
Abstract
Current diagnostic measures for Chronic Kidney Disease (CKD) include detection of reduced estimated glomerular filtration rate (eGFR) and albuminuria, which have suboptimal accuracies in predicting disease progression. The disease complexity and heterogeneity underscore the need for multiplex quantification of different markers. The goal of this study was to determine the association of six previously reported CKD-associated plasma proteins [B2M (Beta-2-microglobulin), SERPINF1 (Pigment epithelium-derived factor), AMBP (Protein AMBP), LYZ (Lysozyme C), HBB (Hemoglobin subunit beta) and IGHA1 (Immunoglobulin heavy constant alpha 1)], as measured in a multiplex format, with kidney function, and outcome. Antibody-free, multiple reaction monitoring mass spectrometry (MRM) assays were developed, characterized for their analytical performance, and used for the analysis of 72 plasma samples from a patient cohort with longitudinal follow-up. The MRM significantly correlated (Rho = 0.5–0.9) with results from respective ELISA. Five proteins [AMBP, B2M, LYZ, HBB and SERPINF1] were significantly associated with eGFR, with the three former also associated with unfavorable outcome. The combination of these markers provided stronger associations with outcome (p < 0.0001) compared to individual markers. Collectively, our study describes a multiplex assay for absolute quantification and verification analysis of previously described putative CKD prognostic markers, laying the groundwork for further use in prospective validation studies.
Similar content being viewed by others


Introduction
Chronic kidney disease (CKD), defined as reduced kidney function and/or evidence of kidney damage, is a major public health problem throughout the world. Major health problems around the globe, with consistent prevalence rates of 10–13% have been reported (depending on reference group and stage)1,2. Disease management is characterized by excessive financial costs (with expenses associated with CKD treatment, exceeding 100 B € in total, annually in Europe)2,3. Common risk factors for CKD include ageing of the population and increased rates of diabetes and hypertension2,4. Of note, patients with CKD have an overall 30-fold increased risk for suffering from Cardio Vascular Disease (CVD) complications, this being the main cause of CKD-associated deaths. Specifically, ~45% of patients with CKD stage 4–5 die from CVD5 and risk of CVD increases with CKD severity, which is already significantly higher in early CKD compared to non-CKD6,7. Early identification of CKD and addressing modifiable risk factors is recommended, as it can reduce the risk of kidney failure and CVD by up to 50%8. Early detection or prediction of complications enable early intervention, thus could increase the chances for higher treatment efficacy9,10,11.
CKD diagnosis is currently based on the detection of reduced estimated glomerular filtration rate (eGFR) and/or albuminuria, as indicators of renal dysfunction12,13. However, these markers have substantial limitations in evaluating CKD progression. Albuminuria quantification and categorization into normalbuminuria (<30 mg/24 hours), microalbuminuria (30–300 mg/24 hours) and macroalbuminuria (>300 mg/24 hours)13,14 is commonly used alone or in combination with eGFR to predict kidney damage15. However, a significant proportion of patients progress without exhibiting significantly increased urinary albumin levels16. Collectively, there is ample room for improvement especially with respect to prognosis of CKD complications, in part due to the fact that the main biomarkers currently used (albuminuria, eGFR) are neither an early indicator nor linked to molecular pathophysiology involved in disease progression, but rather the functional consequences of the already settled pathological modifications.
More accurate evaluation of progression risk could be of significant benefit, since treatment options are presently available9,10,11. As such, biomarker research in the field has been rigorous with ample biomarker data collected until now17,18,19,20. Various plasma proteins have been associated with CKD progression, with CVD, and with patient-relevant outcome [End Stage Renal Disease (ESRD), death]. These include beta-2 microglobulin (B2M), neutrophil gelatinase-associated lipocalin (NGAL), kidney injury molecule-1, liver-type fatty acid binding protein, cystatin-C, FGF23, as prominent examples21,22,23,24,25.
Even though associations with outcome have been reported, the overall suboptimal accuracies of individual markers emphasize the need to establish panels or multi-parametric classifiers, better reflective of the substantial phenotypic and molecular heterogeneity of CKD. Along these lines, a urinary peptide classifier, CKD273, consisting of 273 peptides, fragments of multiple kidney-specific as well as plasma proteins detected by the use of capillary electrophoresis (CE) in combination to mass spectrometry (MS) has been developed, subsequently evaluated26,27,28 and received a letter of support by the US Food and Drug Administration (FDA, USA) for use in early detection of nephropathy in diabetic patients29. CKD273 has been found to predict progression at early CKD stages (eGFR > 70 mL/min/1.73 m2) more accurately than albuminuria30, and has been applied in the proteome-guided intervention trial, PRIORITY31.
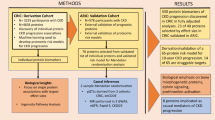
Prediction of progression to ESRD or CVD-related endpoints is critical for disease management26. As a step in this direction, we aimed at establishing absolute quantification assays and performing an initial evaluation of the prognostic value of six plasma proteins, when measured in a multiplex format, for a patient-relevant endpoint, death. The employed method is the targeted mass spectrometry-based multiple reaction monitoring (MRM), relying on the quantification of one or more unique/proteotypic peptides corresponding to a target protein32,33 without using antibodies33. The applied quantification strategy is the Absolute Quantification (AQUA) workflow34, involving stable isotope-labeled peptides, spiked into the sample of interest at predefined amounts and measured simultaneously with the respective endogenous peptides35,36. The selection of the specific markers was guided by the existence of mass spectrometry-based data for these proteins37, their expected abundance levels based on the existent literature, targeting to avoid extensive pre-fractionation, as well as levels of evidence for the association of these proteins with unfavorable outcome. Using the study by Glorieux et al.37 as a basis, involving high resolution LC-MS/MS analysis of plasma proteome from patients with CKD and reporting on associations to outcome, in combination to the aforementioned criteria, the tested panel includes B2M38,39,40,41,42,43,44,45,46 and SERPINF147,48,49,50, having been largely studied in CKD and serving as positive controls for the approach, as well as AMBP, LYZ, HBB, and IGHA1 with some earlier reported associations37, nevertheless not been validated yet in association with disease progression via absolute quantification.
Results
Candidate biomarkers and assay establishment
The markers to be quantified were selected from the list of differentially expressed proteins in plasma of haemodialysis (HD) patients with CKD stage 5 versus CKD stages 2–3 in Glorieux et al.37, further considering availability of proteotypic peptides for MS quantification assays, as described in Methods. To avoid extensive pre-fractionation steps potentially compromising assay reproducibility and applicability, only markers with expected relatively high (>100 ng/ml) plasma abundance levels reported in the literature and existing proteomic databases were considered (Supplementary Table S1). The selected marker candidates were B2M, SERPINF1, AMBP, LYZ, HBB, and IGHA1. Following a development phase, involving testing multiple peptides per marker, as described in Methods, standard curves per selected peptide were generated to ensure reproducibility, minimal or lack of matrix effects and linearity in measurements in the range of expected quantified values. Standard curves using synthetic isotope labeled peptide standards (SIS), specific per marker, were generated using a pooled plasma sample (from patients at CKD stages 2–5, n = 4), as matrix.
As shown in Fig. 1, in all cases, R2 coefficient of at least 0.992 and for a minimum 4-point concentration ranges, spanning the expected abundances were observed. In addition, CVs of the ratio of the abundance of the standard (SIS) to the endogenous (NAT; SIS/NAT) peptide for all dilution points ranged from 0.40–16.06%, (only exception was for the concentration 72.18 ng/mL of LYZ peptide with CV of 20.79%) suggesting good reproducibility and minimal matrix effects for the assays. The LLOQ (Lower Limit of Quantification) ranged from 9.38–167.5 ng/mL, defined as the lowest qualified concentration level of SIS peptide corresponding to SIS/NAT ratio with CV < 20%51,52,53 (with sole exception the LLOQ of LYZ corresponding to SIS/NAT ratio with CV = 20.79%-marginally higher than CV = 20%). Consequently, the LLOD (Lower Limit of Detection) for the peptides was estimated at 6.54–112.52 ng/mL, based on the following formula 3.3*sd/slope: (https://www.ich.org/). (Supplementary Table S2).

Standard curves generated using labeled peptide standards per marker spiked in plasma matrix at different concentrations. The peptides specifically correspond to: (a) DASGVTFTWTPSSGK (IGHA1), (b) LLVVYPWTQR (HBB), (c) LAAAVSNFGYDLYR (SERPINF1), (d) ΤVAACNLPIVR (AMBP), (e) VEHSDLSFSK (B2M), (f) AWVAWR (LYZ). For each dilution point n = 3 replicates were analyzed.
Differential expression analysis for baseline data
Using the established assays, quantification of the selected markers was performed on all baseline samples from 72 patients that were included in the original study. These included patients with CKD stage 5 (n = 33), CKD stage 4 (n = 21), CKD stage 3 (n = 12), and CKD stage 2 (n = 6) at baseline. Thirty-five events (deaths) were observed (mean follow-up time 3.61 ± 2.96 years), with n = 34 patients being alive at the last contact date (mean follow-up time 5.21 ± 2.77 years). For 3 patients, no follow-up information was available. The main clinical data are summarized in Table 1 and more details can be found in Supplementary Table S3.
Using the established assays, high quality spectra were obtained for all but one sample, where the data for SERPINF1, AMBP and B2M did not pass quality control (representative spectra of SIS and NAT peptides per marker are presented in Supplementary Fig. S1). Detailed information on MRM acquisition parameters and measurements per sample are provided in Supplementary Tables S4 and S5, respectively. The mean estimated abundance levels of the 6 proteins per CKD stage are shown in Fig. 2a. A significant change in CKD5 HD versus all other stages was observed for AMBP, B2M, LYZ, SERPINF1 and HBB (p < 0.05 based on Kruskal-Wallis test followed by Mann-Whitney pairwise comparisons), with B2M and LYZ also differing significantly when comparing CKD5 (excluding HD patients; n = 9) to the other stages. The lowest association with CKD stage was observed for IGHA1, found at increased levels only in CKD4 in comparison to CKD5 HD (p < 0.05).

(a) Dot plot graph representation of the MRM quantification data for the six plasma proteins investigated. CKD5 is separated in two groups: (1) CKD5 patients (N = 9) and (2) CKD5 HD (hemodialysis patients, N = 24). *Significant differences of CKD5 HD patients with hemodialysis versus each of the other stages were observed for B2M, LYZ, AMBP, SERPINF1 and HBB with IGHA1 differing only in comparison to CKD 4. An impact of HD on the SERPINF1 levels may be observed with its levels decreasing significantly in comparison to CKD 5 patients. **Significant differences of CKD5 versus each of the other stages were observed for B2M, LYZ with AMBP, SERPINF1 and HBB differing only in comparison to CKD2. ***Significant changes could also be observed between CKD4 versus 3 (B2M, HBB) or CKD4 versus 2 (B2M, HBB, AMBP, LYZ, SERPINF1); ****Significant changes could also be seen for AMBP and SERPINF1 for CKD3 vs 2. (b) Correlation scores between eGFR and the MRM quantified proteins (ng/mL). Spearman correlation coefficients (R score) are shown.
As the investigated markers in the LC-MS/MS analysis by Glorieux et al.37 were compared between early (combined stages 2–3) and advanced CKD (stage 5) with HD, we performed the same comparison with our MRM data (Table 2). An overall agreement with the study by Glorieux et al.37 was observed, with most markers detected at increased abundance in CKD 5 with HD (N = 24) versus combined stages 2–3, except for IGHA1 and SERPINF1, where no significant changes could be detected (Table 2).
B2M, followed by LYZ plasma concentrations showed strong negative correlations to eGFR levels (R = −0.72 and −0.63, respectively) (Fig. 2b). For this analysis, 24 CKD stage 5 patients on HD were excluded due to lack of baseline eGFR values (Supplementary Table S3). Negative correlations were also observed for AMBP, SERPINF1 and HBB with the weakest being observed for the latter (HBB), whereas for IGHA1 significance was not reached (p = 0.057) (Fig. 2b).
Correlation of MRM measurements to ELISA
To further validate the MRM measurements, ELISAs were performed for 5 out of the 6 proteins where specific and well characterized assays were available (IGHA1, SERPINF1, AMBP, B2M, LYZ). Indeed, the correlation of the findings of the 2 methods was to the least moderate (IGHA1 and B2M with Rho = 0.56 and Rho = 0.53, respectively) or in most cases high (for proteins LYZ, SERPINF1 and AMBP with Rho = 0.88, Rho = 0.8 and Rho = 0.64 respectively; Fig. 3) further supporting the validity of the MRM results (Fig. 3). ELISA measurements per sample are presented in Supplementary Table S6.

Spearman correlation analysis of MRM and ELISA data in the same sample cohort.
Association to outcome
To investigate association of the quantified markers to outcome (death), Kaplan Meier analysis was performed. Given its overall poor associations to staging as well as lack of correlation to eGFR, IGHA1 was excluded from further analysis. As shown in Fig. 4, higher (based on a median cut-off) plasma concentrations (ng/mL) of HBB (p = 0.027), AMBP (p = 0.00054), B2M (p < 0.0001) and LYZ (p < 0.0001) and lower plasma concentrations of SERPINF1 (p = 0.00013) were significantly associated with death.

Survival analysis for MRM quantified proteins. Kaplan-Meier plots depicting 7 years’ survival probability for patients with CKD stratified based on median protein concentration (ng/mL) as defined by MRM. Dashed lines indicate confidence intervals at 95% level. Significant differences in survival (p < 0.05) are determined with the Log-rank test.
Establishment of classifier
To assess the added value of combining the measured markers into a single simple classifier we utilized instance-based learning. In that, a k-nearest neighbor (knn) classifier was fed with the MRM data and was trained to distinguish between disease status at 7 years follow up time. The model was validated in predicting risk for mortality by leave one out crossvalidation. As the levels of SERPINF1 were found to be highly affected by HD (Fig. 2a), the marker was excluded and the knn model was developed based on the concentrations (ng/mL) of the remaining 4 proteins, HBB, AMPB, B2M and LYZ. At 7 years’ follow up, using leave-one-out crossvalidation, subjects were predicted either as “deceased” (high risk group) or “censored” (low risk group). Kaplan – Meier analysis of the predicted high/low risk groups illustrated significantly shorter survival time for the high risk group (Fig. 5), suggesting that the combination of HBB, AMBP, B2M and LYZ may efficiently predict survival for patients with CKD.

Survival analysis of high vs low risk groups predicted with k-nearest neighbor classifier. The classifier was trained to distinguish between censored and deceased data from patients with CKD, using leave-one-out crossvalidation.
Furthermore, a similar analysis was performed in 46 patients with available eGFR measurements in plasma, as eGFR is classically used for the evaluation of CKD progression (Fig. 6). The classifier model suggested that the combination of HBB, AMBP, B2M and LYZ may predict more efficiently survival for patients with CKD (p < 0.05) (Fig. 6a) compared to eGFR measurements (p > 0.05), when the cutoff of eGFR was set at 60 mL/min/1.73 m2 (Fig. 6b).

Comparison of survival probability based on a sub-cohort of 46 patients where eGFR measurements are available. (a) Survival analysis in the same group of patients of 46 patients of high vs low risk groups predicted with k-nearest neighbor classifier. The classifier was trained to distinguish between censored and deceased data from patients with CKD, using leave-one-out crossvalidation. (b) Survival probability is evaluated based on eGFR measurements under and over 60 mL/min/1.73 m2. Based on the p value, the KNN classifier seems to predict more effectively the occurrence of death compared to eGFR.
Discussion
Multiple biomarkers for CKD progression towards ESRD and death, widely classified as biomarkers of kidney function and kidney damage, have been described17,54,55,56,57. eGFR and albuminuria are the main clinically used markers, despite the extensively described shortcomings in terms of diagnostic and prognostic accuracies17. Multiple additional proteins, including various tubular markers (such as kidney injury molecule-1, neutrophil gelatinase-associated lipocalin, cystatin C, a-1-microglobulin) have been described in association with the disease17,54,58 but generally are not routinely implemented, yet.
To address disease complexity, multi-parametric, high dimensional classifiers incorporating numerous disease-specific and systemic molecular changes are increasingly being established, as better reflecting the disease molecular heterogeneity59. A prominent example is the multi-peptide urinary classifier CKD273 being used for the detection of nephropathy at early stages26,29.
We employed a multiplex MRM mass spectrometry assay for the quantification of six previously described, exploratory CKD markers. MRM, in contrast to the classically employed highly sensitive ELISA assays, does not require the use of specific antibodies, thus facilitating multiplexing and increasing specificity via eliminating antibody cross-reactivity problems32,33. MRM has been widely applied in plasma for proteomic biomarker validation60 and for various plasma proteins (such as apolipoproteins). It has been demonstrated to provide assay performance equal to well characterized ELISA assays61. In fact, the observed correlations between our MRM data and respective independent immunoassays (Rho = 0.53–0.88) are in line to existing literature and reported acceptable correlations of such assays62,63.
Besides evidence of association with CKD, the selection of the specific markers was driven by the existence of mass spectrometry-based data per protein to ensure detectability, as well as expected abundance levels to eliminate the need for extensive pre-fractionation. The detection limits observed in our study (in the range of ng/mL) are similar to ones regularly reported when using unfractionated plasma64. Applications involving combination of MRM with immunoprecipitation protocols62 or peptide separation by two dimensional liquid chromatography can extent the quantification range from below 100 ng/mL to about 500 pg/mL range65. However, these protocols are substantially more elaborate, and not well suited for routine implementation.
Among the analyzed proteins, B2M is the most well-characterized CKD-associated marker and known uremic solute according to the European Uremic Toxin Work Group66. It is a component of the class I major histocompatibility complex (MHC), synthesized normally by lymphocytes, filtered in the glomerulus and catabolized by the proximal tubular cells67,68. Upon defective renal function, its serum levels increase significantly66,67,68. In line with our study, gradual increase in B2M levels across stages of CKD has been shown38. Furthermore, several associations between B2M levels and death, dialysis or cardiovascular events have been reported39,42,43, in agreement with our findings.
Pigment epithelium-derived factor (SERPINF1), initially characterized as a neuronal differentiation factor in retinoblastoma cells69, was later defined as a member of the serine protease inhibitor family with antiangiogenic, antioxidative, anti-inflammatory, and antitumorigenic activity70, and was recently assigned a renoprotective role71. In line with our analysis, SERPINF1 serum levels were found elevated in ESRD patients compared to healthy controls47. Similarly, SERPINF1 was reported increased in plasma of CKD3 patients in comparison to controls48. In addition, Hui et al., demonstrated that SERPINF1 levels correlated with eGFR (n = 1136), and further associations to CKD progression (n = 1,071 stages 1–3 of which 171 progressed) were supported49. In our study, surprisingly, an association of lower SERPINF1 levels with death was detected. This finding is most likely driven by the HD patients included in the cohort. In fact, if we exclude HD patients from the analysis, higher levels of the protein in CKD5 vs CKD4 can be observed (Fig. 2a).
AMBP has been studied mainly in urine and to a lesser extent in plasma and tissue in the context of CKD. Increased urinary AMBP levels have been associated with kidney damage72 and tubular dysfunction in diabetic nephropathy73. In addition, several shotgun proteomic studies followed by verification via different assays (Western blot, MRM in small sample sizes (n ≤ 11), or ELISA), supported an upregulation of AMPB in patients of various CKD stages compared to healthy controls, in adipose tissue, urine as well as urine exosomes74,75,76. AMBP was also found to progressively increase in plasma of individuals with increasing CKD stage –from CKD1-2 to CKD3-4 and finally to CKD5- based on relative quantification by LC-MS/MS77,78 or MRM77.
AMBP, SERPINF1, B2M, and HBB have been investigated as HDL (High-density lipoprotein) associated proteins by LC-MS/MS, and were found to correlate with severe kidney damage79. This latter study also revealed elevated levels of these proteins in ESRD patients and recipients with poor graft function compared to patients with good graft function, a finding which was further confirmed by Western Blot analysis for SERPINF1 and AMBP79. Our study also supports the progressive up-regulation of AMPB in plasma with increasing CKD stage, negative correlation to eGFR and unfavorable outcome (p = 0.00054).
In comparison to B2M, SERPINF1 and AMBP, the published evidence associating IGHA1, HBB and LYZ to CKD is limited. IGHA1 was found at higher levels in the plasma of patients with CKD5 with HD in comparison to CKD2-3 in the study by Glorieux et al.37. This finding could not be verified in our study, which also failed to demonstrate association of the protein to eGFR. In contrast, the differential abundance of HBB, one of the two polypeptide chains that form Hemoglobin A80, in CKD5 versus earlier stages was verified and a modest association to eGFR was observed. The most pronounced finding of our study is the clear associations of LYZ with all CKD stages, eGFR as well as survival. LYZ is released from leukocytes and macrophages, and has antibacterial properties and an attributed role in myocardial depression and vasodilation81. With the exception of the study by Glorieux et al.37, and an in vitro study suggesting anti-inflammatory properties of LYZ on human proximal tubular epithelial cells (HK-2 cells)82, studies of LYZ in the context of CKD are lacking. Based on our results, such studies seem well justified, which may also expand to the investigation of cardiorenal syndrome, considering reports associating plasma levels of LYZ to coronary artery disease severity82,83.
Collectively, our study describes a multiplex assay for the absolute quantification and verification analysis of previously described putative markers for CKD, laying the groundwork for further investigation in prospective validation studies. As shown, the above markers seem to correlate with CKD stage (Fig. 2a). An extra comparison of protein concentrations between pathological conditions other than CKD (diabetic vs non diabetic as well as hypertensive vs non hypertensive patients, as per availability) was performed for the 6 studied proteins. The overall lack of statistical significance (MW, p > 0.05) in these cases (Supplementary Fig. S2) suggests that our targets could be specific markers for CKD progression. However, the power of the study is too small and further validation is needed to confirm this observation. Overall, the small sample size and restriction to 6 markers are clear limitations. Furthermore, larger scale studies will be required to validate changes per stage including in early disease. The small sample size does not also allow multivariate analysis to be conducted with confidence. An effort to investigate the added value of the classifier on top of clinical variables was performed and even though an independent prognostic value over age could be seen, this was lost with the addition of CKD stage (the output of cox proportional hazard model is summarized in Supplementary Table S7). Nevertheless, the availability of follow-up information allowing preliminary associations to outcome and simplicity of the assay, avoiding large fractionation schemes, provide a strong basis prompting further advancement towards properly sized validation trials.
Methods
Study population
The study population consisted of all 72 patients (outpatient and dialysis unit of Nephrologie Dialyse Saint Guilhem and the Public Hospital of Sète, as well as the Department of Nephrology, Transplantation and Dialysis of the University Hospital of Montpellier) that were recruited for the “Urosysteomics” study26. Of these, 24 were classified as ESRD patients at baseline. The study was approved by the Comité de Protection des Personnes of Montpellier and declared to the French Ministry of Health (reference number DC-2008–417). All methods were performed in accordance with the relevant guidelines and regulations. All individuals gave written informed consent. Pertinent clinical data are provided in Supplementary Table S3. Estimated glomerular filtration rate (eGFR) was calculated using the CKD-EPI equation84.
Selection of peptides for LC-MRM-MS
For the establishment of the MRM assays, proteotypic peptides were selected considering the following criteria: (i) the peptides uniquely represented the target protein, (ii) lysine or arginine was allowed only at the carboxy terminus (no missed tryptic cleavage sites are allowed), (iii) peptide sequence included 6–25 amino acid residues to ensure acceptable ionization and gas-phase fragmentation, and (iv) amino acids that are susceptible to chemical modifications such as cysteine, methionine should be absent. To perform the selection, multiple tools were employed in combination: a spectral library from the National Institute of Standards and Technology (NIST) [“human consensus final true lib”, downloaded from http://www.nist.gov/] on 05062014] was imported into Skyline software. The Skyline indicated proteotypic peptides and respective transitions per peptide which were further validated with additional bioinformatics tools: The PeptidePicker (http://mrmpeptidepicker.proteincentre.com/peptidepicker9/) that predicts proteotypic peptides with similar criteria as Skyline85; The Peptide Tracker (http://tracker.proteincentre.com/) that records transitions and chromatographic properties of specific proteotypic peptides based on previously performed MRM assays86, and the CPTAC Assay Portal (https://assays.cancer.gov/available_assays) that compiles information of existing MRM assays (proteotypic peptides, analytical performance, suggested protocols)87. In addition, shortlisted peptides were evaluated using the Protein Basic Local Alignment Search Tool (BLAST, http://blast.ncbi.nlm.nih.gov) to ensure their proteotypicity.
For the peptides identified (4–7 per biomarker) and their respective transitions (3–5 per peptide), based on the aforementioned procedure and considered for further analysis, initial MRM experiments were conducted using two plasma samples (normal and CKD, respectively) in triplicate. Further shortlisting was performed based on: (i) the quality of the MS/MS spectrum of each peptide in the human spectral library, (ii) score and number of observations in MS-based proteomics experiments, as provided from PeptideAtlas (http://www.peptideatlas.org)88 and (iii) quality and reproducibility of the MS/MS spectrum of the native (NAT) peptide in the two tested plasma samples as defined by: (a) peptide transitions should co-elute and yield Gaussian peak shapes with insignificant fronting and tailing and (b) the same elution profiles should have been observed in the spectral library. Based on these criteria, one peptide per biomarker and 2–5 transitions per peptide were selected for quantification in the clinical samples. A stable isotope-labeled version of each peptide, was synthesized through incorporation of 13C and 15N isotopes into Lysine (K) or Arginine (R) C-terminal residues, for use as internal standards [stable isotope standard (SIS) peptide] (Thermo Scientific, JPT). As the employed labeled peptides were not of highest purity, their purity and final concentrations were defined by MS analysis (MALDI TOF MS for AMBP, HBB, SERPINF1, IGHA1), parallel reaction monitoring (PRM) in comparison to standard/recombinant protein for B2M, or estimation of the labeled peptide amount by correlating 4 different analytical runs (not considered for the definition of the presented clinical associations) to respective ELISAs for LYZ.
Sample preparation and liquid chromatography-multiple reaction monitoring-mass spectrometry (LC-MRM-MS)
Equal volume (2 μL) of plasma samples containing approximately 100 μg of total protein were used for LC-MRM-MS analysis as previously described89,90. Briefly, after protein denaturation (8 M urea), reduction (10 mM dithioerythritol) and alkylation (50 mM iodoacetamide) the samples were digested with trypsin [(1:100 enzyme: protein ratio (w/w)] for 16 hours in the dark (RT). The peptide mixture was desalted with solid phase extraction zip-tips (Thermo Scientific) and the extracted peptides were dried using a vacuum centrifuge. The dried peptides were solubilized in mobile phase A (97.9% H2O, 2% acetonitrile, 0.1% formic acid), pH 3.5 to obtain a final concentration of 0.5 μg/μL. A mixture of the SIS peptides was then added in each sample after drying the peptides as follows: IGHA1: 8000 ng/mL, B2M: 800 ng/mL, HBB: 770 ng/mL, AMBP: 2240 ng/mL, LYZ: 800 ng/mL, SERPINF1: 668 ng/mL. Liquid chromatography was performed using an Eksigent nano-HPLC system, coupled with a C18 analytical column (75 μm × 150 mm, particle size 5 μm, pore size 100 Å) (Thermo Scientific). Peptide separation and elution were performed with a 60 min gradient of 5–90% mobile phase B (80% acetonitrile v/v, 0.1% FA, 19.9% H2O) at a flow rate of 300 nL/min. Samples were injected into the LC system and loaded on the C18 column. Tryptic peptides were analyzed on an AB Sciex 4000QTRAP with a nanoelectrospray ionization source controlled by Analyst 1.5 software (AB Sciex). The mass spectrometer was operated in MRM mode, with the first (Q1) and third quadrupole (Q3) at 0.7 unit mass resolution. Detailed information about the acquisition method and the used parameters are provided in Supplementary Table S4.
Standard curve
To define the range and ensure linearity in measurements, a calibration curve for each peptide was generated spiking SIS peptide at different amounts in a CKD pooled plasma sample (CKD stages 2–5, n = 4), in order to maintain the same background matrix to the analyzed samples. Dilution points were selected to cover reported concentration ranges per marker based on the literature and/or reported MS data and each dilution point was analyzed in triplicate. Samples were run in LC-MRM-MS, as described above.
Data analysis and quantification
Data analysis was performed using the Skyline software91 and all chromatograms were manually inspected to ensure the good quality and accurate peak picking. In particular, manual inspection ensured that the extracted ion chromatograms of the stable isotope-labeled standard (SIS) and endogenous (or natural-NAT) peptides chromatographically co-eluted and exhibited identical peak symmetry, shape, and width. The top signal producing transition was selected as the quantifier transition in all cases, while the remaining transitions were used as qualifier transitions, for accurate peak profile and retention time confirmation. In the case of standard curve, linear regression analysis was performed to define the slope and linearity of measurements.
Finally, the NAT/SIS ratio [Light/Heavy-relative response (RR)] of the quantifier transition was used for quantification (peak area of quantifier transition in NAT peptide/peak area of quantifier transition in SIS peptide). Concentrations were reported to ng/mL by using the protein molecular weights, as determined by Uniprot (https://www.uniprot.org).
ELISA
The same cohort of samples (N = 72) was analyzed with ELISA to investigate the correlation among the two different methods. The ELISA kits that were employed, specifically, included: for protein bikunin/AMBP: DY7744-05/R and D systems;, for SERPINF1: ab213815/Abcam, LYZ: ab108880/Abcam, IGHA1: ab196263/Abcam, and B2M: ab108885/Abcam. Values could not be obtained for n = 1 samples for SERPINF1 and n = 3 samples for B2M.
Statistical analysis (correlation-survival analysis)
Visualization of the stage analysis was performed by GraphPad Prism version 8.0.0 for Windows (GraphPad Software, San Diego, California USA, www.graphpad.com), whereas statistical and survival analysis were conducted in the R (version 3.6.1) environment for Windows utilizing base functions from the packages stats, survival and survminer. Significant changes at the levels of continuous variables across groups were determined with the non-parametric Kruskal-Wallis and Mann-Whitney tests. Kaplan-Meier plots were constructed to compare survival between Low and High protein concentration (ng/mL) groups, defined by a median cut-off point per protein, across samples. Survival curves were compared for significant differences (p < 0.05), from baseline diagnosis to 7 years follow-up time with the Log-rank test. Similarly, correlation analysis between eGFR and MRM quantifications were performed. Linear relationships between the continuous variables were assessed using Spearman’s Rank correlation coefficient and significance was defined at p ≤ 0.05.
Predictive value of the MRM panel
The prognostic value of the quantified proteins was assessed in distinguishing disease status at 7 years follow up time. The k-nearest neighbor classifier was developed with the packages DMwR, and pROC, in the R environment for Windows. In particular, the complete MRM data of 4 proteins (HBB, AMBP, B2M and LYZ) quantified across 69 subjects with CKD were utilized to build the model and parameter optimization was assessed in a 3-fold cross validation setting. In brief, patients were divided into three random partitions (adjusted for a balanced ratio of deceased/censored, stage and class cases), each time using two out of the three partitions for training, and the remaining third as a test set. Summary statistics of the classification performance for the classifier were obtained for the three folds and optimization for most suitable number of k-nearest neighbors was conducted iteratively as a function of maximizing the Area Under the Receiver Operating Characteristic curve across folds. Best fit was recorded for k = 16 neighbors and this number was further utilized to establish the final leave-one-out-crossvalidation (loocv) model. Patient specific probability scores of being labeled as deceased (scores ranging from 0.5 to 1) or censored (scores ranging from 0 to 0.5) were obtained with loocv and were converted to binary groups of “high” and “low risk”, respectively. Significant differences in survival between high and low risk groups were investigated with the Log-rank method, as described. Missing values (1.71% of the data) were replaced with the limit of quantitation for each protein (minimum concentration). Data for the knn classification were subjected to log transformation prior to training. The cox proportional hazard model was developed with the packages survival, survminer and fitting of proportionality was evaluated and examined with the functions cox.zph and ggcoxzph, respectively.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information Files). As described above, all methods were carried out in accordance with relevant guidelines and regulations. Skyline data are available upon request.
References
Hill, N. R. et al. Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. Plos One 11, e0158765 (2016).
Vanholder, R. et al. Reducing the costs of chronic kidney disease while delivering quality health care: a call to action. Nat Rev Nephrol. 13, 393–409 (2017).
Linde, P. G. et al. Overcoming Barriers in Kidney Health-Forging a Platform for Innovation. J Am Soc Nephrol. 27, 1902–1910 (2016).
Ortiz, A. et al. Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet 383, 1831–1843 (2014).
Thompson, S. et al. Cause of Death in Patients with Reduced Kidney Function. J Am Soc Nephrol. 26, 2504–2511 (2015).
Kurella, M., Lo, J. C. & Chertow, G. M. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol. 16, 2134–2140 (2005).
Matsushita, K. et al. Estimated glomerular filtration rate and albuminuria for prediction of cardiovascular outcomes: a collaborative meta-analysis of individual participant data. Lancet Diabetes Endocrinol. 3, 514–525 (2015).
Johnson, D. W. Evidence-based guide to slowing the progression of early renal insufficiency. Intern Med J. 34, 50–57 (2004).
Fink, H. A. et al. Screening for, monitoring, and treatment of chronic kidney disease stages 1 to 3: a systematic review for the U.S. Preventive Services Task Force and for an American College of Physicians Clinical Practice Guideline. Ann Intern Med. 156, 570–581 (2012).
Juncos, L. I. & Caputo, J. Combination therapy in chronic kidney disease? Ther Adv. Cardiovasc Dis. 9, 147–152 (2015).
Bonadonna, R. C., Borghi, C., Consoli, A. & Volpe, M. Novel antidiabetic drugs and cardiovascular risk: Primum non nocere. Nutr Metab Cardiovasc Dis. 26, 759–766 (2016).
KDOQI Clinical Practice Guideline for Diabetes and CKD: 2012 Update. Am J Kidney Dis. 60, 850–886 (2012).
Eckardt, K. U. et al. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet 382, 158–169 (2013).
Collins, A. J. et al. ‘United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. Am J Kidney Dis. 59(A7), e1–420 (2012).
Gansevoort, R. T., Nauta, F. L. & Bakker, S. J. Albuminuria: all you need to predict outcomes in chronic kidney disease? Curr Opin Nephrol Hypertens. 19, 513–518 (2010).
Perkins, B. A., Ficociello, L. H., Roshan, B., Warram, J. H. & Krolewski, A. S. In patients with type 1 diabetes and new-onset microalbuminuria the development of advanced chronic kidney disease may not require progression to proteinuria. Kidney Int. 77, 57–64 (2010).
Mischak, H., Delles, C., Vlahou, A. & Vanholder, R. Proteomic biomarkers in kidney disease: issues in development and implementation. Nat Rev Nephrol. 11, 221–232 (2015).
Francois, H. & Chatziantoniou, C. Renal fibrosis: Recent translational aspects. Matrix Biol. 68–69, 318–332 (2018).
Pena, M. J., Mischak, H. & Heerspink, H. J. Proteomics for prediction of disease progression and response to therapy in diabetic kidney disease. Diabetologia 59, 1819–1831 (2016).
Petra, E., Zoidakis, J. & Vlahou, A. Protein biomarkers for cardiorenal syndrome. Expert Rev Proteomics 16, 325–336 (2019).
Bhavsar, N. A., Kottgen, A., Coresh, J. & Astor, B. C. Neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule 1 (KIM-1) as predictors of incident CKD stage 3: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Kidney Dis. 60, 233–240 (2012).
Hojs, R., Ekart, R., Bevc, S. & Hojs, N. Biomarkers of Renal Disease and Progression in Patients with Diabetes. J Clin Med. 4, 1010–1024 (2015).
Krolewski, A. S. et al. Early progressive renal decline precedes the onset of microalbuminuria and its progression to macroalbuminuria. Diabetes Care 37, 226–234 (2014).
Looker, H. C. et al. Biomarkers of rapid chronic kidney disease progression in type 2 diabetes. Kidney Int. 88, 888–896 (2015).
Rebholz, C. M. et al. Serum fibroblast growth factor-23 is associated with incident kidney disease. J Am Soc Nephrol. 26, 192–200 (2015).
Argiles, A. et al. CKD273, a new proteomics classifier assessing CKD and its prognosis. Plos One 8, e62837 (2013).
Good, D. M. et al. Naturally occurring human urinary peptides for use in diagnosis of chronic kidney disease. Mol Cell Proteomics 9, 2424–2437 (2010).
Pontillo, C. et al. A urinary proteome-based classifier for the early detection of decline in glomerular filtration. Nephrol Dial Transplant. 32, 1510–1516 (2017).
Woodcock, J. Biomarker letter of support, https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/UCM508790.pdf (2016).
Critselis, E. & Lambers Heerspink, H. Utility of the CKD273 peptide classifier in predicting chronic kidney disease progression. Nephrol Dial Transplant. 31, 249–254 (2016).
Lindhardt, M. et al. Proteomic prediction and Renin angiotensin aldosterone system Inhibition prevention Of early diabetic nephRopathy in TYpe 2 diabetic patients with normoalbuminuria (PRIORITY): essential study design and rationale of a randomised clinical multicentre trial. BMJ Open 6, e010310 (2016).
Ebhardt, H. A., Root, A., Sander, C. & Aebersold, R. Applications of targeted proteomics in systems biology and translational medicine. Proteomics 15, 3193–3208 (2015).
Mermelekas, G., Vlahou, A. & Zoidakis, J. SRM/MRM targeted proteomics as a tool for biomarker validation and absolute quantification in human urine. Expert Rev Mol Diagn. 15, 1441–1454 (2015).
Gerber, S. A., Rush, J., Stemman, O., Kirschner, M. W. & Gygi, S. P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci USA 100, 6940–6945 (2003).
Percy, A. J., Chambers, A. G., Parker, C. E. & Borchers, C. H. Absolute quantitation of proteins in human blood by multiplexed multiple reaction monitoring mass spectrometry. Methods Mol Biol. 1000, 167–189 (2013).
Percy, A. J., Mohammed, Y., Yang, J. & Borchers, C. H. A standardized kit for automated quantitative assessment of candidate protein biomarkers in human plasma. Bioanalysis 7, 2991–3004 (2015).
Glorieux, G. et al. New insights in molecular mechanisms involved in chronic kidney disease using high-resolution plasma proteome analysis. Nephrol Dial Transplant. 30, 1842–1852 (2015).
Liabeuf, S. et al. Plasma beta-2 microglobulin is associated with cardiovascular disease in uremic patients. Kidney Int. 82, 1297–1303 (2012).
Wu, H. C., Lee, L. C. & Wang, W. J. Associations among Serum Beta 2 Microglobulin, Malnutrition, Inflammation, and Advanced Cardiovascular Event in Patients with Chronic Kidney Disease. J Clin Lab Anal. 31 (2017).
Liu, X. et al. Non-GFR Determinants of Low-Molecular-Weight Serum Protein Filtration Markers in CKD. Am J Kidney Dis. 68, 892–900 (2016).
Foster, M. C. et al. Non-GFR Determinants of Low-Molecular-Weight Serum Protein Filtration Markers in the Elderly: AGES-Kidney and MESA-Kidney. Am J Kidney Dis. 70, 406–414 (2017).
Rebholz, C. M. et al. Risk of ESRD and Mortality Associated With Change in Filtration Markers. Am J Kidney Dis. 70, 551–560 (2017).
Foster, M. C. et al. Serum beta-Trace Protein and beta2-Microglobulin as Predictors of ESRD, Mortality, and Cardiovascular Disease in Adults With CKD in the Chronic Renal Insufficiency Cohort (CRIC) Study. Am J Kidney Dis. 68, 68–76 (2016).
Foster, M. C. et al. Filtration markers as predictors of ESRD and mortality in Southwestern American Indians with type 2 diabetes. Am J Kidney Dis. 66, 75–83 (2015).
Rebholz, C. M., Grams, M. E., Matsushita, K., Selvin, E. & Coresh, J. Change in novel filtration markers and risk of ESRD. Am J Kidney Dis. 66, 47–54 (2015).
Astor, B. C. et al. Novel markers of kidney function as predictors of ESRD, cardiovascular disease, and mortality in the general population. Am J Kidney Dis. 59, 653–662 (2012).
Motomiya, Y., Yamagishi, S., Adachi, H. & Abe, A. Increased serum concentrations of pigment epithelium-derived factor in patients with end-stage renal disease. Clin Chem. 52, 1970–1971 (2006).
Shiga, Y., Miura, S., Mitsutake, R., Yamagishi, S. & Saku, K. Significance of plasma levels of pigment epithelium-derived factor as determined by multidetector row computed tomography in patients with mild chronic kidney disease and/or coronary artery disease. J Int Med Res. 39, 880–890 (2011).
Hui, E. et al. Elevated circulating pigment epithelium-derived factor predicts the progression of diabetic nephropathy in patients with type 2 diabetes. J Clin Endocrinol Metab. 99, E2169–2177 (2014).
Terawaki, H. et al. Pigment epithelium-derived factor as a new predictor of mortality among chronic kidney disease patients treated with hemodialysis. Ther Apher Dial. 17, 625–630 (2013).
Percy, A. J., Chambers, A. G., Yang, J., Hardie, D. B. & Borchers, C. H. Advances in multiplexed MRM-based protein biomarker quantitation toward clinical utility. Biochim Biophys Acta 1844, 917–926 (2014).
Guidance for Industry Bioanalytical Method Validation, https://www.fda.gov/downloads/Drugs/Guidance/ucm070107.pdf (2001).
Bioanalytical Method Validation Guidance for Industry, https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf (2018).
Uwaezuoke, S. N., Ayuk, A. C., Muoneke, V. U. & Mbanefo, N. R. Chronic kidney disease in children: Using novel biomarkers as predictors of disease. Saudi J Kidney Dis Transpl. 29, 775–784 (2018).
Bidin, M. Z., Shah, A. M., Stanslas, J. & Seong, C. L. T. Blood and urine biomarkers in chronic kidney disease: An update. Clin Chim Acta 495, 239–250 (2019).
George, J. A. & Gounden, V. Novel glomerular filtration markers. Adv Clin Chem. 88, 91–119 (2019).
Fassett, R. G. et al. Biomarkers in chronic kidney disease: a review. Kidney Int. 80, 806–821 (2011).
Castillo-Rodriguez, E. et al. Kidney Injury Marker 1 and Neutrophil Gelatinase-Associated Lipocalin in Chronic Kidney Disease. Nephron 136, 263–267 (2017).
Schanstra, J. P. & Mischak, H. Proteomic urinary biomarker approach in renal disease: from discovery to implementation. Pediatr Nephrol. 30, 713–725 (2015).
Kontostathi, G., Makridakis, M., Zoidakis, J. & Vlahou, A. Applications of multiple reaction monitoring targeted proteomics assays in human plasma. Expert Rev Mol Diagn. 19, 499–515 (2019).
Agger, S. A., Marney, L. C. & Hoofnagle, A. N. Simultaneous quantification of apolipoprotein A-I and apolipoprotein B by liquid-chromatography-multiple- reaction-monitoring mass spectrometry. Clin Chem. 56, 1804–1813 (2010).
Lin, D., Alborn, W. E., Slebos, R. J. & Liebler, D. C. Comparison of protein immunoprecipitation-multiple reaction monitoring with ELISA for assay of biomarker candidates in plasma. J Proteome Res. 12, 5996–6003 (2013).
Klont, F. et al. Quantification of surfactant protein D (SPD) in human serum by liquid chromatography-mass spectrometry (LC-MS). Talanta 202, 507–513 (2019).
Percy, A. J. et al. Protocol for Standardizing High-to-Moderate Abundance Protein Biomarker Assessments Through an MRM-with-Standard-Peptides Quantitative Approach. Adv Exp Med Biol. 919, 515–530 (2016).
Percy, A. J., Simon, R., Chambers, A. G. & Borchers, C. H. Enhanced sensitivity and multiplexing with 2D LC/MRM-MS and labeled standards for deeper and more comprehensive protein quantitation. J Proteomics 106, 113–124 (2014).
Vanholder, R. et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 63, 1934–1943 (2003).
Bernier, G. M. beta 2-Microglobulin: structure, function and significance. Vox Sang. 38, 323–327 (1980).
Duranton, F. et al. Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol. 23, 1258–1270 (2012).
Tombran-Tink, J., Chader, G. G. & Johnson, L. V. PEDF: a pigment epithelium-derived factor with potent neuronal differentiative activity. Exp Eye Res. 53, 411–414 (1991).
Tanabe, K., Sato, Y. & Wada, J. Endogenous Antiangiogenic Factors in Chronic Kidney Disease: Potential Biomarkers of Progression. Int J Mol Sci. 19 (2018).
He, X. et al. Pigment epithelium-derived factor, a noninhibitory serine protease inhibitor, is renoprotective by inhibiting the Wnt pathway. Kidney Int. 91, 642–657 (2017).
Dajak, M., Ignjatovic, S., Stojimirovic, B., Gajic, S. & Majkic-Singh, N. Evaluation of renal damage by urinary beta-trace protein in patients with chronic kidney disease. Clin Lab. 57, 29–36 (2011).
Shore, N., Khurshid, R. & Saleem, M. Alpha-1 microglobulin: a marker for early detection of tubular disorders in diabetic nephropathy. J Ayub Med Coll Abbottabad 22, 53–55 (2010).
Gertow, J. et al. Altered Protein Composition of Subcutaneous Adipose Tissue in Chronic Kidney Disease. Kidney Int Rep. 2, 1208–1218 (2017).
Liao, W. L. et al. Urinary Proteomics for the Early Diagnosis of Diabetic Nephropathy in Taiwanese Patients. J Clin Med. 7 (2018).
Zubiri, I. et al. Diabetic nephropathy induces changes in the proteome of human urinary exosomes as revealed by label-free comparative analysis. J Proteomics 96, 92–102 (2014).
Luczak, M. et al. Deeper insight into chronic kidney disease-related atherosclerosis: comparative proteomic studies of blood plasma using 2DE and mass spectrometry. J Transl Med. 13, 20 (2015).
Luczak, M. et al. Chronic kidney disease-related atherosclerosis - proteomic studies of blood plasma. Proteome Sci. 9, 25 (2011).
Kopecky, C. et al. Restoration of renal function does not correct impairment of uremic HDL properties. J Am Soc Nephrol. 26, 565–575 (2015).
Thein, S. L. The molecular basis of beta-thalassemia. Cold Spring Harb Perspect Med. 3, a011700 (2013).
Gotes, J., Kasian, K., Jacobs, H., Cheng, Z. Q. & Mink, S. N. Lysozyme, a mediator of sepsis that deposits in the systemic vasculature and kidney as a possible mechanism of acute organ dysfunction. Shock 41, 256–265 (2014).
Gallo, D. et al. Human recombinant lysozyme downregulates advanced glycation endproduct-induced interleukin-6 production and release in an in-vitro model of human proximal tubular epithelial cells. Exp Biol Med. (Maywood) 239, 337–346 (2014).
Abdul-Salam, V. B. et al. Identification and assessment of plasma lysozyme as a putative biomarker of atherosclerosis. Arterioscler Thromb Vasc Biol. 30, 1027–1033 (2010).
Florkowski, C. M. & Chew-Harris, J. S. Methods of Estimating GFR - Different Equations Including CKD-EPI. Clin Biochem Rev. 32, 75–79 (2011).
Mohammed, Y. et al. PeptidePicker: a scientific workflow with web interface for selecting appropriate peptides for targeted proteomics experiments. J Proteomics 106, 151–161 (2014).
Mohammed, Y. et al. PeptideTracker: A knowledge base for collecting and storing information on protein concentrations in biological tissues. Proteomics 17 (2017).
Whiteaker, J. R. et al. Using the CPTAC Assay Portal to Identify and Implement Highly Characterized Targeted Proteomics Assays. Methods Mol Biol. 1410, 223–236 (2016).
Desiere, F. et al. The PeptideAtlas project. Nucleic Acids Res. 34, D655–658 (2006).
Bitsika, V. et al. High-Throughput LC-MS/MS Proteomic Analysis of a Mouse Model of Mesiotemporal Lobe Epilepsy Predicts Microglial Activation Underlying Disease Development. J Proteome Res. 15, 1546–1562 (2016).
Kontostathi, G. et al. Development and Validation of Multiple Reaction Monitoring (MRM) Assays for Clinical Applications. Methods Mol Biol. 1959, 205–223 (2019).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Acknowledgements
This research has been co‐financed by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (mELISA-CKD, project code:T1EDK-03551). Also supported by EU COST action CliniMARK (CA16113).
Author information
Authors and Affiliations
Contributions
G.K. wrote the manuscript, performed sample preparation, Skyline data analysis, performed ELISA experiments and interpretation of the data. M.M. assisted in writing of the manuscript, performed sample preparation, mass spectrometry analysis, data collection and interpretation. E.P. performed Skyline data analysis and standard curve data analysis. R.S. developed the classifier, performed the survival and correlation analysis, as well as the statistical analysis. V.L. performed ELISA experiments. S.F. performed clinical correlations and assisted in writing. F.D. provided the clinical samples and respective clinical information. H.M. contributed to conception and design of the study and critically revised the manuscript. A.A. led the clinical study and data interpretation in clinical context and critically revised the manuscript. J.Z. contributed to study design, performed data interpretation and critical revision of the manuscript. A.V. conceived and designed the study, performed data interpretation and critically revised the manuscript. All authors took part in revision process and gave the final approval of the version to be published.
Corresponding author
Ethics declarations
Competing interests
H.M. is the co-owner of Mosaiques Diagnostics. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Makridakis, M., Kontostathi, G., Petra, E. et al. Multiplexed MRM-based protein quantification of putative prognostic biomarkers for chronic kidney disease progression in plasma. Sci Rep 10, 4815 (2020). https://doi.org/10.1038/s41598-020-61496-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61496-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
