
Abstract
Epigenetic changes precede the development of diabetes by many years, providing clues to its pathogenesis. We explored whether the epigenetic markers, circulating microRNAs (miRNAs), were associated with incident diabetes in Japanese Americans. We conducted a pilot study (n = 10) using plasma from age- and sex-matched participants who did or did not develop diabetes in the Japanese American Community Diabetes Study, an observational study of diabetes risk factors. Extraction and high-throughput sequencing of miRNAs were performed using samples collected at baseline. Regression models were fit comparing circulating miRNAs (N = 1640) among individuals who did or did not develop incident diabetes at 10-year follow-up. Participants averaged 51.7 years of age at baseline; 60% were male. We identified 36 miRNAs present at different (10 higher and 26 lower) levels in individuals who developed diabetes compared to those who did not (log2fold change ≥1.25 and false discovery rate ≤5%). These included miRNAs with functions in skeletal muscle insulin metabolism (miR-106b and miR-20b-5p) and miRNAs with functions in both skeletal muscle insulin metabolism and cell cycle regulation in endocrine pancreas (miR-15a and miR-17). Circulating miRNAs were associated with subsequent development of diabetes among Japanese Americans over 10 years of follow-up. Results are preliminary. Large-scale miRNA sequencing studies could inform our understanding of diabetes pathogenesis and development of therapies, based on gene expression regulation, that target diabetes.
Similar content being viewed by others


Introduction
A better understanding of tissue level changes in gene expression and regulation occurring early in the course of type 2 diabetes could provide information about its pathogenesis and inform the development of predictive biomarkers. Unfortunately, tissue-specific biomarkers are rarely measured in longitudinal observational studies because tissue-collection protocols are invasive, expensive and time consuming, limiting our understanding of tissue-level abnormalities that precede diabetes. Circulating microRNAs (miRNAs) are short non-coding RNAs that regulate tissue gene expression1. They are actively secreted into the circulation2, where they are markers of tissue regulation of gene expression as well as mediators of inter-organ cross-talk3. Using a real-time polymerase chain reaction (RT-PCR), approach two studies have identified altered patterns in circulating miRNAs that precede the development of diabetes by up to 15 years in Europeans4,5. To our knowledge, no prior study has been conducted in Japanese Americans, a group at high risk of type 2 diabetes. In addition, prior studies involved candidate miRNA profiling, limiting potential discovery that follows comprehensive miRNA profiling. Thus, we used next-generation RNA sequencing to examine whether circulating miRNAs preceded the development of diabetes in Japanese Americans after 10 years of follow-up.
Methods
Study setting and study population
Study participants were selected from the Japanese American Community Diabetes Study (JACDS), a community-based cohort of individuals of 100% Japanese ancestry. Selection and recruitment procedures have been described previously6. For this analysis, we randomly selected five individuals from among those without prevalent diabetes at baseline and incident diabetes at the year 10 follow-up visit and five controls individually matched by age and sex who remained free from diabetes for 10 years. The study was approved by the University of Washington Human Subjects Division. Participants provided written informed consent, and all research was performed in accordance with relevant guidelines and regulations.
Data collection
Data and sample collection were done at the General Clinical Research Center at the University of Washington, Seattle, by trained staff. Blood specimens were drawn at enrollment and at a follow-up visit at 10–11 years after at least a 10-hour fast. Plasma was isolated and stored at −80 °C. Presence of diabetes was assessed at baseline and follow-up using a two-hour oral glucose tolerance test (75 gram load) and defined as fasting glucose ≥126 mg/dl (7.0 m mol/L) and/or 2-h glucose ≥200 mg/dl (11.1 mmol/L)7, or use of diabetes medication.
Pre-processing, extraction and profiling of circulating miRNAs
Thawed samples were spun at ~2500 RPMs for five minutes to completely clear plasma of cells. Small RNAs were extracted from 500 μL plasma aliquots using the Exiqon (now Qiagen) miRCURYTM RNA Biofluids Isolation Kit (Exiqon, Woburn, MA). Integrity, purity and quantity of purified miRNA was assessed using spectrophotometry and an Agilent 2100 Bioanalyzer capillary electrophoresis system (Agilent Technologies Inc, Palo Alto, CA). The Qiagen QIAseq miRNA NGS Library Kit was used for library preparation. MiRNAs were sequenced using an Illumina sequencer. Lab personnel were blinded to participant outcomes. Additional technical details are provided in Supplemental Materials.
Statistical and bioinformatics analyses
Number (%) and mean (standard deviation) describe study population characteristics. Analyses were conducted in R version 3.4.0. Samples were normalized using a weighted trimmed mean of M-values (TMM), which calculates a normalization factor that is used to scale the library sizes. Because count data are not normally distributed and may have transcripts with zero counts8, a linear model based on the negative binomial distribution in the Bioconductor edgeR package9 using quasi-likelihood F-tests10 was used. Exploratory principal component analysis showed an apparent large batch effect captured by the first principal component; therefore, the first principal component was included as an adjustment variable, along with two surrogate variables detected using the Bioconductor sva package11. To protect against choosing miRNA transcripts that may be differentially expressed at a statistically significant but low-fold–change level that is not biologically meaningful, miRNAs with at least a 25% difference12 in expression as well as a <5% false discovery rate (FDR) were identified as significantly different, as has been previously reported13. We used the Bioconductor sizepower package to estimate study power, post hoc. The average standard error was 0.999, and we tested for a 1.25-log2fold difference. We found 36 out of 1640 miRNAs significant. Accepting 10 false positives, we calculate we had 31% power to identify differences in circulating miRNAs between the groups. The Core Analysis feature of the Ingenuity Pathway Analysis (IPA) software program (Built version–486617 M; Content version–33559992; Ingenuity Systems, A Qiagen Company, Redwood City, CA) was used to identify transcriptional networks using microRNAs that were present at different levels in cases than in controls14. IPA’s microRNA Target Filter to identify mRNA targets was also used, restricting the search to “experimentally validated” targets. IPA Core Analysis was performed on the lists of targets that were identified as described above by IPA’s microRNA Target Filter feature.
Ethics approval and consent to participate
This study was approved by the institutional review board at the University of Washington as # STUDY00001823, and all participants provided written informed consent.
Results
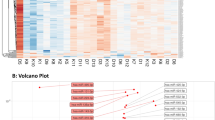
Baseline characteristics of the cohort are shown in Table 1. After excluding miRNA transcripts with mean log counts per million (CPM) ≤ 2.5, 1640 transcripts were carried forward. Table 2 and Fig. 1 show log2fold-change differences (logFC), log counts per million (logCPM), and false discovery rates (FDR) for circulating miRNAs measured at baseline that differ between individuals with and without incident diabetes after 10 years of follow-up (n = 36; FDR < 0.05, ≥1.25-fold change). Identified miRNAs included miR-20b-5p (logFC −2.48, FDR 0.0042), miR-363-3p (logFC −2.06, FDR 0.0042), miR-7-5p (logFC −2.32, FDR 0.0042), miR-144-3p (logFC −2.44, FDR 0.0074), miR-20a (logFC −1.98, FDR 0.0074), miR-451b (logFC −2.06, 0.0074), miR-15b-5p (logFC −2.02, FDR 0.0109), and miR-15a-5p (logFC −1.71, FDR 0.0156), which were all lower in individuals who developed diabetes compared to those who did not. A pathway analysis suggested identified miRNAs may play a role in cell-cycle regulation (Supplementary Table 1, Supplementary Fig. 1). The top network represented by experimentally validated gene (mRNA) targets of microRNAs was “Cell cycle, embryonic development.”

Barplot of log fold changes for each miRNA. For aid in interpretation we have sorted the miRNA transcripts by fold change.
Discussion
In this exploratory analysis using archival samples from a community-based study of diabetes risk factors in Japanese Americans, we identified 36 miRNAs (FDR < 0.05, >1.25-fold change) present at different levels in plasma of individuals who did versus did not develop diabetes up to 10 years later. Although the sample size is small, this report establishes new findings and extends previous findings in this area in several important ways. First, we extended the study of circulating miRNAs with long-term follow-up for incident diabetes to a new population, Asian-Americans. Second, by using a comprehensive sequencing approach, we identified circulating miR-106b, miR-17 and miR-20b-5p not reported in previous long-term longitudinal studies. These miRNAs have putative roles in the pathogenesis of diabetes via actions in skeletal muscle and/or pancreatic islet cells. Lastly, we replicated previous observations4,5 that lower baseline levels of miR-15a were associated with incident diabetes, suggestingits potential as a predictive diabetes biomarker.
In the Spanish CORDIOPREV cohort (n = 462), higher levels of miR-150 and miR-30a-5p and lower levels of miR-15a and miR-375 were associated with higher diabetes risk at 60 months4. In the Bruneck (Italy) cohort, individuals with incident diabetes at 10 years (n = 19) had lower baseline plasma levels of miR-15a, miR-29b, miR-126 and miR-223 and higher levels of miR-28-3p than matched controls5. In our sample, except for miR-15a, the other associations were not replicated. A secondary analysis of the Practicing Restorative Yoga vs. Stretching for the Metabolic Syndrome study examined associations of circulating miRNAs with incident diabetes over 12 months of follow-up15. As in our study, baseline levels of miR-106b, miR-20b, miR-363, miR-486, miR-532 and miR-93 were associated with incident diabetes, as well as with response to a lifestyle intervention15.
In skeletal muscle from monozygotic twins discordant for type 2 diabetes, expression of miR-106b, miR-15b, miR-17, miR-20a, miR-20b, miR-25, miR-363, miR-451, miR-93 (identified in the current study), and others were downregulated in twins with type 2 diabetes compared to twins without diabetes16. In a community-based sample, miR-106b, miR-144, miR-15b, miR-451, and miR-93 were upregulated and miR-15a downregulated in skeletal muscle of individuals with type 2 diabetes compared to individuals with normal glucose tolerance17. In another study, higher levels of both skeletal muscle miR-15a and miR-15b were associated with higher fasting and 2-hour glucose in adults with and without diabetes18. Inconsistency of identified miRNAs in previous as well as the current study could be due to racial and ethnic differences in the study populations, heterogeneous mechanisms contributing to diabetes pathogenesis across the populations studied,or differences in methods (sample source, processing, and profiling). Carefully designed studies incorporating repeated measurement of miRNAs over time in cohorts at high diabetes risk will be needed to clarify these relationships.
Identified miRNAs including miR-17, miR-106b and miR-20b-5p have functions in skeletal muscle insulin metabolism. In skeletal muscle, miR-1719 and miR-106b20 downregulate components of the glucose transporter 4 pathway, while overexpression of miR-20b-5p impairs insulin signaling and suppresses genes in pathways related to immune function (AKTIP and STAT3)21. Some identified miRNAs have functions in islet cells as well. In pancreatic cell lines, miR-17 downregulates thioredoxin-interacting protein, a pro-apoptotic regulator of beta-cells in diabetes22. In mouse islets, miR-7 (miR-7a) targets multiple components of the mTOR signaling pathway, and its inhibition promotes adult beta-cell replication23. In rats, miR-25 negatively regulates expression of insulin I messenger transcripts24. Last, islet cell expression of miR-15a is upregulated in the presence of brief (1 h) exposure to hyperglycemia but depressed after a longer period of exposure25. At the same time, overexpression of miR-15a increases insulin levels and its repression inhibits insulin synthesis by inhibiting uncoupling protein-2 (UCP-2) gene expression25. Five miRNAs identified in this study are members of the highly conserved polycistronic miR-17~92 family (miR-17, miR-20, miR-25, miR-93, and miR-106b)26, which may be important in cell-cycle regulation, as suggested by our bioinformatics analysis results.
An important limitation of our pilot study is that it was underpowered to detect an exhaustive list of circulating miRNAs associated with incident diabetes. We used conservative correction for multiple testing to mitigate risk of type II error. Results are preliminary and need to be replicated in other populations. In addition, at baseline the groups differed in distribution of impaired fasting glucose (IFG) and impaired glucose tolerance (IGT). To further characterize baseline glycemic status, we used 30-, 60-, 120-, and 180-minute values of glucose and insulin from baseline oral glucose tolerance tests to calculate Matsuda and insulinogenic indices (IGI) for study participants. These values were very similar between the groups (Matsuda 3.1 ± 0.7 vs. 3.2 ± 1.3, p = 0.879; IGI 0.8 ± 1.0 vs. 0.7 ± 0.4, p = 0.883), suggesting that differences in baseline miRNA levels were unlikely to be attributable to differences in glucose tolerance at baseline, despite the differing distributions of IFG and IGT.
This analysis demonstrates the utility of measuring circulating miRNAs with RNA sequencing from observational and interventional studies of type 2 diabetes to characterize changes that precede the development of diabetes. Results can be carried forward in the development of predictive biomarkers of future type 2 diabetes.
Data availability
The datasets generated during and/or analysed during the current study are not publicly available due because this provision was not made in the original IRB application.
References
Mirra, P. et al. The Destiny of Glucose from a MicroRNA Perspective. Front. Endocrinol. 9, 46, https://doi.org/10.3389/fendo.2018.00046 (2018).
Lin, X. et al. MiR-155 Enhances Insulin Sensitivity by Coordinated Regulation of Multiple Genes in Mice. PLoS Genet. 12, e1006308, https://doi.org/10.1371/journal.pgen.1006308 (2016).
Ying, W. et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 171, 372–384e312, https://doi.org/10.1016/j.cell.2017.08.035 (2017).
Jimenez-Lucena, R. et al. A plasma circulating miRNAs profile predicts type 2 diabetes mellitus and prediabetes: from the CORDIOPREV study. Exp. Mol. Med. 50, 168, https://doi.org/10.1038/s12276-018-0194-y (2018).
Zampetaki, A. et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circulation Res. 107, 810–817, https://doi.org/10.1161/CIRCRESAHA.110.226357 (2010).
Fujimoto, W. Y. et al. Prevalence of diabetes mellitus and impaired glucose tolerance among second-generation Japanese-American men. Diabetes 36, 721–729 (1987).
Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 20, 1183-1197 (1997).
Anders, S. et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 8, 1765–1786, https://doi.org/10.1038/nprot.2013.099 (2013).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, https://doi.org/10.1093/bioinformatics/btp616 (2010).
Lund, S. P., Nettleton, D., McCarthy, D. J. & Smyth, G. K. Detecting differential expression in RNA-sequence data using quasi-likelihood with shrunken dispersion estimates. Statist. Applicat. Genet. Molecul. Biol. 11 https://doi.org/10.1515/1544-6115.1826 (2012).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883, https://doi.org/10.1093/bioinformatics/bts034 (2012).
McCarthy, D. J. & Smyth, G. K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 25, 765–771, https://doi.org/10.1093/bioinformatics/btp053 (2009).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Kramer, A., Green, J., Pollard, J. Jr & Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530, https://doi.org/10.1093/bioinformatics/btt703 (2014).
Flowers, E. et al. Preliminary evidence supports circulating microRNAs as prognostic biomarkers for type 2 diabetes. Obes. Sci. Pract. 3, 446–452, https://doi.org/10.1002/osp4.134 (2017).
Bork-Jensen, J. et al. Glucose tolerance is associated with differential expression of microRNAs in skeletal muscle: results from studies of twins with and without type 2 diabetes. Diabetologia 58, 363–373, https://doi.org/10.1007/s00125-014-3434-2 (2015).
Gallagher, I. J. et al. Integration of microRNA changes in vivo identifies novel molecular features of muscle insulin resistance in type 2 diabetes. Genome Med. 2, 9, https://doi.org/10.1186/gm130 (2010).
Houshmand-Oeregaard, A. et al. Increased expression of microRNA-15a and microRNA-15b in skeletal muscle from adult offspring of women with diabetes in pregnancy. Hum. Mol. Genet. 27, 1763–1771, https://doi.org/10.1093/hmg/ddy085 (2018).
Xiao, D. et al. MicroRNA-17 impairs glucose metabolism in insulin-resistant skeletal muscle via repressing glucose transporter 4 expression. Eur. J. Pharmacol. 838, 170–176, https://doi.org/10.1016/j.ejphar.2018.08.036 (2018).
Zhou, T. et al. Regulation of Insulin Resistance by Multiple MiRNAs via Targeting the GLUT4 Signalling Pathway. Cell Physiol. Biochem. 38, 2063–2078, https://doi.org/10.1159/000445565 (2016).
Katayama, M. et al. Circulating Exosomal miR-20b-5p is Elevated in Type 2 Diabetes and Could Impair Insulin Action in Human Skeletal Muscle. Diabetes, https://doi.org/10.2337/db18-0470 (2018).
Hong, K., Xu, G., Grayson, T. B. & Shalev, A. Cytokines Regulate beta-Cell Thioredoxin-interacting Protein (TXNIP) via Distinct Mechanisms and Pathways. J. Biol. Chem. 291, 8428–8439, https://doi.org/10.1074/jbc.M115.698365 (2016).
Wang, Y., Liu, J., Liu, C., Naji, A. & Stoffers, D. A. MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic beta-cells. Diabetes 62, 887–895, https://doi.org/10.2337/db12-0451 (2013).
Setyowati Karolina, D., Sepramaniam, S., Tan, H. Z., Armugam, A. & Jeyaseelan, K. miR-25 and miR-92a regulate insulin I biosynthesis in rats. RNA Biol. 10, 1365–1378, https://doi.org/10.4161/rna.25557 (2013).
Sun, L. L. et al. MicroRNA-15a positively regulates insulin synthesis by inhibiting uncoupling protein-2 expression. Diabetes Res. Clin. Pract. 91, 94–100, https://doi.org/10.1016/j.diabres.2010.11.006 (2011).
Tanzer, A. & Stadler, P. F. Molecular evolution of a microRNA cluster. J. Mol. Biol. 339, 327–335, https://doi.org/10.1016/j.jmb.2004.03.065 (2004).
Acknowledgements
This manuscript is dedicated to the memory of Marguerite J. McNeely who was an active investigator in the JACDS. Her critical contributions will be missed. We are grateful to the King County Japanese-American Community for their involvement and support in this and prior studies. VA Puget Sound supported the participation of Drs. Kahn and Boyko in this research. This work was supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award number DK103945, as well as NIH grants DK031170, HL049293, DK002654, DK017047, DK035816, P30ES00703 and RR000037. VA Puget Sound supported the participation of Drs. Kahn and Boyko in this research.
Author information
Authors and Affiliations
Contributions
P.L.W. conceived of the proposal and wrote the first draft of the manuscript. D.A.E. and E.J.B. provided significant contributors to the design of the studyand the initial manuscript draft. T.K.B., S.S. and J.M. performed the analyses. E.J.B., S.E.K., D.L. and W.Y.F. were instrumental in the design and conduct of the parent study from which the data and samples were taken. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wander, P.L., Enquobahrie, D.A., Bammler, T.K. et al. Short Report: Circulating microRNAs are associated with incident diabetes over 10 years in Japanese Americans. Sci Rep 10, 6509 (2020). https://doi.org/10.1038/s41598-020-63606-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-63606-3
This article is cited by
-
MiR-20b-5p contributes to the dysfunction of vascular smooth muscle cells by targeting MAGI3 in hypertension
Journal of Molecular Histology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
