
Abstract
Water level fluctuations are an inherent feature regulating the ecological structures and functions of lakes. It is vital to understand the effects of water level fluctuations on bacterial communities and metabolic characteristics in freshwater lakes in a changing world. However, information on the microbial community structure and functional properties in permanently and seasonally flooded areas are lacking. Poyang Lake is a typical seasonal lake linked to the Yangtze River and is significantly affected by water level fluctuations. Bottom water was collected from 12 sampling sites: seven inundated for the whole year (inundated areas) and five drained during the dry season (emerged areas). High-throughput 16S rRNA gene sequencing was used to identify the bacterial communities. The results showed that the taxonomic structure and potential functions of the bacterial communities were significantly different between the inundated and emerged areas. Cyanobacteria was dominant in both areas, but the relative abundance of Cyanobacteria was much higher in the emerged areas than in the inundated areas. Bacterial communities were taxonomically sensitive in the inundated areas and functionally sensitive in the emerged areas. Nitrogen, phosphorus, and dissolved organic carbon concentrations and their ratios, as well as dissolved oxygen, played important roles in promoting the bacterial taxonomic and functional compositional patterns in both areas. According to the metabolic predictions based on 16S rRNA gene sequences, the relative abundance of functional genes related to assimilatory nitrate reduction in the emerged areas was higher than in the inundated areas, and the relative abundance of functional genes related to dissimilatory nitrate reduction in the inundated areas was higher. These differences might have been caused by the nitrogen differences between the permanently and seasonally flooded areas caused by intra-annual water level fluctuations. The relative abundance of functional genes associated with denitrification was not significantly different in the inundated and emerged areas. This study improved our knowledge of bacterial community structure and nitrogen metabolic processes in permanently and seasonally flooded areas caused by water level fluctuations in a seasonal lake.
Similar content being viewed by others

Introduction
Microbial assemblages are fundamental components of aquatic ecosystems and play important roles in driving global energy fluxes and biogeochemical cycling1. Microbial communities have high species diversity and genetic diversity2,3,4,5, which make them sensitive to environmental perturbations6,7. The relative balance of external nutrient loading influences microbial functional genes and associated metabolic processes in lake ecosystems8. Understanding the taxonomic and functional compositions of microbial communities is essential to elucidating the responses of natural bacterial communities to environmental perturbations, such as hydrological fluctuations and nutrient pollution9,10,11.
Poyang Lake (PYL) is a seasonal lake characterized by recurrent wet-dry phases12,13,14. Precipitation, evaporation, catchment inflows, and the Three Gorges Dam control the hydrological processes of PYL by regulating its water level15, which plays a crucial role in determining hydrodynamic processes16. In PYL, some low-lying areas (inundated areas) are inundated with water throughout the whole year, while some higher areas (emerged areas) are only inundated for a few weeks or months during the wet season17,18. During the dry season from October to March, the water level decreases and emerged areas dry. The water level increases in the wet season and the whole lake area is inundated13,14. During the wet season, some dissolved and particulate organic matter and nutrients are released from the sediment, affecting the chemical characteristics of the water bodies19. Previous studies have suggested that water level fluctuations could regulate the transformation of nutrients by changing bacterial activities in active erosion and transport zones17,20,21,22,23,24. In seasonal lakes, drying and inundation cycles within a year can influence nitrogen cycling by regulating nitrogen metabolism pathways25,26,27, such as anammox, nitrification, denitrification, and nitrogen fixation28,29,30.
Determining metabolic functions related to the nitrogen cycle of the microbial communities in seasonal lakes is essential for understanding their roles in biogeochemical processes related to nutrient cycles2,3,31,32 and their response to water level fluctuations24,27,33,34. Previous study indicated that bacterial communities are distinct taxonomically and functionally in the dry-season and wet-season in Poyang Lake24. Based on the previous study, we hypothesize that changing the water level may also affect the bacterial community composition in permanently and seasonally flooded areas in PYL. In this study, we compared the taxonomic composition and predicted functional traits (especially nitrogen metabolism pathways) of bacterial communities between permanently (inundated areas) and seasonally (emerged areas) flooded areas and explored their relationships with variations in environmental factors. Our aim was to study the differences in bacterial community structure and potential functions between permanently and seasonally flooded areas caused by water level fluctuations in PYL.
Results
Environmental variables
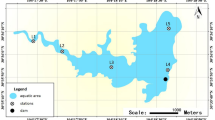
The Environmental Fluid Dynamics Code (EFDC) was applied to simulate daily water depths at each sampling site in 2016, and the results are visualized in Fig. 1b,c. According to daily water depths, the sampling sites were divided into two groups: inundated areas (PY01, PY02, PY04, PY05, PY06, PY07, and PY10) and emerged areas (PY03, PY08, PY09, PY11, and PY12) (Fig. 1). Turbidity, total phosphorus (TP), phosphate (PO4−), and ammonium (NH4+) were significantly higher in the emerged areas than in the inundated areas (p < 0.05), while the C/P and N/P ratios were significantly higher in the inundated areas than in the emerged areas (Table 1, t-test, p < 0.05). Dissolved organic carbon (DOC) was not significantly different between the two areas (p > 0.05).

(a) Study area, (b) drought days at the sampling sites, and (c) daily water depth at the sampling sites in 2016.
Bacterial community structure
According to t-tests, the relative abundances of the phyla differed dramatically between the inundated and emerged areas (Fig. 2). The OTUs shared by the two groups were 61.6% of the total number of OTUs. The unique OTUs of the inundated and emerged areas was 17.23% and 21.17% of the total number of OTUs, respectively. In the inundated and emerged areas, the dominant phylum was Cyanobacteria (50.52% and 61.54%), followed by Actinobacteria (14.25% and 14.27%) and Proteobacteria (13.94% and 9.60%, respectively). The relative abundance of Cyanobacteria in the emerged areas was significantly higher than in the inundated areas (p < 0.05), while the relative abundances of Proteobacteria and Planctomycetes were significantly higher in the inundated areas (p < 0.05).

Relative abundances of bacterial phyla in the inundated areas and emerged areas. Only phyla with a relative abundance >1% in inundated and emerged areas are shown; “others” represent the unassigned operational taxonomic units (OTUs) and the phyla with a relative abundance <1%. The comparison of bacterial community composition in inundated and emerged areas was assessed using t-tests. “**” indicates levels of statistical significance at p < 0.01, and “*” indicates p < 0.05.
Heatmaps and analysis of similarity (ANOSIM) were conducted to reveal the differences of taxonomic composition and potential metabolic functions between bacterial communities in the two areas. A heatmap showed that the bacterial communities in the inundated areas were clustered apart from the communities in the emerged areas according to taxonomic composition, while there were no significant differences of potential metabolic functions between the two areas (Fig. S1). ANOSIM analysis showed that bacterial communities were significantly different in taxonomic composition between the two groups (r = 0.252, p < 0.05), while the potential functions were not significantly different (r = 0.087, p > 0.05). The differences in the taxonomic and functional alpha diversities were not significant between the two groups (Table S1, t-test, p > 0.05).
Bacterial co-occurrence
We constructed two networks based on samples from the inundated and emerged areas (Fig. 3) and calculated nine topological parameters to assess the interactions between the OTUs in the two networks (Table 2). The bacterial network for the inundated areas contained 500 nodes and 4218 edges (Fig. 3a), and the network for the emerged areas contained 477 nodes and 4158 edges (Fig. 3b). The proportions of positive OTUs correlations in the bacterial networks of the inundated and emerged areas were 97.7% and 98.20%, respectively. Network diameter, network centralization, network heterogeneity, and characteristic path length values were significantly higher in the emerged areas than in the inundated areas. Compared with the emerged areas, the network density, clustering coefficient, and modularity values were much higher in the inundated areas. Microbial assemblages in the two groups exhibited modular structures, and the modularity was notably higher in the inundated areas than in the emerged areas. Nodes with high degrees, high closeness centrality, and low betweenness centrality were considered keystone taxa. In the inundated areas, 40% of the top 10 keystone taxa were Proteobacteria and 20% were Cyanobacteria. In the emerged areas, Proteobacteria and Cyanobacteria represented 40% and 50% of the top 10 keystone taxa, respectively.

Co-occurrence networks of bacterial communities in the (a) emerged and (b) inundated areas. Modularity networks of (a1) emerged and (b1) inundated areas are shown. Circular nodes represent OTUs with a relative abundance higher than 0.01%. Edges represent Spearman’s correlations (Spearman’s r > 0.9 or r < −0.9, p < 0.05). The grey and blue lines indicate positive and negative correlations, respectively.
Potential functional properties
The Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was used to predict the potential functions of the bacterial communities based on 16S rRNA sequences. According to the KEGG database, the potential metabolic functions were classified into Genes and Genomes orthologies (KOs) at three different pathway levels (Fig. S2). The nearest sequenced taxon index (NSTI) was determined for each sample to assess the accuracy of the functional prediction. The mean value of NSTI was 0.09 + 0.02 for all samples, indicating the high accuracy of the predicted metabolic functions in our study. To investigate the differences between the metabolic pathways related to core resources, genes associated with energy metabolism, amino acid metabolism, and carbohydrate metabolism (at level-3) were compared. The relative abundances of the functional genes associated with nitrogen metabolism were higher in the emerged areas than in the inundated areas.
The correlations between the dissimilarities of the potential functions, the taxonomic composition of the bacterial community, and environmental factors were assessed by Mantel tests. There was a significantly positive relationship between potential functional genes’ dissimilarity and environmental distance in the emerged areas (Fig. 4a, r = 0.401, p < 0.05), while it was not significant in the inundated areas (Fig. 4b, r = 0.026, p > 0.05). Mantel tests revealed a significantly positive relationship between potential functional genes’ dissimilarity and taxonomic dissimilarity in the inundated areas (Fig. 4d, r = 0.688, p < 0.05), while was not significant in the emerged areas (Fig. 4c, r = 0.412, p > 0.05). Redundancy analysis (RDA) was applied to analyze the relationships between bacterial community distributions and environmental factors (Fig. 5). The first two axes accounted for 56.24% of the variance (RDA 1: 31.43%; RDA 2: 24.81%). According to the Monte Carlo test (p < 0.05), conductivity (Cond), dissolved oxygen (DO), water depth (WD), DOC, NH4+, TP, PO4−, C/N, C/P, and N/P were associated with bacterial community distribution of the two groups.

Mantel tests assessing the relationships between functional gene dissimilarity matrices based on Bray-Curtis distance and environmental distance matrices based on Euclidean distance in (a) emerged and (b) inundated areas and the relationships between functional and taxonomic composition of the bacterial community dissimilarity in (c) emerged and (d) inundated areas. One point represents one sample pair. The Pearson correlation coefficient (r) and statistical significance (p) of linear regression are shown. Blue dotted lines denote the 95% confidence interval.

Redundancy analysis plots revealing the association of microbial communities and environmental factors. Only environmental factors that were significantly correlated with the microbial communities (Monte Carlo test, p < 0.05) are shown as solid black lines.
Potential nitrogen metabolism
We analyzed the relative abundance of the potential functional genes encoding pathways related to nitrogen cycling. The relative abundance of potential functional genes associated with assimilatory nitrate reduction (ANR) was significantly higher in the emerged areas than in the inundated areas, while the relative abundance of potential functional genes associated with dissimilatory nitrate reduction (DNR) was higher in the inundated areas (Fig. 6). We conducted Spearman correlations between the relative abundance of potential functional genes related to nitrogen metabolism pathways and environmental factors (Table 3). In the emerged areas, ANR was negatively correlated with pH and NO3− but positively correlated with turbidity and NH4+ (p < 0.05). DNR was positively correlated with pH, TN, and NO3− (p < 0.05). Potential denitrification was positively associated with TN, NO3−, and DOC. Potential nitrification was negatively correlated with turbidity. Moreover, potential anammox was negatively linked with TP (p < 0.05). In the inundated areas, DNR was positively correlated with TN, NO3− and DOC. ANR was positively correlated with turbidity and negatively correlated with TN and DOC (p < 0.05). Potential denitrification was positively correlated with TN, NO3−, and DOC. Potential anammox was positively correlated with turbidity (p < 0.05).

Relative abundances of the functional genes encoding the enzymes that catalyse nitrogen cycling pathways based on the KEGG database. “*” indicates p < 0.05 level.
Discussion
Microbial assemblages and co-occurrence network
Small changes in water levels may have crucial effects on bacterial distribution and structure by influencing microhabitat conditions21,35,36,37. This study showed that Cyanobacteria was the dominant phylum in the inundated and emerged areas in PYL, while the relative abundance of Cyanobacteria was much higher in the emerged areas than in the inundated areas (Fig. 2). Previous studies have indicated that phosphorus and nitrogen are the main causes of freshwater eutrophication, and long-lasting Cyanobacteria blooms are tightly associated with nutrient dynamics38,39,40,41. In the inundated areas, dissolved phosphorus and nitrogen can be released into sedimentary pore water and lake water in a short time18,21 and used in microbial growth in the water42. In our study, the emerged areas had higher turbidity, NH4+, and TP than that of the inundated areas (Table 1), and these nutrients were crucial factors influencing the microbial assemblages in the studied areas (Fig. 5). Previous studies also have determined that these nutrients are the dominant factors influencing the structure and composition of microbial communities in erosion areas43,44.
Microbial co-occurrence patterns were analyzed to assess community assembly rules and interaction networks in highly complex systems31,45,46. Compared with that in the emerged areas, the bacterial co-occurrence network in the inundated areas was more complex, which could have been caused by the continuous hydrological connection in the inundated areas during the whole year47,48. Previous studies have also indicated that positive correlations between nodes in co-occurrence networks of desert soil bacterial communities could be the result of functional interdependencies among bacterial taxa under extreme environmental conditions49,50,51. In our study, high proportions of positive correlations between nodes in the two networks suggested the interdependencies among bacterial taxa under environmental disturbance. Topological parameters provide important information to help us to understand microbial community structure2,3,47,52. Higher values of network centralization and heterogeneity in the emerged areas than in the inundated areas suggested that there were many tightly connected bacterial modules (subnetworks in the whole network) in the emerged areas. Species interactions are more frequent and intense in a module than in the remainder of a community47. Thus, a minor disturbance in a main module of a bacterial network could have a large impact on the whole network of microbial communities. Moreover, higher mean degree and high closeness centrality, as well as lower betweenness centrality, could be used collectively to identify bacterial keystone taxa53,54, which were mainly classified as Proteobacteria and Cyanobacteria in the emerged and inundated areas, respectively. These keystone taxa could exert considerable influence on the structure and function of bacterial communities54. Removal of those strongly connected taxa in a network would cause the collapse of the freshwater ecosystem structure and function55,56.
Potential functional genes and nitrogen metabolism
Seasonal water level fluctuations are one of the dominant forces controlling lake ecosystem functions, and any changes in water level could affect microbial metabolic functions21,36,37,57. To gain more insights into the effects of the inundated conditions on microbial functions, we calculated the relative abundance of potential functional genes related to metabolism pathways at level-1, level-2, and level-3 in the emerged and inundated areas. According to the previous studies58,59,60, the low NSTI mean values (0.09 + 0.02) indicated high accuracy of the predicted potential metabolic functions in our study. In general, the functional genes’ composition is strongly correlated with taxonomic composition in freshwater ecosystems2,3,61,62,63,64. In our study, taxonomic dissimilarity drove the potential metabolic functions of the bacterial communities in the inundated areas. Moreover, environmental variables were vital factors influencing the potential metabolic functions of the bacterial community in the emerged areas (Fig. 4). In the wet season, the re-flooding process in the emerged areas could cause the release of sedimentary nutrients42, and these nutrients might determine bacterial functional attributes14,65,66.
Metabolic functions related to the nitrogen cycle are particularly susceptible to environmental fluctuations because there are large differences in nitrogen metabolic processes with different forms of nitrogen67,68. Nitrogen in multiple chemical forms is cycled by a suite of coupled biogeochemical processes catalyzed by microbe-derived enzymes1,69,70. In our study, the relative abundances of the functional genes related to ANR were higher in the emerged areas, while the functional genes related to DNR were higher in the inundated areas (Fig. S2c). In general, ANR processes are much more prevalent than DNR processes in natural ecosystems3,71, and high concentrations of NH4+ could stimulate assimilatory pathways67,71,72. The reduction processes of NO3− to NO2− and then to NH4+ are part of the ANR processes, which usually occur in anaerobic environments such as sediment and soil73. In the wet season, a large amount of mineralized nitrogen (especially NH4+) could be easily dissolved in pore water and be rapidly released into lake water, and these mineralized nitrogen types could be used by bacterial communities as an energy source3,71,74. DNR is another nitrogen catabolic pathway that can retain nitrogen in the system in a bioavailable form for further biological processes71,75. Bacterial communities tend to use mineralized nitrogen as an energy source for growth in the emerged areas while as a nitrogen source for cell biosynthesis in the inundated areas. In addition, denitrification is another important pathway related to nitrogen metabolism. Through denitrification, nitrogen can be transformed into N2O or N2 and released into the atmosphere71. The addition of nitrate plus phosphate might affect microbial community structure, stimulate microbial enzyme activities, and promote denitrification rates18. Thus, during the wet season, reflooding processes could influence the nitrogen metabolism of bacterial communities by stimulating the release of sedimentary dissolved nitrogen and phosphate, which might promote the release of N2 and N2O over a short period in the emerged areas. The actual measurements of N2 and N2O at the sediment-water interface were important for understanding the nitrogen cycle driven by microorganisms under water level fluctuations. Although there was no data for N2 and N2O in our study now, the actual measurements will be considered for future research.
In our study, we explored the differences in bacterial communities and potential metabolic functions between the inundated and emerged areas during the wet season. The study of water level fluctuations in different seasons is crucial to elucidating the effects of inundation duration on microbial community structure in future studies. Metabolic functions were predicted from 16S rRNA sequencing data using the PICRUSt approach in our study. Although metagenomics reflects potential rather than actual functional capacity, our data offer a window into the poorly understood bacterial metabolism in seasonal lakes affected by water level fluctuations. The actual measurements of such functions will be considered in our future research.
Conclusion
Water level fluctuations and duration of inundation could affect the microbial communities and their potential metabolic functions in Poyang Lake. Cyanobacteria dominated the bacterial communities in the inundated and emerged areas, while the relative abundance of Cyanobacteria was higher in the emerged areas than in the inundated areas. The redundancy analysis revealed that nitrogen, phosphorus, and carbon played important roles in driving taxonomic and functional genes’ composition of bacterial communities. The relative abundance of the functional genes related to potential assimilatory nitrate reduction (ANR) was higher in the emerged areas than in the inundated areas, while the relative abundance of functional genes related to potential dissimilatory nitrate reduction (DNR) was higher in the inundated areas. NH4+ and turbidity were the crucial factors promoting the potential ANR processes in the emerged areas. The variations in DOC might influence potential denitrification processes, which could impact the production of N2 and N2O. Overall, this study increased our knowledge of the impacts of water level fluctuations on bacterial communities in permanently and seasonally flooded areas of seasonal lakes.
Methods
Study area
Poyang Lake (115°47′–116°45′E, 28°22′–29°45′N) is China’s largest freshwater lake, and its catchment is located on the south bank of the middle and lower reaches of the Yangtze River76. Poyang Lake shows typical interlacing changes among water and land due to the influence of water level fluctuations14,15. During the dry season, there is only a narrow and meandering channel in PYL due to the decrease in the water level66,77,78. With water drawdown, many inundated and emerged areas emerge in the dry seasons15,34. During the wet season, many inundated areas are connected with emerged areas to form the whole lake due to the increase in the water level2,13,14,66,76,77,78.
Sampling and physicochemical analyses
Bottom water was collected from inundated and emerged areas in Poyang Lake in August 2016, and the distance of each sampling site to sediment was the same (Table S2). The study area and sampling site distribution are shown on the map (Fig. 1). The map was created in ArcGIS 10.2 (http://desktop.arcgis.com/en/arcmap/) using ASTER GDEM data downloaded from the United States Geological Survey [ASTER GDEM is a product of the Ministry of Economy, Trade, and Industry (METI) and the National Aeronautics and Space Administration (NASA)] (Fig. 1a). The Environmental Fluid Dynamics Code (EFDC)79 was used to simulate the daily water depth of PYL in 2016 (Fig. 1b,c). A total of 600 ml of water was filtered through Whatman nylon membrane filters (pore size: 0.2 μm), and these membrane filters were immediately stored at −80 °C for subsequent DNA extraction. At each sample site, the water temperature (Temp), dissolved oxygen (DO), pH, turbidity, and conductivity (Cond) were measured in situ using a YSI Model 80 meter (Yellow Springs Instruments, Yellow Springs, Ohio, USA). Water depth (WD) was measured using a digital ultrasonic echosounder (Umwelt and Wissenschafts Technik, Mondsee, Austria). Water samples were acid fixed and transported to the laboratory at 4 °C for chemical analysis. Total nitrogen (TN) was analyzed by ion chromatography after persulfate oxidation. Nitrate (NO3−) was determined by ion chromatography. NH4+ was analyzed using the indophenol colorimetric method. TP and PO4− were quantified using the ammonium molybdate method after oxidation. DOC was analyzed using a Shimadzu TOC Analyzer (TOC-VCPH, Shimadzu Scientific Instruments, Columbia, Maryland).
DNA extraction, PCR, and sequencing
Bacterial 16S rRNA genes were analyzed to determine microbial community structures. Genomic DNA of microbial samples was extracted following the manufacturer protocols using the PowerSoil DNA Isolation Kit (MoBio, Carlsbad, CA, USA). The V3 to V4 regions of the 16S rRNA genes were amplified using primers 806R (GGACTACHVGGGTWTCTAAT) and 338F (ACTCCTACGGGAGGCAGCA) (Invitrogen, Vienna, Austria). Polymerase chain reaction (PCR) was performed using a thermal cycler (model 2720, ABI, USA) following standard procedures: 1 min hot start at 80 °C, 5 min of initial denaturation at 94 °C, followed by 30 cycles of denaturation at 94 °C for 30 s, followed by annealing at 52 °C for 30 s, and 90 s of extension at 72 °C, with a final extension step at 72 °C for 10 min. Amplified DNA samples were verified by 1.0% agarose gel electrophoresis with 1x TAE buffer and purified using the gel extraction kit (Qiagen, Hilden, Germany). The final sequencing process was run on a MiSeq sequencing platform (Illumina, USA).
Sequence analysis and functional genes prediction
In total, 657,365 raw sequences (available from the National Center for Biotechnology Information database under the BioProject number PRJNA436872 and the accession number SRP133903) were processed using the QIIME pipeline80. The forward and reverse reads were merged and assigned to samples based on the barcodes and truncated by removing the barcode and primer sequences. Quality filtering on merged sequences was performed and sequences that did not meet the following criteria were discarded: sequence length <200 bps, no ambiguous bases, and mean quality score>20. The sequences were compared with a reference database (RDP Gold database) using the UCHIME algorithm to detect chimeric sequences81 that were removed in QIIME. After quality filtration, 261,458 reads were clustered into operational taxonomic units (OTUs) with a complete linkage algorithm at 97% sequence identity level, and representative sequences from each OTU were identified by the Greengenes database. Potential functions were predicted from 16S rRNA data using PICRUSt59,82. The normalized OTU table was obtained by dividing the OTU table counts by marker gene copy numbers to estimate the abundance of each OTU. After data normalization, we obtained 151,116 sequences in the normalized OTU table to predict the potential functional genes using the KEGG database. The nearest sequenced taxon index (NSTI), the average branch length separating OTUs in each sample from the reference genome59, was calculated to assess the accuracy of the functional prediction. The predicted functional genes were further classified into metabolic pathways at different levels (level 1–3).
Statistical analysis
We compared environmental factors and the taxonomic profiles of microbial communities between the emerged and inundated areas. To determine whether the relative abundance of the phyla and functional genes were significantly different between the two areas, SPSS (Version 12.0) was applied to conduct bootstrap t-tests. We used heatmap (using Heatplus and Gplots packages in R version 3.5.1) and analysis of similarity (ANOSIM, PAST 3.0) to determine the differences in bacterial community compositions and potential metabolic functions between the two areas. Network analyses were conducted to reveal the co-occurrence patterns of the bacterial communities. OTUs with an average relative abundance higher than 0.01% and with a presence in more than half of the samples were used. The pairwise correlations between OTUs were calculated using the Spearman correlation in R (version 3.3.2 and Hmisc package 4.0–1), and p-values were adjusted using the Benjamin-Hochberg procedure. Only strong (Spearman’s r > 0.9 or r < −0.9 and p < 0.05) correlations were considered. The network was visualized using Cytoscape (version 3.6.1). An edge-weighted and spring-embedded network was applied to display the co-occurrence patterns of the OTUs. The topological and node/edge metrics were calculated to analyze the interactions between the nodes in the networks. The modular structural analysis of each network was conducted using ClusterMaker in Cytoscape. Redundancy analysis (RDA) was applied to analyze the spatial distribution of the bacterial communities with respect to various environmental factors using R (version 3.3.2 and Vegan package 2.4). Monte Carlo permutations (p < 0.05) were used to select a set of environmental factors that had significant effects on the microbial distribution. Environmental factors with high partial correlation coefficients (r > 0.5, p < 0.05) and variance inflation factors >20 were eliminated from the final RDA. Mantel tests were run to assess the correlations between the dissimilarities of the functional and taxonomic composition of the bacterial community based on Bray–Curtis distance and correlations between the dissimilarities of function (Bray–Curtis distance) and environmental factors (Euclidean distance). Correlation analyses were conducted to assess the relationships between metabolic pathways and abiotic factors using the Spearman correlation in SPSS software (Version 12.0).
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
References
Falkowski, P. G., Fenchel, T. & Delong, E. F. The microbial engines that drive Earth’s biogeochemical cycles. Science. 320, 1034–1039 (2008).
Ren, Z. et al. Taxonomic and Functional Differences between Microbial Communities in Qinghai Lake and Its Input Streams. Front. Microbiol. 8, 2319 (2017).
Ren, Z., Gao, H., Elser, J. J. & Zhao, Q. Microbial functional genes elucidate environmental drivers of biofilm metabolism in glacier-fed streams. Sci. Rep. 7, 12668 (2017).
Peter, H. & Sommaruga, R. Shifts in diversity and function of lake bacterialcommunities upon glacier retreat. Isme J. 10, 1545–1554 (2016).
Levi, P. S. et al. Microbial community diversity and composition varies with habitat characteristics and biofilm function in macrophyte-rich streams. Oikos. 126, 398–409 (2017).
Amann, R. & Rossello-Mora, R. The Underestimation of Global Microbial Diversity. MBio 7, e01623–16 (2016).
Peter, H. & Sommaruga, R. Shifts in diversity and function of lake bacterial communities upon 655 glacier retreat. Isme J. 10, 1545–1554 (2016).
Eiler, A. & Bertilsson, S. Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environ Microbiol. 6(1228–1243), 1228–1243 (2004).
Van der Gucht, K. et al. The power of species sorting_ local factors drive bacterial community composition over a wide range of spatial scales. Proc. Natl. Acad. Sci. U.S.A. 51, 20404–20409 (2007).
Green, J. L., Bohannan, B. J. & Whitaker, R. J. Microbial biogeography: from taxonomy to traits. Science. 320, 1039–1043 (2008).
Fierer, N., Lauber, C. L. & Ramirez, K. S. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. Isme J. 6, 1007–1017 (2012).
Soldatova, E. et al. Sources and behaviour of nitrogen compounds in the shallow groundwater of agricultural areas (Poyang Lake basin, China). J Contam Hydrol. 202, 59–69 (2017).
Lu, J., Chen, X., Zhang, L., Sauvage, S. & Sánchez-Pérez, J.-M. Water balance assessment of an ungauged area in Poyang Lake watershed using a spatially distributed runoff coefficient model. J. Hydroinform. 5, 1009–1024 (2018).
Wan, R., Yang, G., Dai, X., Zhang, Y. & Li, B. Water Security-based Hydrological Regime Assessment Method for Lakes with Extreme Seasonal Water Level Fluctuations: A Case Study of Poyang Lake, China. Chin. Geogr. Sci. 28, 456–469 (2018).
Liang, Y. et al. Impact of high water level fluctuations on stable isotopic signature of POM and source identification in a floodplain lake—Bang Lake (Poyang Lake). Environ. Earth Sci. 75, 255 (2016).
Li, Y., Yao, J. & Zhang, L. Investigation into mixing in the shallow floodplain Poyang Lake (China) using hydrological, thermal and isotopic evidence. Water Sci. Technol. 11, 2582–2598 (2016).
Baldwin, D. S. & Mitchell, A. M. The effects of drying and re‐flooding on the sediment and soil nutrient dynamics of lowland river–floodplain systems: a synthesis. Regul River. 16, 457–467 (2000).
Zhang, L. et al. Influence of long-term inundation and nutrient addition on denitrification in sandy wetland sediments from Poyang Lake, a large shallow subtropical lake in China. Environ. Pollut. 219, 440–449 (2016).
Tzoraki, O., Nikolaidis, N. P., Amaxidis, Y. & Skoulikidis, N. T. In-stream biogeochemical processes of a temporary river. Environ. Sci. Technol. 41, 1225–1231 (2007).
Kanson, L. H., Parparov, A. & Hambright, K. D. Modelling the impact of water level fluctuations on water quality (suspended particulate matter) in Lake Kinneret. Ecol. Model. 128, 101–125 (2000).
Hambright, K. D., Eckert, W., Leavitt, P. R. & Schelske, C. L. Effects of historical lake level and land use on sediment and phosphorus accumulation rates in Lake Kinneret. Environ. Sci. Technol. 38, 6460–6467 (2004).
Bayley, P. B. & Sparks, R. E. The flood pulse concept in river-floodplain systems. Can. Spec. Publ. Fish. Aquat. Sci. 106, 110–127 (1989).
Barbault, R. et al. Towards a metabolic theory of ecology. Ecology 85, 1771–1789 (2004).
Ren, Z., Qu, X., Zhang, M., Yu, Y. & Peng, W. Distinct bacterial communities in wet and dry seasons during a seasonal water level fluctuation in the largest freshwater lake (Poyang Lake) in China. Front. Microbiol. 10, 1167 (2019).
Butturini, A. et al. Influences of the stream groundwater hydrology on nitrate concentration in unsaturated riparian area bounded by an intermittent Mediterranean stream. Water Resour. Res. 39, 1–3 (2003).
Romaní, A. M., Vázquez, E. & Butturini, A. Microbial availability and size fractionation of dissolved organic carbon after drought in an intermittent stream: biogeochemical link across the stream-riparian interface. Microb. Ecol. 52, 501–512 (2006).
Ma, Y. et al. Bacterial and Fungal Community Composition and Functional Activity Associated with Lake Wetland Water Level Gradients. Sci. Rep. 8, 760 (2018).
Peterson, B. J. et al. Control of nitrogen export from watersheds by headwater streams. Science. 292, 86–90 (2001).
Mulholland, P. J. et al. Stream denitrification across biomes and its response to anthropogenic nitrate loading. Nature. 452, 202–205 (2008).
Zeglin, L. H. Stream microbial diversity in response to environmental changes: review and synthesis of existing research. Front. Microbiol. 6, 454 (2015).
Freedman, Z. & Zak, D. Atmospheric N deposition alters connectance, but not functional potential among saprotrophic bacterial communities. Mol. Ecol. 24, 3178–3180 (2015).
Wang, K. et al. Regional variations in the diversity and predicted metabolic potential of benthic prokaryotes in coastal northern Zhejiang, East China Sea. Sci. Rep. 6, 38709 (2016).
Zoppini, A., Amalfitano, S., Fazi, S. & Puddu, A. Dynamics of a benthic microbial community in a riverine environment subject to hydrological fluctuations (Mulargia River, Italy). Hydrobiogia. 657, 37–51 (2010).
Liang, Y. et al. Variation in sources of inorganic nitrogen under different hydrological conditions in a floodplain lake: a case study of Bang Lake (Poyang Lake, Jiangxi Province, China). Inland Waters. 8, 176–185 (2018).
Dinka, M., Agoston-Szabo, E., Berczik, A. & Kutrucz, G. Influence of water level fluctuation on the spatial dynamic of the water chemistry at Lake Ferto/Neusiedler See. Limnological. 34, 48–56 (2004).
Leira, M. & Cantonati, M. Effects of water-level fluctuations on lakes: An annotated bibliography. Hydrobiologia. 613, 171–184 (2008).
Wang, Y. et al. Potential influence of water level changes on energy flows in a lake food web. Chinese Sci. Bull. 56, 2794–2802 (2011).
Kangur, K., Möls, T., Milius, A. & Laugaste, R. Phytoplankton response to changed nutrient level in Lake Peipsi (Estonia) in 1992–2001. Hydrobiologia 506, 265–272 (2003).
Huisman, J. et al. Cyanobacterial blooms. At. Rev. Microbiol. 16, 471–483 (2018).
Kramer, B. J. et al. Nitrogen limitation, toxin synthesis potential, and toxicity of cyanobacterial populations in Lake Okeechobee and the St. Lucie River Estuary, Florida, during the 2016 state of emergency event. PLoS One. 13, e0196278 (2018).
Wejnerowski, L., Rzymski, P., Kokocinski, M. & Meriluoto, J. The structure and toxicity of winter cyanobacterial bloom in a eutrophic lake of the temperate zone. Ecotoxicology. 27, 752–760 (2018).
Borken, W. & Matzner, E. Reappraisal of drying and wetting effects on C and N mineralization and fluxes in soils. Global Change Biol. 15, 808–824 (2009).
Li, J., Hansson, L.-A. & Persson, K. Nutrient Control to Prevent the Occurrence of Cyanobacterial Blooms in a Eutrophic Lake in Southern Sweden, Used for Drinking Water Supply. Water. 10, 919 (2018).
Guildford, S. J. & Hecky, R. E. Total nitrogen, total phosphorus, and nutrient limitation in lakes and oceans: Is there a common relationship? Limnol. Oceanogr. 45, 1213–1223 (2000).
Gotelli, N. J. & Mccabe, D. J. Species Co-occurrence: A Meta-Analysis of J. M. Diamond’s Assembly Rules Model. Ecology. 83, 2091–2096 (2002).
Fuhrman, J. Microbial community structure and its functional implications. Nature. 459, 193–199 (2009).
Newman, M. E. J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 103, 8577–8582 (2006).
Kara, E. L., Hanson, P. C., Hu, Y. H., Winslow, L. & McMahon, K. D. A decade of seasonal dynamics and co-occurrences within freshwater bacterioplankton communities from eutrophic Lake Mendota, WI, USA. ISME J 7, 680–684 (2013).
Neilson, J. W. et al. Significant Impacts of Increasing Aridity on the Arid Soil Microbiome. MSystems. 2, e00195–00116 (2017).
Van Goethem, M. W., Makhalanyane, T. P., Cowan, D. A. & Valverde, A. Cyanobacteria and Alphaproteobacteria May Facilitate Cooperative Interactions in Niche Communities. Front. Microbiol. 8, 2099 (2017).
Mandakovic, D. et al. Structure and co-occurrence patterns in microbial communities under acute environmental stress reveal ecological factors fostering resilience. Sci. Rep. 8, 5875 (2018).
Qu, X. et al. Influences of anthropogenic land use on microbial community structure and functional potentials of stream benthic biofilms. Sci. Rep. 7, 15117 (2017).
Berry, D. & Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 5, 219 (2014).
Banerjee, S., Schlaeppi, K. & van der Heijden, M. G. A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 16, 567–576 (2018).
Montoya, J. M., Pimm, S. L. & Sole, R. V. Ecological networks and their fragility. Nature. 442, 259–264 (2006).
Saavedra, S., Stouffer, D. B., Uzzi, B. & Bascompte, J. Strong Contributors to Network Persistence are the Most Vulnerable to Extinction. Nature. 478, 233–235 (2011).
Coops, H., Beklioglu, M. & Crisman, T. L. The role of water-level fluctuations in shallow lake ecosystems – workshop conclusions. Hydrobiologia. 506–509, 23–27 (2003).
Koo, H. et al. Microbial Communities and Their Predicted Metabolic Functions in Growth Laminae of a Unique Large Conical Mat from Lake Untersee, East Antarctica. Front. Microbiol. 8, 1347 (2017).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013).
Staley, C. et al. Core functional traits of bacterial communities in the Upper Mississippi River show limited variation in response to land cover. Front. Microbiol. 5, 414 (2014).
Louca, S. et al. High taxonomic variability despite stable functional structure across microbial communities. Nat. Ecol. Evol. 1, 15 (2016).
Fierer, N. et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 109, 21390–21395 (2012).
Allison, S. D., Lu, Y. & Weihe, C. Microbial abundance and composition influence litter decomposition response to environmental change. Ecology. 94, 714–725 (2013).
Dopheide, A., Lear, G., He, Z., Jizhong, Z. & Lewis, G. D. Functional Gene Composition, Diversity and Redundancy in Microbial Stream Biofilm Communities. PloS One. 10, e0123179 (2016).
Sheng, P. et al. Bacterial diversity and distribution in seven different estuarine sediments of Poyang Lake, China. Environ. Earth Sci. 75, 479 (2016).
Zhang, M. et al. Determining the macroinvertebrate community indicators and relevant environmental predictors of the Hun-Tai River Basin (Northeast China): A study based on community patterning. Sci. Total. Environ. 634, 749–759 (2018).
Gruber, N. & Galloway, J. N. An Earth-system perspective of the global nitrogen cycle. Nature. 451, 293–296 (2008).
Ollivier, J. et al. Nitrogen turnover in soil and global change. FEMS Microbiol. Ecol. 78, 3–16 (2011).
Nelson, M. B., Berlemont, R., Martiny, A. C. & Martiny, J. B. Nitrogen Cycling Potential of a Grassland Litter Microbial Community. Appl. Environ. Microbiol. 81, 7012–7022 (2015).
Ye, F. et al. Soil properties and distribution in the riparian zone: the effects of fluctuations in water and anthropogenic disturbances. European Journal of Soil Science, https://doi.org/10.1111/ejss.12756 (2019).
Zhu, Y., Jin, X., Tang, W., Meng, X. & Shan, B. Comprehensive analysis of nitrogen distributions and ammonia nitrogen release fluxes in the sediments of Baiyangdian Lake, China. J Environ Sci (China) 76, 319–328 (2019).
Zumft, W. G. Cell biology and molecular basis of denitrification. Microbiology and Molecular Biology Reviews 61, 533–616 (1997).
Lamba, S. et al. Organization of biogeochemical nitrogen pathways with switch-like adjustment in fluctuating soil redox conditions. R. Soc. Open. Sci. 4, 160768 (2017).
Frey, S. D., Knorr, M., Parrent, J. L. & Simpson, R. T. Chronic nitrogen enrichment affects the structure and function of the soil microbial community in temperate hardwood and pine forests. For. Ecol. Manage. 196, 159–171 (2004).
Rice, C. W. & Tiedje, J. M. Regulation of nitrate assimilation by ammonium in soils and in isolated soil microorganisms. Soil Biol. Biochem. 21, 597–602 (1989).
Feng, L. et al. Assessment of inundation changes of Poyang Lake using MODIS observations between 2000 and 2010. Remote Sens. Environ. 121, 80–92 (2012).
Zhang, X., Qin, H., Wang, H., Wan, A. & Liu, G. Effects of water level fluctuations on root architectural and morphological traits of plants in lakeshore areas of three subtropical floodplain lakes in China. Sci. Pollut. Res. 25, 34583–34594 (2018).
Shankman, D., Keim, B. D. & Song, J. Flood frequency in China’s Poyang Lake region: trends and teleconnections. Int. J. Climatol. 26, 1255–1266 (2006).
Zhou, J., Falconer, R. A. & Lin, B. Refinements to the EFDC model for predicting the hydro-environmental impacts of a barrage across the Severn Estuary. Renew. Energy. 62, 490–505 (2014).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7, 335 (2010).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 27, 2194–2200 (2011).
Minoru, K. & Susumu, G. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1, 27–30 (2000).
Acknowledgements
This study was supported by the State Key Laboratory of Simulation and Regulation of Water Cycle in River Basin (SKL2018CG02), the National Natural Science Foundation of China (No. 51439007 and No. 41671048), and the IWHR Research and Development Support Program (WE0145B052018 and WE0145B532017).
Author information
Authors and Affiliations
Contributions
Liu, Ren, and Qu performed analyses and prepared the manuscript; Peng reviewed and revised the manuscript. Peng, Qu and Ren designed the study. Qu, Zhang, Yu, and Zhang performed the field work and laboratory analysis; Peng provided suggestions during the whole study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Ren, Z., Qu, X. et al. Microbial community structure and functional properties in permanently and seasonally flooded areas in Poyang Lake. Sci Rep 10, 4819 (2020). https://doi.org/10.1038/s41598-020-61569-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61569-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
