response. Several gene families might be involved in hair diameter regulation, including growth factors, immune cytokines, solute carrier families, cellular respiration and glucose transport. Proteomic analysis also identified several differentially expressed proteins. This comprehensive study could serve as a starting point of further investigations leading to a better understanding of the molecular mechanisms regulating wool diameter and to the identification of new strategies leading to the production of fine wool.
Methods
Animals and sample preparation
Animal handling was performed according to the animal protocols defined by national and local animal welfare bodies. All animal work was approved by the Shandong Province Biological Studies Animal Care and Use Committee28.
In August 2010, one male and two female sheep of the Aohan fine wooland small tail Han sheep (one-year-old) breeds respectively, were used in this study. Animals from each breed were half siblingssharing the same male parent. Full-thickness whole skin (including epidermis and dermis) was sampled from the body side skin(more wool growing) of these six animal sunder local anesthesia. These samples were used for both transcriptome and proteome analysis. The skin area sampled was approximately 1 cm2. After removal, all samples were immediately stored in liquid nitrogen for RNA and protein extraction28. A total of 15, 208 probes encoding proteins were spotted on this Agilent Sheep Gene Expression Microarray (Santa Clara, CA, USA)28.
RNA extraction and microarray hybridization
Total RNA was extracted using TRIzol (Invitrogen). The extraction was performed according to the manufacturer’s protocol28. RNA integrity and concentration were evaluated by Nanodrop. Our experiments were accomplished at 2010, when Agilent 2100 Bioanalyzer were not available in China. So we deployed Nanodrop and agarose gel electrophoresis to detect the quality of RNA samples. The hybridization of the RNA samples to the Agilent Sheep Gene expression Microarray (Santa Clara, CA, USA)28 was performed by the Kangchen Biotechnology Limited Company (Shanghai, China).
Microarrays data analysis
After hybridization and washing, microarray slides were scanned with the GenePix 4000B microarray scanner (Molecular Devices, LLC., USA) 28. The resulting text files extracted from Agilent Feature Extraction Software (version 9.5.3) were imported into the Agilent Gene Spring GX software (version 7.3) for further analysis28. Differentially expressed genes were screened bystudent’s T-test. False discovery rate (FDR) values were generated using permutations of the repeated measurements to estimate the percentage of genes identified by chance61.A P valueof0.05 and a FDR value of 0.05 were set as a threshold. Clustering analysis of all differentially expressed genes was performed using Cluster 3.062,63 to analyze the similarity in the expression patterns among different species/breeds28. The functional annotation of differentially expressed genes was performed by the DAVID (The Database for Annotation, Visualization and Integrated Discovery) gene annotation tool (http://david.abcc.ncifcrf.gov/)28,64.
qPCR confirmation
Total RNA samples prepared for microarray analysis were also used for qPCR confirmation28. Reverse transcription was performed using RevertAid First Strand cDNA Synthesis Kit (MBI Fermentas, Vilnius, Lithuania) according to the manufacturer’s protocols28. Primers were designed with the Primer-BLAST program (https://www.ncbi.nlm.nih.gov/tools/primer-blast/)28. Primer sequences, melting temperatures and product sizes are described in Table 1.
Tissue protein extraction
For protein extraction by homogenization, 1% protein inhibitor cocktail and 2% IPG-buffer were added in advance to lysis buffer (42% Urea, 15.2% Thiourea, 4% CHAPS, 1% DTT)28. This buffer is then added onto skin tissue previously cut into small pieces by ophthalmic scissors28 at the ratio of 1:7 (weight/volume). After tissue homogenization, the tissue slurry is placed at 4 °C for 1 h, vortexing it every 15 min. Following this incubation, samples are centrifuged at 40,000 g for 30 min. Supernatant is separated and store it at −80 °C. Protein concentration was determined by the Bradford method.
One-dimensional electrophoresis
Add 0.5% IPG-buffer into 150 μg protein sample (400–600 μL), and place it into One-dimensional electrophoresis instrument28. The programme is as below:
Step-n-hold
S130 V 6 h
S2 60 V 6 h
Gradient
S3 500 V 1 h
S4 1000 V 1 h
S5 3000 V 3 h
S6 8000 V 3 h
Step-n-hold
S7 8000 V 20 h
2-dimensional (2-D) SDS-PAGE preparation
Tris-HCl (PH = 8.8), Monomer storage (30% Acrylamid and 0.8% NN′-methylenebisacrylamid), 10×electrophoresis buffer (3.03% Tris-Base, 14.4% Glycine, 1% SDS), balanced solution (36.05% Urea, 5% Tris-HCl, 2% SDS, 34.5% Glycerine)28.
The electrophoresis programme used for the second dimension SDS-PAGE is as follows28:
Transfer: Voltage 300 v, Current 50 mA, Time 1 h.
Separation: Voltage 300 v, Current 200~250 mA, Time 4~5 h.
Fixative preparation: 40% Ethanol and 10% Acetic acid.
Fixation: take out the rubber strip and put it into Fixative for 1 h.
Staining and comparison of expression levels
Gels were silver stained, scanned and analysed using Image Master TM 2D platinum software (Version 5.0, GE Healthcare, San Francisco, CA, USA)28. The expression level was determined by the relative volume of each spot in the gel and expressed as %Vol (%Vol = [spot volume/Σvolumes of all spots resolved in the gel])28. We calculated the means and standard deviations of both sample groups and assessed statistical significance with Student’s t-tests using Image Master TM 2D platinum software28. P values < 0.05 were considered statistically significant28.
Identification of altered proteins by mass spectrometry (MS)
Protein spots with significant differences between the two groups were excised, dehydrated in acetonitrile, and dried at room temperature28. Gel pieces were denatured, alkylated, trypsin digested and analysed by an Ultraflex II MALDI-TOF-TOF mass spectrometer (Bruker Daltonics GmbH, Bremen, Germany) under the control of FlexControl TM 2.4 software (Bruker Daltonics GmbH)28. Acquired peptide mass fingerprint (PMF) were processed using the software Flex AnalysisTM 3.0 (Bruker Daltonics, Bremen, Germany)28. The peak detection algorithm was: SNAP (Sort Neaten Assign and Place); S/N threshold: 1.5; Quality Factor Threshold: 50. The tryptic auto-digestion ion picks (trypsin [108–115] 842.5094 Da, trypsin [58–77] 2211.104 Da) were used as internal standards28. The resulting peptide mass lists were used to search the Matrix science database (http://www.matrixscience.com)28. The following search parameter criteria were used65: mass tolerance 100 ppm, miss cleavage ≤ 1, modification comprises Carbamidomethyl and methionine oxidation28. Matched peptides number between experimental PMF and theoretical PMF ≥ 528.
References
Purvis, I. W. & Franklin, I. R. Major genes and QTL influencing wool production and quality: a review. Genet. Sel. Evol. 37(Suppl 1), S97–107 (2005).
Bidinost, F. et al. Wool quantitative trait loci in Merino sheep. Small Rumin. Res. 74, 113–118 (2008).
Itenge, T. O., Hickford, J. G. H., Forrest, R. H. J., McKenzie, G. W. & Frampton, C. M. Improving the quality of wool through the use of gene markers. South. Afr. J. Anim. Sci. 39, 219–223 (2009).
Beh, K. J. et al. A genome scan for QTL affecting fleece and wool traits in Merino sheep. Wool. Technol. Sheep Breed. 49, 88–89 (2001).
Cano, E. M. et al. QTL affecting fleece traits in Angora goats. Small Rumin. Res. 71, 158–164 (2007).
Schmidt-Ullrich, R. & Paus, R. Molecular principles of hair follicle induction and morphogenesis. Bioessays 27, 247–261 (2005).
Galbraith, H. Fundamental hair follicle biology and fine fibre production in animals. Animal 4, 1490–1509 (2010).
McGrice, H. A. Molecular Characterisation of Primary Wool Follicle Initiation in Merino Sheep PhD thesis, University of Adelaide, (2010).
Rhee, H., Polak, L. & Fuchs, E. Lhx2 maintains stem cell character in hair follicles. Science 312, 1946–1949 (2006).
Janich, P. et al. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature 480, 209–214 (2011).
Ohyama, M. et al. Characterization and isolation of stem cell-enriched human hair follicle bulge cells. J. Clin. Invest. 116, 249–260 (2006).
Cotsarelis, G. Gene expression profiling gets to the root of human hair follicle stem cells. J. Clin. Invest. 116, 19–22 (2006).
Ollier, S., Robert-Granie, C., Bernard, L., Chilliard, Y. & Leroux, C. Mammary transcriptome analysis of food-deprived lactating goats highlights genes involved in milk secretion and programmed cell death. J. Nutr. 137, 560–567 (2007).
Faucon, F. et al. Terminal differentiation of goat mammary tissue during pregnancy requires the expression of genes involved in immune functions. Physiol. Genomics 40, 61–82 (2009).
Bongiorni, S. et al. A Tool for Sheep Product Quality: Custom Microarrays from Public Databases. nutrients, 235–250 (2009).
Norris, B. J., Bower, N. I., Smith, W. J. M., Cam, G. R. & Reverter, A. Gene expression profiling of ovine skin and wool follicle development using a combined ovine-bovine skin cDNA microarray. Australian J. Exp. Agriculture 45, 867–877 (2005).
Smith, W. J. et al. A genomics-informed, SNP association study reveals FBLN1 and FABP4 as contributing to resistance to fleece rot in Australian Merino sheep. BMC Vet. Res. 6, 27 (2010).
Penagaricano, F., Zorrilla, P., Naya, H., Robello, C. & Urioste, J. I. Gene expression analysis identifies new candidate genes associated with the development of black skin spots in Corriedale sheep. J Appl Genet (2012).
MacKinnon, K. M., Burton, J. L., Zajac, A. M. & Notter, D. R. Microarray analysis reveals difference in gene expression profiles of hair and wool sheep infected with Haemonchus contortus. Vet. Immunol. Immunopathol. 130, 210–220 (2009).
Keane, O. M. et al. Gene expression profiling of naive sheep genetically resistant and susceptible to gastrointestinal nematodes. BMC Genomics 7, 42 (2006).
Wenguang, Z., Jianghong, W., Jinquan, L. & Yashizawa, M. A subset of skin-expressed microRNAs with possible roles in goat and sheep hair growth based on expression profiling of mammalian microRNAs. OMICS 11, 385–396 (2007).
Liu, G. et al. Identification of microRNAs in wool follicles during anagen, catagen, and telogen phases in Tibetan sheep. PLoS One 8, e77801 (2013).
Yuan, C. et al. Discovery of cashmere goat (Capra hircus) microRNAs in skin and hair follicles by Solexa sequencing. BMC Genomics 14, 511 (2013).
Geng, R., Yuan, C. & Chen, Y. Exploring differentially expressed genes by RNA-Seq in cashmere goat (Capra hircus) skin during hair follicle development and cycling. PLoS One 8, e62704 (2013).
Dong, Y. et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 31, 135–141 (2013).
Jiang, Y. et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 344, 1168–1173, https://doi.org/10.1126/science.1252806 (2014).
Yue, Y. et al. Exploring Differentially Expressed Genes and Natural Antisense Transcripts in Sheep (Ovis aries) Skin with Different Wool Fiber Diameters by Digital Gene Expression Profiling. PLoS One 10, e0129249 (2015).
Liu, N. et al. Identification of skin-expressed genes possibly associated with wool growth regulation of Aohan fine wool sheep. BMC Genet. 15, 144 (2014).
Liu, N. et al. Differential expression of genes and proteins associated with wool follicle cycling. Mol. Biol. Rep. 41, 5343–5349 (2014).
Yu, Z. D. et al. Micro-arrays as a discovery tool for wool genomics. Proc. N. Zealand Soc. Anim. Prod. 66, 129–133 (2006).
Yu, Z. D. et al. Gene expression profiling of wool follicle growth cycles by cDNA microarray. Proc. N. Zealand Soc. Anim. Prod. 68, 39–42 (2008).
Zhao, J. et al. Identification of genes and proteins associated with anagen wool growth. Anim. Genet. 48, 67–79 (2017).
Adelson, D. L., Cam, G. R., DeSilva, U. & Franklin, I. R. Gene expression in sheep skin and wool (hair). Genomics 83, 95–105 (2004).
Rogers, G. E. Biology of the wool follicle: an excursion into a unique tissue interaction system waiting to be re-discovered. Exp. Dermatol. 15, 931–949 (2006).
Nagorcka, B. The reaction-diffusion (RD) theory of wool (hair) follicle initiation and development. I. Primary follicles. Australian J. Agric. Res. 46, 333–355 (1995).
Nagorcka, B. The reaction-diffusion (RD) theory of wool (hair) follicle initiation and development. II. Original secondary follicles. Australian J. Agric. Res. 46, 357–378 (1995).
Sick, S., Reinker, S., Timmer, J. & Schlake, T. WNT and DKK determine hair follicle spacing through a reaction-diffusion mechanism. Science 314, 1447–1450 (2006).
Gibbs, S. et al. Molecular characterization and evolution of the SPRR family of keratinocyte differentiation markers encoding small proline-rich proteins. Genomics 16, 630–637, https://doi.org/10.1006/geno.1993.1240 (1993).
Ishida-Yamamoto, A., Kartasova, T., Matsuo, S., Kuroki, T. & Iizuka, H. Involucrin and SPRR are synthesized sequentially in differentiating cultured epidermal cells. J. Invest. Dermatol. 108, 12–16 (1997).
Candi, E. et al. Biochemical, structural, and transglutaminase substrate properties of human loricrin, the major epidermal cornified cell envelope protein. J. Biol. Chem. 270, 26382–26390 (1995).
Wang, L. & Baldwin, R. L. t. & Jesse, B. W. Identification of two cDNA clones encoding small proline-rich proteins expressed in sheep ruminal epithelium. Biochem. J. 317(Pt 1), 225–233 (1996).
Akiyama, M., Smith, L. T., Yoneda, K., Holbrook, K. A. & Shimizu, H. Transglutaminase and major cornified cell envelope precursor proteins, loricrin, small proline-rich proteins 1 and 2, and involucrin are coordinately expressed in the sites defined to form hair canal in developing human hair follicle. Exp. Dermatol. 8, 313–314 (1999).
Bodo, E. et al. Dissecting the impact of chemotherapy on the human hair follicle: a pragmatic in vitro assay for studying the pathogenesis and potential management of hair follicle dystrophy. Am. J. Pathol. 171, 1153–1167 (2007).
Yoshikawa, Y. et al. Upregulation of genes orchestrating keratinocyte differentiation, including the novel marker gene ID2, by contact sensitizers in human bulge-derived keratinocytes. J. Biochem. Mol. Toxicol. 24, 10–20 (2010).
Aubert, J. et al. Gene expression profiling in psoriatic scalp hair follicles: clobetasol propionate shampoo 0.05% normalizes psoriasis disease markers. J. Eur. Acad. Dermatol. Venereol. 24, 1304–1311 (2010).
French, A. T. et al. Up-regulation of intelectin in sheep after infection with Teladorsagia circumcincta. Int. J. Parasitol. 38, 467–475 (2008).
Mistry, D. H. & Medrano, J. F. Cloning and localization of the bovine and ovine lysophosphatidic acid acyltransferase (LPAAT) genes that codes for an enzyme involved in triglyceride biosynthesis. J. Dairy. Sci. 85, 28–35 (2002).
Karnik, P. et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J. Invest. Dermatol. 129, 1243–1257 (2009).
Rowe, J. M. et al. Illuminating role of CYP1A1 in skin function. J. Invest. Dermatol. 128, 1866–1868 (2008).
Liu, B. et al. A Microarray-Based Analysis Reveals that a Short Photoperiod Promotes Hair Growth in the Arbas Cashmere Goat. PLoS One 11, e0147124 (2016).
Christoph, T. et al. The human hair follicle immune system: cellular composition and immune privilege. Br. J. Dermatol. 142, 862–873 (2000).
Osaka, N. et al. ASK1-dependent recruitment and activation of macrophages induce hair growth in skin wounds. J. Cell Biol. 176, 903–909 (2007).
Kawano, M. et al. Comprehensive analysis of FGF and FGFR expression in skin: FGF18 is highly expressed in hair follicles and capable of inducing anagen from telogen stage hair follicles. J. Invest. Dermatol. 124, 877–885 (2005).
Powell, B. C. & Beltrame, J. S. Characterization of a hair (wool) keratin intermediate filament gene domain. J. Invest. Dermatol. 102, 171–177 (1994).
Horner, M. E., Parkinson, K. E., Kaye, V. & Lynch, P. J. Dowling-Degos disease involving the vulva and back: case report and review of the literature. Dermatol. Online J. 17, 1 (2011).
Rosenquist, T. A. & Martin, G. R. Fibroblast growth factor signalling in the hair growth cycle: expression of the fibroblast growth factor receptor and ligand genes in the murine hair follicle. Dev. Dyn. 205, 379–386 (1996).
Menzies, M., Stockwell, S., Brownlee, A., Cam, G. & Ingham, A. Gene expression profiles of BMP4, FGF10 and cognate inhibitors, in the skin of foetal Merino sheep, at the time of secondary follicle branching. Exp. Dermatol. 18, 877–879 (2009).
Botchkarev, V. A. et al. Noggin is a mesenchymally derived stimulator of hair-follicle induction. Nat. Cell Biol. 1, 158–164 (1999).
Yuan, C. et al. The up-regulation of 14-3-3 proteins in Smad4 deficient epidermis and hair follicles at catagen. Proteomics 8, 2230–2243 (2008).
Hammond, N. L., Headon, D. J. & Dixon, M. J. The cell cycle regulator protein 14-3-3sigma is essential for hair follicle integrity and epidermal homeostasis. J. Invest. Dermatol. 132, 1543–1553 (2012).
Zhou, P. et al. Molecular characterization of transcriptome-wide interactions between highly pathogenic porcine reproductive and respiratory syndrome virus and porcine alveolar macrophages in vivo. Int. J. Biol. Sci. 7, 947–959 (2011).
Tang, Z. et al. LongSAGE analysis of skeletal muscle at three prenatal stages in Tongcheng and Landrace pigs. Genome Biol. 8, R115 (2007).
Nacht, M. et al. Molecular characteristics of non-small cell lung cancer. Proc. Natl Acad. Sci. USA 98, 15203–15208 (2001).
Dennis, G. Jr. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3 (2003).
Berndt, P., Hobohm, U. & Langen, H. Reliable automatic protein identification from matrix-assisted laser desorption/ionization mass spectrometric peptide fingerprints. Electrophoresis 20, 3521–3526 (1999).
Acknowledgements
This project was funded by First Class Grassland Science Discipline Program of Shandong Province (China), National Natural Science Foundation of China (31572383, 31301936), National Hair Sheep Industry Technology System (CARS-40), the Special Fund for Agro-scientific Research in the Public Interest (201403071) and Projects of Qingdao People’s Livelihood Science and Technology (13-1-3-88-nsh, 14-2-3-45-nsh, 19-6-1-68-nsh).
Author information
Authors and Affiliations
Contributions
J.Z. and H.L. conceived the experiments. H.L., H.Q., J.X. and N.L. conducted the experiments. J.Z., H.L., N.L., R.H. and F.P.C. analysed the results. J.Z. and H.L. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J., Qin, H., Xin, J. et al. Discovery of genes and proteins possibly regulating mean wool fibre diameter using cDNA microarray and proteomic approaches. Sci Rep 10, 7726 (2020). https://doi.org/10.1038/s41598-020-64903-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-64903-7
This article is cited by
-
Genetics of the phenotypic evolution in sheep: a molecular look at diversity-driving genes
Genetics Selection Evolution (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
