
Abstract
This study was initiated to identify genomic regions conferring resistance to Karnal Bunt (KB) disease in wheat through a genome-wide association study (GWAS) on a set of 179 pre-breeding lines (PBLs). A GWAS of 6,382 high-quality DArTseq SNPs revealed 15 significant SNPs (P-value <10−3) on chromosomes 2D, 3B, 4D and 7B that were associated with KB resistance in individual years. In particular, two SNPs (chromosome 4D) had the maximum R2 values: SNP 1114200 | F | 0–63:T > C at 1.571 cM and R2 of 12.49% and SNP 1103052 | F | 0–61:C > A at 1.574 cM and R2 of 9.02%. These two SNPs displayed strong linkage disequilibrium (LD). An in silico analysis of SNPs on chromosome 4D identified two candidate gene hits, TraesCS4D02G352200 (TaNox8; an NADPH oxidase) and TraesCS4D02G350300 (a rhomboid-like protein belonging to family S54), with SNPs 1103052 | F | 0–61:C > A and 1101835 | F | 0–5:C > A, respectively, both of which function in biotic stress tolerance. The epistatic interaction analysis revealed significant interactions among 4D and 7B loci. A pedigree analysis of confirmed resistant PBLs revealed that Aegilops species is one of the parents and contributed the D genome in these resistant PBLs. These identified lines can be crossed with any elite cultivar across the globe to incorporate novel KB resistance identified on 4B.
Similar content being viewed by others

Introduction
Wheat (Triticum aestivum L.) is consumed worldwide as a key component of the human diet. The global wheat production for 2018–19 was 730.9 million tons, which is approximately 29 million tons lower than production in the preceding year (http://www.fao.org/worldfoodsituation/csdb/en/). With the global population estimated to be 9 billion in 2050, wheat demand is expected to increase by 60%. Increases in the annual wheat yield must grow from the current level of less than 1% to at least 1.6% to meet the demand (https://www.wheatinitiative.org/about-us/wheat-research-food-security).
Rusts, smuts, bunts, mildews and foliar blights are major biotic factors that significantly affect wheat production and productivity. Karnal bunt (KB), which is caused by Neovossia indica, is one of five bunt and smut diseases of wheat and a serious concern to the grain industry due to quarantine regulations that restrict the international movement and trade of affected stocks. KB directly attacks the economic part, i.e., seed of the crop (Supplementary Fig. S1). After the attack, the fungus forms black teliospores in sori and deteriorates the grain quality by generating an unpleasant, and foul smell attributed to trimethylamine produced by the fungus1. The infection of approximately 4% of kernels is sufficient for the grain to become unfit for human consumption due to a low palatability2. Internationally, approximately 70 countries have placed an embargo on the import of Karnal bunt-infected wheat3. The European and Mediterranean Plant Protection Organization (EPPO) has categorized N. indica as a quarantine pest (zero tolerance), which forced the adoption of strict laws under the Sanitary and Phytosanitary (SPS) agreement4.
The management of KB is challenging due to long-lived teliospores5. As a seed-, soil- and air-borne fungus, the control of the disease through cultural practices and/or seed treatment is not very effective6. The chemical control of KB by spraying fungicides at the anthesis stage is possible but not cost-effective for farmers; thus, conventional methods are not sufficient to cope with KB2,7.
The identification and development of resistant genotypes is a viable option to manage KB8. However, the lack of fast, simple and environment-independent methods for screening wheat germplasm that resist the pathogen is a bottleneck during the development of resistant varieties. Researchers are managing the problem with available screening methods, but because KB is a disease of the mature grain, the identification of resistant donors is only possible in the next generation, which also decelerates the breeding progress. Because of these limitations, marker-aided selection/screening (MAS) is another opportunity for efficient introgression of resistance into elite genotypes8.
Linkage analyses and genome-wide association studies (GWAS) are two powerful statistical genetic methods that have been successfully deployed for the genetic dissection of quantitative traits9. The former relies on trait segregation in a bi-parental cross population and has been exploited for the identification of QTLs for KB resistance in a range of studies3,8. GWAS is a population-based method that exploits the linkage disequilibrium phenomenon between a trait and a genetic marker10. This mapping population-independent method relies on a population consisting of a set of unrelated accessions and has been successfully used to identify numerous traits in many crops, such as Arabidopsis thaliana11, rice12, barley13 and wheat14. However, only two GWAS studies have been reported to date with the aim of determining marker-trait associations (MTAs) for KB resistance in elite wheat lines15,16. Both these studies were carried out with wheat germplasm collection. Here, we report the very first association analysis of KB in the diverse wheat pre-breeding lines derived from exotic x elite crosses and elucidate the genetic components of KB resistance.
Results and Discussion
KB is a quarantine disease (zero percent tolerance) that is indirectly responsible for economic losses in more than 40 countries by affecting the free import-export of wheat grains. Additionally, infected grains are not fit for human and animal consumption17. Thus, the identification and/or development of KB resistance genotypes(s) that minimize the probability of establishment or decrease the inoculum spread rate into newer areas are urgently needed17. The development of resistant cultivars through breeding programs is an appropriate strategy to achieve zero tolerance to the disease.
Evaluation of pre-breeding germplasm for KB resistance
The sporadic incidence of KB in natural hotspots and the role of the environment in disease development make studies of KB resistance more challenging and time-consuming. Screening of wheat germplasm against the pathogen has been attempted by various researchers. Until 1979, screening experiments were conducted at KB hotspot locations. Subsequently, researchers developed an effective screening system, and since then, a large number of wheat germplasm and allied species have been screened under artificial epiphytotic conditions18. The severity of KB incidence in the germplasm of 179 pre-breedling lines from different environments ranged from 1.43–58.34%, whereas infection with KB-SUS (check) ranged from 53.38 to 98.84% (mean = 86.08 ± 12.49; Fig. 1). The mean severity was 26.09 ± 13.76%, indicating a significant difference in KB resistance among the studied genotypes at the genetic level. A summary of the results from the detailed phenotypic analysis is presented in Table 1 and Supplementary Table S1. The frequency distribution of the disease in the population lines is presented in Fig. 2 and indicated a trend towards a normal distribution. The severity of infection ranged from 0 to 69.03% in E-1 and from 0–56.92% in E-2. The mean value for infection with the disease in E-1 (28.26%) was greater than in E-2 (23.91%). The variation in disease resistance in the currently analyzed germplasm suggested that KB resistance was reinforced in different genetic backgrounds of the most appropriate high yielding varieties/genotypes. Therefore, PBLs with KB resistance can be used to create immunity for the disease19. This magnitude of genetic variation in infection indicated that a GWAS would likely reveal the underlying QTLs. During KB screening, breeders select genotypes that exhibit an infection rate less than 5% and exploit them in wheat improvement program. In current study with more stringency of selection, seven PBLs exhibited an infection severity of less than 4.0% at both E-1 and E-2 (Table 2 and Fig. 1). Variable levels of disease incidence for KB have been reported in different germplasm specimens by various researchers; for example, 1 to 48% and 2 to 95% disease infection rates were recorded in spring and durum wheat, respectively20. A previous study reported a percentage of disease incidence ranging from 0.2–63.1% among 150 wheat genotypes21. A significant correlation (0.81**) between KB infection and the year indicated that the disease pressure was substantial in both years and phenotypic data were suitable for the GWAS analysis (Supplementary Table S1).

The severity of KB disease in seven highly resistant genotypes (green bar; 1.43–3.99% infection), seven highly susceptible genotypes (red bar; 53.38–58.34% infection) and KB-SUS check (purple bar; 86% infection). X-axis = name of the genotypes and check and Y-axis = KB infection (%).

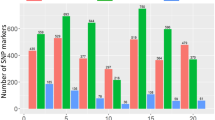
Genome-wide scan (GWAS analysis) of SNP markers associated with Karnal bunt resistance in 179 PBLs in environment 2 (2017–18). The plots show SNP-based Manhattan plots with the names of the most significant SNPs. The chromosomes are shown on the X-axis and the genome-wide scan −log10 (P-values) are shown on the Y-axis.
The analysis of variance suggested that both the genotype and year exerted a significant effect on KB (Supplementary Table S2). Similar results were recorded during QTL mapping for KB resistance in two different studies2,8. The heritability of trait was high in both years (91% and 87% in E-1 and E-2, respectively) (Supplementary Table S3), suggesting that KB resistance can be mapped through QTL mapping and GWAS2,15. The phenotyping protocols established at CENEB, Cd. Obregon and reported in a previous study22 that limit environmental or unexplained variations account for the high heritability observed in the present and previous studies. The disease is highly influenced by environmental conditions, but in current investigation the environmental effect was not significant and high heritability of the disease was observed. The high level of genetic variation and high heritability recorded in the current study will be very useful for selection to genetically improve KB resistance in wheat, and highly resistant genotypes may be shared with partners to exploit them in KB-resistant breeding programs.
Distribution frequency of KB disease in PBLs
The distribution frequency of disease infection was continuous in both environments, suggesting that KB resistance in wheat displays a quantitative inheritance pattern (Supplementary Fig. S2). The continuum of resistance against KB in the germplasm of wheat is well-established in previous studies8,23 and our study corroborates these findings. Moreover, the distribution of KB infections was skewed towards low disease severity, indicating that some major effect-related genes or genes involved in epistatic effects controlled KB resistance in the population investigated.
Distribution of SNP markers, Genetic Diversity and Population Structure
A total 6,382 of high-quality SNPs were obtained from the 179 PBLs and were subjected to genetic diversity, population structure and GWAS analyses. The analysis of the genotype data revealed differences in the distribution of polymorphic DArT markers among the A, B, and D genomes (Supplementary Fig. S3). The B genome contained the greatest number of SNPs (2349, 36.80%), followed by the D (2038, 31.93%) and A genomes (1995, 31.25%). The D genome in wheat contains comparatively fewer polymorphic markers than the A and B genomes due to low recombination attributed to its evolutionary and domestication history. Consistent with previous findings, the B genome contains a greater number of polymorphisms than the A and D genomes, which is attributed to a greater number of effective recombination events in the B genome24,25,26. The average number of SNPs per chromosome was 304, and the value ranged from 147 (4B) to 500 (7D) (Supplementary Fig. S4).
The ratio of the number of SNPs in the B and A genomes was 1.17, 1.15 in the B and D genomes, and 0.98 in the A and D genomes. Based on these ratios, the number of SNPs in the D genome was approximately equal to the number of SNPs in the A genome and only slightly less than the number of SNPs in the B genome. A similar ratio was also recorded between the A and D genomes27. Many researchers have reported a high value for the ratios between D and A or B genomes, but this finding was not corroborated in the current study, indicating the high variability of D genome in PBLs. As described in a previous study, unique genetic variations and wild relative genes can be used to broaden the genetic base of wheat, extend the genetic gain and weaken the genetic bottlenecks associated with the D genome28.
The mean genetic diversity in current sets of germplasm was 0.32 (range 0.035–0.71), which is higher than previous studies examining SNP markers29,30. The high value of genetic diversity may be attributed to the exploitation of diverse exotic parental lines to generate PBLs. The mean genetic diversity value was close to the value reported for the Wheat Association Mapping Initiative (WAMI) population (0.31), which comprises many synthetically derived lines. The PIC (0.28; range 0.01–0.59) of the current study was comparatively higher than values (0.21, 0.16) reported in previous studies31,32.
The population structure can distort the MTA by revealing false positive associations in a GWAS. Therefore, information on the population structure is a prerequisite33. The largest delta K was observed at K = 6, suggesting the presence of six sub-populations (Supplementary Fig. S5). The maximum number of genotypes were grouped in sub-population 3 (83 PBLs) followed by sub-population 5 (44 PBLs). Sub-population 4 included two PBLs, while sub-populations 1, 2 and 6 contained 10, 13 and 27 PBLs, respectively.
Two elite lines (Baj #1 and Super 152) in the parentage dominated sub-population 1, as 80% of the population lines had these parents. Seventy percent of the lines in subpopulation 2 shared three elite parents, Wiblli, Kukuna and Tacupeto F2001, while the remaining lines shared two parents, Baj #1 and Super152. Similarly, sub-populations 3, 4, 5 and 6 were dominated by parents Baj#1, Super152/Villa Juarez F2009, Kachu and Kiritati, respectively. Based on these results, sub-populations largely corresponded to the elite parents in each cross of exotic/elite1/elite2.
Linkage disequilibrium analysis and genome-wide association study of KB resistance
The extent of LD decay in this panel at cut off r2 = 0.1 has been reported to 2.5 cM for whole genome34, which suggests higher genetic diversity of the investigated panel as compared to the wheat panels used in previous studies31,35,36,37. This panel has been drawn from a large set of pre-breeding lines (PBLs) developed by a three way crossing scheme (exotic/elite1//elite2) among exotics and elites38. Each pre-breeding line acquired approximately 25% of the exotic genome and 75% of the elite genome at an early stage thus allowing recombination between exotic and elite genomes. Further, LD decay in the three genomes was at 2.5, 5.0, and 2.5 cM for A, B, and D genomes, respectively. These results indicate a faster LD decay in the D genome, almost comparable to A genome, than reported in previous studies. This might be due to the use of synthetics for developing PBLs, driving more recombination in the D genome.
With MLM (PCA + K), 15 significant SNPs on chromosomes 2D (1 SNPs), 3B (4 SNPs), 4D (7 SNPs), 5B (1 SNP), 6A (1 SNP) and 7B (1 SNP) were identified with P-values <10−3 in the joint analysis (Supplementary Fig. S6) and explained 5.48 to 12.49% of the phenotypic variation (Table 3, Supplementary Table S4). Significant marker-trait associations at the 0.05 FDR threshold in E1, E2, and joint analysis are presented in Table 3. Consistent and common associations were observed on chromosomes 2D, 3B, 4D and 7B based on FDR values. In the current study, the year exerted a significant effect on disease expression and the same trend was also recorded in the analysis of marker-trait associations, as many of SNPs were not shared between the years. In individual years, 13 significant SNPs were detected in E-1 (Fig. 3), while 10 significant SNPs were detected in E-2 (Fig. 2). Among E-1, E-2 and the joint analysis, five SNPs (2D/0.139 cM, 3B/0.562 cM, 4D/1.496 cM, 4D/1.571 cM, and 7B/0.952 cM) were confirmed to be common and explained between 7.5 to 12.49% of the variation (Table 3).

Genome-wide scan (GWAS analysis) of SNP markers associated with Karnal bunt resistance in 179 PBLs in environment 1 (2016–17). Plots show SNP-based Manhattan plot with the names of the most significant SNPs. The chromosomes are shown on the X-axis and the genome-wide scan −log10 (P-values) are shown on the Y-axis.
Previous studies reported the presence of KB resistance loci scattered on many chromosomes in all three genomes of wheat15,16. Hence, cultivar pyramiding/stacking of favourable alleles from multiple loci is a prerequisite to develop KB resistance22.
The results of the current study validated the consensus region on chromosome 3B reported in previous studies2,15 (Fig. 4). Likewise, QTLs for KB resistance on chromosomes 5B and 2D8,15 were also reported previously in RIL populations and a GWAS panel, respectively. A marker-trait association was also detected on chromosome 7B, which might be the same locus that was previously reported23. Interestingly, seven SNPs were detected on chromosome 4D. Two of these SNPs had maximum R2 values: SNP (1114200 | F | 0–63:T > C) at position 1.571 cM with an R2 of 12.49% and SNP (1103052 | F | 0–61:C > A) at position 1.574 cM with an R2 of 9.02%. The physical positions of SNPs, sequence reads of the SNPs were blasted to the reference genome of RefSeq V.1.0 with E value <10−8 and identity >95%38. These two SNPs were in strong LD with each other, confirming that they are part of the same genomic region on chromosome 4D (Supplementary Fig. S7). No previous studies have reported loci for KB resistance on chromosome 4D, suggesting that the D genome should be targeted for an in-depth investigation to map new KB resistant loci.

Effect of alleles of significant SNPs (common in three datasets) on KB severity (green bar = favorable allele providing resistance; red bar = unfavorable allele; yellow bar = % increase in resistance if favorable allele introgressed). X-axis = name of SNPs and Y-axis = infection level (%).
The donor of the D genome, i.e., Aegilops tauschii, in hexaploid wheat has been a source of useful genes, for example, rust resistance genes23. In the current study, two KB resistant loci were located on the D genome (chromosomes 2D and 4D) (Fig. 4). The wider genetic variation derived from synthetics and the diverse genetic make-up of PBLs obtained from populations used in previous studies may be the probable explanation for the identification of novel KB resistance loci on the D genome.
Chromosome 3B appears to be a hot spot for various disease resistance genes, as it is the location of QTLs for KB resistance24 and resistance to tan spot39, yellow rust39, leaf rust24 and crown rot26. Genomic regions inherited as multi-disease resistance loci are not uncommon in wheat39. Therefore, researchers should focus on chromosome 3B for future introgression breeding to create cultivars with simultaneous resistance to multiple diseases. Likewise, the current GWAS panel may be screened for pathogens causing devastating diseases, such as a blast to identify major effect QTLs and/or to reveal genomic hot spot(s) for multi-disease resistance.
During the detection of highly tolerant PBLs based on phenotypic data, a set of 7 lines were confirmed to be resistant (Table 2). An analysis of the pedigree of these 6 PBLs revealed that Aegilops species are one parent used during the creation of synthetic wheat and contributed the D genome to these resistant PBLs. The analysis of these same lines for the SNP 1114200 marker (chromosome 4D) indicated that 6 of 7 PBLs contained the ‘T’ allele (favorable allele contributing to resistance). The role of the D genome in KB resistance suggested that the D genome must be further enriched with additional polymorphic SNPs to exploit more QTLs for various diseases viewed as upcoming threats to wheat cultivation.
During the study, SNPs on chromosome 4D were not associated with the Rht-D1 dwarfing locus, as the frequency of the dwarf allele was 0.1% (2 of 179 PBLs exhibited wild allele, i.e., G), indicating that the Rht-D1 gene was fixed in the current KB panel of PBLs and the KB resistance locus on chromosome 4D is novel (Supplementary Table S5).
Significance of chromosome 4D
Seven SNP markers on chromosome 4D were associated with KB infection in the joint analysis (Table 3). Noticeably, all but one SNP were present in strong LD (Supplementary Fig. S7). We divided the panel into two groups. Group 1 (carrying all favourable alleles of 7 markers) contained 109 PBLs and group 2 (carrying unfavourable alleles for any of the 7 markers) contained 70 PBLs. The mean KB% was 12.16% higher than in group 1. This comparison further confirms that the allelic profile of group 1 PBLs is associated with resistance to KB (Supplementary Fig. S8). However, an in-depth analysis of a set of diverse exotic and elite accessions should improve our understanding of this genomic region on chromosome 4D.
The identified markers were subjected to BLAST search in Ensembl Plants server (https://plants.ensembl.org/Triticum_aestivum/Info/Index) to obtain additional insights into the genes on chromosome 4D that are involved in KB resistance. Of the 8 associated SNPs located on chromosome 4D, three showed direct hits with candidate genes and three were located in the vicinity (2 Mb) (Supplementary Table S6). Of the three direct hits, two hits with markers 1103052 | F | 0–61:C > A (TraesCS4D02G352200) and 1101835 | F | 0–5:C > A (TraesCS4D02G350300) were interesting, belonging to RBOHB and Rhomboid-like protein gene families40,41. The gene TraesCS4D02G352200 showed the closest relationship to the AET4Gv20828800 gene in Aegilops tauschii (Supplementary Fig. S9). One of the molecular functions of TraesCS4D02G352200 is NAD(P)H oxidase activity. As key producers of reactive oxygen species, NADPH oxidases (NOXs), which are also known as respiratory burst oxidase homologs (RBOHs), play crucial roles in various biological processes in plants. Two TaNOX genes have been reported to confer resistance to brown rust infection40. To date, 15 TaNOXs have been reported to be co-expressed with different sets of other genes that participate in several critical intracellular processes such as cell wall biosynthesis, defence response, and signal transduction, suggesting vital but diverse roles for these genes in regulating plant growth and stress responses in wheat43. Two of these genes (TaNOX8 and TaNOX9) were mapped to chromosome 4DL. TaNOX8 is specifically present on the membrane and is co-expressed with TaNOX13AL on chromosome 5AL. Thus, we speculate that the marker 1103052 | F | 0–61:C > A might represent the TaNOX8 gene. The other candidate gene, TraesCS4D02G350300, is a Rhomboid-like protein, which belongs to a family of serine proteases (family S54) that cleave substrates within transmembrane domains44. Rhomboid protease family member S54 has proven to be active during fungal-plant interactions45.
Furthermore, these two markers, 1103052 | F | 0–61:C > A and 1101835 | F | 0–5:C > A, on chromosome 4D were significantly involved in epistatic interactions with other loci on chromosome 4D and with the associated locus on chromosome 7B (1072740 | F | 0–54:G > C) (Fig. 5), contributing an additional 8% to the variation. Therefore, KB resistance is controlled by minor additive and epistatic loci. Previous studies using bi-parental populations and a GWAS panel have reported additive QTLs for KB resistance2,15,16, but information on epistatic QTL is lacking. This report is the first to identify additive and epistatic QTL for KB resistance in a GWAS panel. A similar level of interaction was also reported for APR loci for stem rust in wheat46.

Epistatic interactions among loci associated with KB resistance in wheat. The magnitude of the marker effect (F value) is represented in shades of blue (dark blue indicates a stronger interaction). The magnitude of the epistatic interaction is presented with colors ranging from yellow (stronger interaction) to red (weaker interaction).
Materials and methods
Plant material
The plant material used in the current investigation comprised 179 pre-breeding lines (PBLs). A detailed description of the development of these PBLs has been reported in Singh et al.38. Briefly, a set of 179 outstanding PBLs were selected from a larger set of 984 TC1F5:6 based on their tolerance to biotic and abiotic stresses, namely, heat, drought, yellow rust (Puccinia striiformis f. sp. tritici), and powdery mildew (Blumeria graminis f. sp. tritici (Bgt)), character and yield performance in Mexico. The pedigree information for these lines is presented in Supplementary Table S7. KB-SUS was used as a susceptible line and grown around all of the experimental blocks.
Phenotyping PBLs for Karnal bunt disease through artificial inoculation methods
The phenotypic screening was conducted at Ciudad Obregon, Mexico over two years (2016–17 was referred to as E-1 and 2017–18 was referred to as E-2) using a randomized complete block design (RCBD). For the preparation of the inoculation, one-year-old teliospores were scraped off infected grains with a dissecting needle and incubated in a water-Tween 20 solution for 24 h; then, the suspension was filtered through a 60 µM nylon sieve and centrifuged at 3000 rpm. After discarding the supernatant, 0.5% sodium hypochlorite was applied as the active ingredient for 2 min to disinfect teliospores and the suspension was centrifuged again. Teliospores were then rinsed twice with sterile distilled water, followed by centrifugation. Teliospores were resuspended in sterile distilled water in the centrifuge tube. One milliliter of the teliospore suspension was spread on Petri dishes containing 2% water-agar (AA) and incubated at 18–22 °C in the dark. After 6 to 9 days, teliospore germination was evaluated using a compound microscope at 10x magnification. Pieces of AA with germinated teliospores were removed and placed upside down on the lid of Petri dishes containing potato-dextrose-agar (PDA). After 10 to 14 days, 2 to 3 mL of sterile distilled water was added to the plates and the colonies were scraped gently using a sterile spatula. Hyphae and sporidia were inoculated onto other plates with PDA using a sterile syringe, and the plates were incubated at 18–22 °C in the dark for approximately nine days. After the incubation, pieces of PDA with the different fungal propagules were transferred and placed upside down on the lids of sterile glass Petri dishes to induce the production of allantoid secondary sporidia47,48. Three milliliters of sterile distilled water were added to the bottom of the plates.
Water was collected from the plates every 24 h; secondary allantoid sporidia were collected and counted using a hemocytometer. Then, the concentration was adjusted to 10,000 sporidia per mL. Five heads of each experimental line at the boot stage were inoculated by injecting 1 mL of the allantoid sporidial suspension during the boot stage. Stems of the inoculated heads were identified with a piece of red plastic. An automatic mist irrigation system was used to water the plants during the period of inoculation (January-March) for 20 min five times per day, and the area was covered with nets to prevent bird damage. Grains were harvested manually, and the healthy and infected grains were counted by a visual inspection to calculate the percentage of infection (infected grains). Standard agronomic practices were used during the crop season. The mean disease incidence (%) was estimated from five inoculated spikes per plot. The phenotyping for KB screening was based on ratings reported as percentages to identify tolerant and susceptible genotypes2.
The mean for each PBL was calculated to reduce the environmental variance in the data recorded through the partitioning of phenotypic variance using the maximum residual likelihood (ReML) algorithm. The KB infection data were subjected to an ANOVA to reveal the effects of the genotype, year and their interaction.
High-throughput genotyping
A modified CTAB (cetyltrimethylammonium bromide) method was used to extract genomic DNA from leaf samples49. Leaves were collected from each PBL (TC1F5) 25 days after germination. The extracted DNA was quantified with a NanoDrop 8000 spectrophotometer V 2.1.0, while intactness was analyzed using agarose gel electrophoresis. The DNA concentration was adjusted to 50–100 ng µL−1 in a volume of 30 µL. Genotypic characterization used DArTseq technology (http://www.diversityarrays.com/dart-application-dartseq) at the Genetic Analysis Service for Agriculture(SAGA) service unit at CIMMYT headquarters (Texcoco, Mexico). The main parameters to select markers were call rate (the proportion of samples with genotypic score and not recorded as missing data) and average reproducibility (the proportion of technical replicate assay pairs for which the marker score was consistent). More details on SNP filtering are given in Ledesma-Ramírez34. A total of 12,071 SNP markers belonging to 10,111 sequence tags were identified based on above criteria. Chromosome location, marker order and genetic distances were defined based on a 64,000-marker DArT-seq consensus map released by Diversity Arrays Technology Pty Ltd. (DArT) (http://www.diversityarrays.com/sequence-maps). The methodology described by Vikram et al.49 was followed to capture 6,382 high quality SNP markers on 179 PBLs. These high-quality DArTseq SNPs were used for the population structure and association analyses (Supplementary Table S8).
Population Structure and Linkage Disequilibrium (LD) Analysis
The structure was analyzed using K-values ranging from 1 to 8 for the entire population with 6,382 SNPs markers with STRUCTURE software51. Five independent analyses were performed for each K-value, with 100,000 set as the length of the burn-in time and number of Markov Chain Monte Carlo replications. The correct estimation of K was provided by an ad hoc statistic deltaK52 that was calculated using the program STRUCTURE HARVESTER53. The linkage disequilibrium analysis was conducted on this panel using GAPIT version 2.034. Birefly, a squared correlation coefficient r2 was estimated for all pairwise comparisons. Pattern of LD decay was then visualized by plotting pair-wise r2 values against the genetic distance for A, B, and D genomes separately and for whole genome. For the LOESS estimation of LD decay, genetic distance was estimated as the point where the LOESS curve first crosses the baseline r2 of 0.1.
Association analysis
Phenotypic data from two years were analyzed separately and combined for the association analysis. TASSEL software v5.0154 was used to calculate associations between the markers and KB score with the mixed linear model (MLM)55. MLM takes advantage of the population structure (Q) and kinship (K) matrix as covariates during a GWAS to avoid spurious associations56. Both of these matrices were also generated using TASSEL 5.01. To correct for multiple testing, the step up procedure of Benjamini and Hochberg29 which controls the false discovery rate was used with a cut-off value of 0.05. ANOVA analysisand estimates ofcorrelations and heritability were conducted in SAS 9.4. The KB scoing data of both environments separately and mean over the environments (Supplementary Table S9) was used for the association mapping.
Epistatic interactions
Two- and three-locus interactions among associated loci were studied using an in-house script executed in R, as described previously31,42,50. A significant threshold of P < 0.001 represented significant marker-marker interactions.
In silico analysis
In silico analysis of the significant loci was conducted in Ensembl Plants sequence database (https://plants.ensembl.org/Triticum_aestivum/Info/Index) by using BLASTN algorithm with functional annotations from IWGSC v1.1 RefSeq annotation. Precisely, to find the candidate genes, the physical starting point of the marker along with chromosome name was put in Ensembl Plants and around 300 bp were added before and after the SNP. For some markers, SNPs fell within gene sequences and hence were classified as direct gene hits in Supplementary Table S6. However, for markers where SNPs did not fall within a gene, potential candidate genes were picked 2 Mb upstream and downstream of the SNPs which were related to pathogenic process or known to regulate induction of genes related to pathogenesis. Sometimes annotations were not available in Triticum aestivum genome and for such cases orthologous genes in related species were screened with known predicted functions using the comparative genomics tool in Ensembl.
Conclusions
In the present study, an association mapping panel consisting of 179 PBLs genotyped with 6382 high-quality DArTseq SNP markers was utilized to obtain an understanding of the genetics of Karnal bunt resistance in wheat (Supplementary Fig. S10). Fifteen significant SNPs were identified on chromosomes 2D, 3B, 4D and 7B in a joint analysis of data collected from 2 years. This report is the first to describe QTLs/genes associated with KB resistance on chromosome 4D. The SNPs associated with KB resistance might be exploited through marker-assisted selection to facilitate the breeding of resistant wheat. Eventually, PBLs carrying more favourable alleles coupled with superior agronomic performance could be utilized as excellent parental materials to produce improved wheat lines that resist this stress. These results provide new information for further research aiming to improve KB resistance in common wheat. Moreover, the 7 best identified resistant lines have become part of the breeding programs of many Asian countries (India and Pakistan) where KB is a major problem.
References
Bonde, M. R., Peterson, G. L., Schaad, N. W. & Smilanick, J. L. Karnal bunt of wheat. Plant. Dis. 81, 1370–1377 (1997).
Brar, G. S. et al. Genetic mapping of resistance in hexaploid wheat for a quarantine disease: Karnal Bunt. Front. Plant. Sci. 9, 1497 (2018).
Chunneja, P., Kaur, S., Singh, K. & Dhaliwal, S. Evaluation of Aegilops tauschii (Coss.) germplasm for Karnal bunt resistance in a screen house with simulated environmental conditions. Plant. Genet. Res. 6, 79–84 (2008).
Kumar, A., Singh, U. S., Kumar, J. & Garg, G. K. Application of molecular and immune diagnostic tools for detection, surveillance and quarantine regulation of Karnal bunt (Tilletia indica) of wheat. Food Agr. Immunol. 19, 293–311 (2008).
Stein, J., Maples, H. & Rush, C. Epidemiology of Tilletia indica teliospores in regulated wheat fields in Texas. Plant. Dis. 89, 828–833 (2005).
Shukla, D., Srivastava, J. & Tiwari, P. Efficacy of fungicides for the management of Karnal bunt of wheat. J Pharmacog Phytochem. 7, 3941–3942 (2018).
Carris, L. M., Castlebury, L. A. & Goates, B. J. Nonsystemic bunt fungi—Tilletia indica and T. horrida: a review of history, systematics, and biology. Annu. Rev. Phytopathol. 44, 113–133 (2006).
Singh, S. et al. Molecular mapping of QTLs for Karnal bunt resistance in two recombinant inbred populations of bread wheat. Theor. Appl. Genet. 116, 147–154 (2007).
Zhu, C., Gore, M., Buckler, E. S. & Yu, J. Status and prospects of association mapping in plants. The Plant. Genome. 1, 5–20 (2008).
Jannink, J. L., Mink, M. C. & Jansen, R. C. Using complex pedigrees to map valuable genes. Trends Plant. Sci. 6, 337–342 (2001).
Aranzana, M. J. Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet. 1(5), e60 (2005).
Agrama, H., Eizenga, G. & Yan, W. Association mapping of yield and its components in rice cultivars. Mol. Breed. 19, 341–356 (2007).
Kraakman, A., Martinez, F., Mussiraliev, B., Euwijk, V. & Niks, R. Linkage disequilibrium mapping of morphological, resistance, and other agronomically relevant traits in modern spring barley cultivars. Mol. Breed. 17, 41–58 (2006).
Neumann, K., Kobiljski, B., Dencic, S., Varshney, R. & Boerner, A. Genome-wide association mapping: a case study in bread wheat (Triticum aestivum L.). Mol. Breed. 27, 37–58 (2011).
Emebiri L., et al Unravelling the complex genetics of Karnal bunt (Tilletia indica) resistance in common wheat (Triticum aestivum) by genetic linkage and genome-wide association analyses. G3-Genes Genome Genet., g3–400103, (2019).
Gupta, V. et al. Genome wide association study of karnal bunt resistance in a wheat germplasm collection from Afghanistan. Int. J. Mol. Sci. 20, 3124 (2019).
Rush, C. M. Status of Karnal bunt of wheat in the United States 1996 to 2004. Plant. Dis. 89, 212–223 (2005).
Aujla S., Grewal A. & Sharms I. Artificial creation of Karnal bunt disease of wheat. Cereal Res Commun., 171–176 (1982).
Singh et al. The emergence of Ug99 races of the stem rust fungus is a threat to world wheat production. Ann Review. Phytopathology 49, 465–481 (2011).
Riccioni, L. et al. Susceptibility of European bread and durum wheat cultivars to Tilletia indica. Plant. Pathol. 57, 612–622 (2008).
Kumar, S., Singh, D. & Pandey, V. K. Research Note Field screening of wheat genotypes against Karnal bunt caused by Neovossia indica (Mitra) Mund. Electron. J. Plant. Breed. 5, 602–604 (2014).
Sharma, I., Bains, N. & Nanda, G. Inheritance of Karnal bunt-free trait in bread wheat. Plant. Breed. 123, 96–97 (2004).
Singh, S. et al. Mapping of a resistance gene effective against Karnal bunt pathogen of wheat. Theor. Appl. Genet. 106, 287–292 (2003).
Alipour, H. et al. Genotyping-by-sequencing (GBS) revealed molecular genetic diversity of Iranian wheat landraces and cultivars. Front. Plant. Sci. 8, 1293 (2017).
Eltaher, S. et al. Genetic Diversity and Population Structure of F3:6 Nebraska Winter Wheat Genotypes Using Genotyping-By-Sequencing. Front. Genet. 9, 76 (2018).
Erginbas-Orakci, G. et al. Identification of novel Quantitative Trait Loci linked to crown rot resistance in spring wheat. Intl J. Mol. Sci. 19, 2666 (2018).
Bhatta, M., Morgounov, A., Belamkar, V., Poland, J. & Baenziger, P. S. Unlocking the novel genetic diversity and population structure of synthetic hexaploid wheat. BMC Genomics. 19, 591 (2018).
Ogbonnaya, F. C. et al. Synthetic hexaploids: harnessing species of the primary gene pool for wheat improvement. Plant. Breed. Rev. 37, 35–122 (2013).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Wang, S. et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant. Biotech. J. 12, 787–796 (2014).
Sehgal, D. et al. Identification of genomic regions for grain yield and yield stability and their epistatic interactions. Sci. Rep. 7, 41578 (2017).
Chao, S. et al. Population-and genome-specific patterns of linkage disequilibrium and SNP variation in spring and winter wheat (Triticum aestivum L.). BMC Genomics. 11, 727 (2010).
Qaseem, M. F. et al. Genome-wide association mapping in bread wheat subjected to independent and combined high temperature and drought stress. PLoS one. 13, 0199121 (2018).
Ledesma-Ramírez, L. et al. GWAS to Identify Genetic Loci for Resistance to Yellow Rust in Wheat Pre-Breeding Lines Derived From Diverse Exotic Crosses. Front. Plant. Sci. 10, 1390 (2019).
Würschum, T. et al. Population structure, genetic diversity and linkage disequilibrium in elite winter wheat assessed with SNP and SSR markers. Theor. Appl. Genet. 126, 1477–1486 (2013).
Edae, E. A., Byme, P. F., Haley, S. D., Lopes, M. S. & Reynolds, M. P. Genome-wide association mapping of yield and yield components of spring wheat under contrasting moisture regimes. Theor. Appl. Genet. 127, 791–807 (2014).
Zegeye, H., Rasheed, A., Makdis, F., Badebo, A. & Ogbonnaya, F. C. Genome-wide association mapping for seedling and adult plant resistance to stripe rust in synthetic hexaploid wheat. PLoS One 9(8), e105593 (2014).
Singh, S. et al. Harnessing genetic potential of wheat germplasm banks through impact-oriented- pre-breeding for future food and nutritional security. Sci. Rep. 8, 12527 (2018).
Singh, S. et al. Multi-trait and multi-environment QTL analyses for resistance to wheat diseases. PLoS One 7(6), e38008 (2012).
Fonseca-García, C. et al. Transcriptome analysis of the differential effect of the NADPH oxidase gene RbohB in Phaseolus vulgaris roots following Rhizobium tropici and Rhizophagus irregularis inoculation. BMC Genomics 20, 1–18. (2019).
García-Lorenzo, M., Sjödin, A., Jansson, S. & Funk, C. Protease gene families in Populus and Arabidopsis. BMC Plant. Biol. 6, 30 (2006).
Dmochowska-Boguta, M., Nadolska-Orczyk, A. & Orczyk, W. Roles of peroxidases and NADPH oxidases in the oxidative response of wheat (Triticum aestivum) to brown rust (Puccinia triticina) infection. Plant. Pathol. 62, 993–1002 (2013).
Hu C.H., et al. Genome-wide identification and functional analysis of NADPH oxidase family genes in wheat during development and environmental stress responses. Front Plant. Sci. 9, 906 (2018).
Tripathi, R. & Snowdhamini, R. Cross genome comparisons of serine proteases in Arabidopsis and rice. BMC Genomics. 7, 200 (2006).
Larriba, E. et al. Sequencing and functional analysis of the genome of a nematode egg-parasitic fungus, Pochonia chlamydosporia. Fungal Genet. Biol. 65, 69–80 (2014).
Yu, L. X. et al. Identification of Ug99 stem rust resistance loci in winter wheat germplasm using genome-wide association analysis. Theor. Appl. Genet. 125, 749–758 (2012).
Dhaliwal, D. & Singh, D. V. Up-to-date life cycle of Neovossia indica (Mitra) Mundkur. Curr. Sci. 57, 675–677 (1988).
Fuentes-Dávila, G. et al. Resultados de inoculaciones artificiales del 5◦ vivero de selección para Resistencia a Tilletia indica Mitra. Sci. Fung 3, 57–65 (1993).
Vikram, P. et al. Unlocking the genetic diversity of Creole wheats. Sci. Rep. 6, 23092 (2016).
Sansaloni, C. et al. Diversity Arrays Technology (DArT) and next generation sequencing combined: genome wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proceed 26(Suppl 7), 54 (2011).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics. 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. & VonHoldt, B. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4(2), 359–361 (2012).
Bradbury, P. J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 23, 2633–2635 (2007).
Yu, J. et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genetics. 38, 203 (2006).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature. 38, 904 (2006).
Acknowledgements
This study was implemented by CIMMYT as part of the Seeds of Discovery, Sustainable Modernization of Traditional Agriculture (Mas-Agro), in collaboration with INIFAP, and made possible by the generous support of the Secretariat of Agriculture and Rural Development (SADER), Mexico. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the institutions.
Author information
Authors and Affiliations
Contributions
Conceived and designed the study: S.S. Performed the study: S.S., P.V. and G.F. Analyzed the data: S.S., S.K., D.S., M.A.R.A. and P.V. Contributed materials, genotyping/analysis tools: S.S., C.S., P.V. and C.O. Interpretation of results: S.S., S.K., D.S., P.V. and M.A.R.A. Wrote first draft of the paper: S.S. and S.K. Revised the manuscript: S.S., D.S., P.V., S.K. and M.A.R.A.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, S., Sehgal, D., Kumar, S. et al. GWAS revealed a novel resistance locus on chromosome 4D for the quarantine disease Karnal bunt in diverse wheat pre-breeding germplasm. Sci Rep 10, 5999 (2020). https://doi.org/10.1038/s41598-020-62711-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62711-7
This article is cited by
-
Understanding the Rice Fungal Pathogen Tilletia horrida from Multiple Perspectives
Rice (2022)
-
Meta-analysis reveals consensus genomic regions associated with multiple disease resistance in wheat (Triticum aestivum L.)
Molecular Breeding (2022)
-
Comprehensive evaluation of mapping complex traits in wheat using genome-wide association studies
Molecular Breeding (2022)
-
Direct introgression of untapped diversity into elite wheat lines
Nature Food (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}