
Abstract
Aluminium (Al) toxicity is the single most important contributing factor constraining crop productivity in acidic soils. Hydroponics based screening of three rice genotypes, a tolerant (ARR09, AR), a susceptible (IR 1552, IR) and an acid soil adapted landrace (Theruvii, TH) revealed that AR accumulates less Al and shows minimum decrease in shoot and root biomass under Al toxicity conditions when compared with IR. Transcriptome data generated on roots (grown in presence or absence of Al) led to identification of ~1500 transcripts per genotype with percentage annotation ranging from 21.94% (AR) to 29.94% (TH). A total of 511, 804 and 912 DEGs were identified in genotypes AR, IR and TH, respectively. IR showed upregulation of transcripts involved in exergonic processes. AR appears to conserve energy by downregulating key genes of glycolysis pathway and maintaining transcript levels of key exergonic step enzymes under Al stress. The tolerance in AR appears to be as a result of novel mechanism as none of the reported Al toxicity genes or QTLs overlap with significant DEGs. Components of signal transduction and regulatory machinery like transcripts encoding zinc finger protein, calcieurin binding protein and cell wall associated transcripts are among the highly upregulated DEGs in AR, suggesting increased and better signal transduction in response to Al stress in tolerant rice. Sequencing of NRAT1 and glycine-rich protein A3 revealed distinct haplotype for indica type AR. The newly identified components of Al tolerance will help in designing molecular breeding tools to enhance rice productivity in acidic soils.
Similar content being viewed by others


Introduction
Soil acidity affects 40% of the global arable land1. Forty nine million hectares of the total land area of India is acidic in nature2, a majority of which is in the North-Eastern Region, with an estimated 65% of area having pH below 5.53.
Aluminium (Al) is the most abundant metal and non-essential to plants. Enhanced soluble soil Al at low pH is the most important limiting factor in 67% of the acidic soils4. Under acidic condition, Al becomes more soluble. When pH drops, the soil bound Al is released in soluble forms such as Al(OH)2+ and Al(H2O)63+, (commonly known as Al3+) and becomes phytotoxic to the plants. When present in micromolar levels, Al hinders root elongation affecting uptake of water and nutrients5. Al toxicity causes fixation of phosphorous in soils and on root surfaces. As a result of which the crop productivity, especially in the upland conditions (where Al toxicity predominantly exists), gets severely affected.
Differential tolerance to Al toxicity attributed to variation in structure and function of roots. Short exposure to low Al concentration may only inhibit cell elongation. However, continuous exposure to high Al concentration inhibits both cell elongation and division and causes abnormal root morphology4,5. The mechanism of inhibition of root growth and aluminium tolerance is still not well understood.
The term Al resistance is used for plants which retain sizeable yields in soils having Al toxicity6. Among the small grained cereal crops, rice is the most Al tolerant with japonica varieties being more tolerant than indica varieties7. The Al toxicity tolerance in rice is a multigenic trait having complex genetics as supported by identification of 32 non-overlapping quantitative trait loci (QTLs) from different bi-parental studies8,9,10,11,12, and identification of 48 novel loci for Al resistance by genome-wide association mapping13. Association study of Chinese rice landraces using genome-wide simple sequence repeats (SSRs) has suggested several genomic regions associated with Al toxicity tolerance14. Certain genes/loci of vital importance for better understanding have been suggested for Al toxicity tolerance15.
Several genes (OsSTAR1 (Sensitive to Al Rhizotoxicity 1), OsSTAR2 (Sensitive to Al Rhizotoxicity 2)), transcription factors (OsART1 (Aluminium rhizotoxicity 1)) and transporters (ALMT, OsNRAT1 (Nramp (natural resistance-associated macrophage protein) aluminum transporter 1), OsFRDL4 (FERRIC REDUTASE DEFECTIVE LIKE 1), OsALS1 (Al-Sensitive 1)) associated with Al toxicity resistance have been identified till date6,16. Aluminium activated Malate Transporter (ALMT) was the first aluminium toxicity tolerance gene to be identified in wheat7,17. ALMT protein is involved in efflux of malate from roots6. A MATE (Multidrug And Toxic compound Extrusion) ortholog of SbMATE (Sorghum bicolor MATE) in rice is FRDL418, which transports citrate transporter and can form complexes with Al. An aluminium transporter localized in the PM, NRAT1, is responsible for transporting Al across PM19. A vacuolar ABC (ATP Binding Cassette) transporter, ALS1, sequesters Al into vacuoles20, and in this sequestration process NRAT1 works together with ALS16. The Al responsive genes, STAR1 and STAR2 were isolated by mutant screen and these together work as an efflux ABC transporter to transport UDP-glucose into cell wall21,22. This modification of cell wall by altering carbohydrate composition results in reduce Al binding and thereby, accumulation. A zinc finger transcription factor, ART1, was characterized as an ortholog of Arabidopsis transcription factor, AtSTOP123,24,25. OsART1 regulates at least 30 down-stream genes including STAR1, STAR2, NRAT1, FRDL4 and ALS mostly via a specific cis-element26. Apart from the aforementioned genes, two rice genes located on PM, an Mg uptake transporter (OsMGT1; Magnesium Transporter 1) and a small peptide (OsCDT3; Cadmium Tolerance 3), stop entry of Al inside roots by binding to it27,28.
The above mentioned studies indicate that there may be multiple genes and molecular mechanisms affecting Al toxicity tolerance in rice. However, a comprehensive understanding of nature and role of such genes in rice is lacking. It has also been suggested that high level of rice Al resistance is due to synergy of mechanisms involving multiple genes6.
The current study was undertaken to gain a better understanding of Al toxicity tolerance in Indica rice genotypes adapted to upland acidic soils of North Eastern India, where aluminum toxicity is a major problem. The aim of this study was to generate seedling stage root transcriptome (RNAseq) data on three different rice genotypes, a tolerant, a susceptible and an adapted landrace, to identify genes differentially regulated under Al toxic conditions. The selection of genotypes ARR09 (AR), Theruvii (TH) and IR1552 (IR) was based on (a) differential response in upland acidic field conditions (pH < 4.2) and (b) genetic diversity29. While AR is an upland genotype from North eastern state of Arunachal Pradesh, TH is an acidic soil adapted landrace from Meghalaya and IR is an international check for Al sensitivity. During field evaluation for performance under upland acidic field conditions, AR showed good performance (infact used as tolerant local check) while IR showed very poor performance (unpublished data). The genotype, TH, is a landrace with low yield. The analysis of transcriptome has led to identification of Al responsive genes and pathways associated with Al tolerance, which in addition to better insight, would lead to development of novel markers and molecular breeding tools for enhancing rice productivity in Al toxic acidic soils.
Results
Selection of rice genotypes for transcriptome study
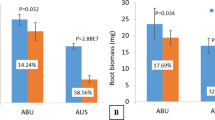
Hydroponics experiment conducted for aluminium toxicity tolerance (0.54 mM) proved that 5 days treatment is able to distinguish between tolerant (AR), susceptible genotype (IR) and acidic soil adapted landrace (TH) based on root thickness, lateral roots (Fig. 1A) and differential haematoxylin staining (Fig. 1C–E). Our previous experiments using background SSR markers had suggested that these three rice genotypes are genetically diverse29. The comparison of Al3+ accumulation, as indicated by aluminum estimation suggests that susceptible genotype IR accumulates significant amounts of Al in its roots whereas AR and TH do not. Hematoxylin staining (Fig. 1A), with RRG (relative root growth) data, showed that root inhibition and Al3+ accumulation in roots are related. RRG value was least in TH followed by IR and AR (Table 1).

Morphological variation for 10 days old three rice genotypes grown in modified Magnavaca nutrient in presence or absence of Al3+ Variation in the root growth pattern (A), aluminium content in root (b) and shoot (C), 2% haematoxylin staining (D–F) of 10 days old rice genotypes grown under control (0 Al3+) and treatment (0.54 mM of Al³+) for five days (G) root and shoot biomass of treated plants expressed as respective percentage of plants grown under control condition. The encircled portions (D–F) represent the root portion where Al3+ gets accumulated Values are means (n = 3), and bars indicate ± standard errors. The significant values are indicated by *Names of the genotypes are given on the X-axis.
Data generation and assembly of transcriptome
The transcriptome data generated on the three genotypes was checked for quality (QC) (Supplemental File S1) and the QC passed reads ranged from 0.41 million reads (AR_T) (Table 2) to 2.86 million reads (AR_C) (Table 2). These QC passed transcripts were then assembled. For assembly without reference reads>200 bp length were considered. The MIRA assembler provided a better integrated assembly of transcriptome, as compared to TRINITY (Tables 3 and 4). Also, the number of transcripts that were >200 bp in length was ~1500 per genotype for MIRA. A merged transcriptome assembly was done for each of the genotype (eg. ARC_ART denotes both samples of genotype AR merged to form the assembly) and this was used for further annotation of transcripts.
Mapping statistics and annotation of transcripts
The QC passed transcripts were considered as expressed. A total of 8,506, 24,725 and 25,120 transcripts passed the cut-off for TH, IR and AR, respectively (Fig. 2A). The percentage annotation ranged from 21.94% (AR), 23.54% (IR) to 29.94% (TH) (Fig. 2A, Supplementary Tables S1–S3) suggesting transcriptionally active regions which are uncharacterized till date. The transcripts that passed QC were checked for differential expression in presence or absence of Al3+. A cut off p value (<0.05 and a minimum read count of 10) was used to identify differentially expressed genes (DEGs). Significant DEGs (>2 log2 fold expression) were also identified for each of the three genotypes. As shown in Fig. 2B, 511 DEGs were identified in AR out of which 99 and 100 transcripts were significantly upregulated and downregulated, respectively. Similarly, 804 DEGs were identified in IR, out of which 219 were upregulated and 368 were downregulated. In the case of TH, out of a total of 912 DEGs, only 37 transcripts showed significant upregulation in presence of Al3+ (Fig. 2B).

Differential gene expression under Al stress condition. (A) The total, unannotated and annotated transcripts identified for the three rice genotypes AR, IR and TH is shown in the bar graph. (B) Bar diagram showing the number of up- and down- regulated genes in response to Al toxicity in the roots of AR, IR and TH. The total number of genes differentially expressed under each condition is given on the top of each bar (C). The correlation of gene expression results obtained from qPCR analysis and RNA-seq for fifteen selected genes.
Validation of selected up-regulated DEGs by Q-PCR
A set of 15 genes were randomly selected and validated by qRT–PCR analysis in three biological replicates for confirming the results obtained from DEG analysis. The analysis revealed similar expression pattern for all the selected genes in qPCR analysis as observed from RNA-seq data. The statistical analysis also showed significant association (r2 = 0.78) between the results of qPCR and RNA-seq data analyses (Fig. 2C).
Analysis of stress-responsive genes reveals genes specific for Al toxicity conditions
Using rice Nipponbare genome as reference, transcripts were assigned gene ontology (GO) terms in three different categories: biological process, molecular function and cellular component. Using top 10 GO terms in each of the three categories, the figure generated for the tolerant and susceptible genotypes revealed a clear difference. In the case of biological activity, while the tolerant genotype AR showed increase in GO terms associated with metabolic processes, oxidation and reduction and cellular processes (Fig. 3A), IR showed increase in DNA replication along with metabolic processes and oxidation and reduction processes (Fig. 3B). The genotype TH under Al toxicity conditions shows increase in GO terms associated with DNA replication, proteolysis and endonucleolytic activity (Fig. 3C). For molecular function GOSlim terms, the largest number of transcripts for AR belonged to catalytic activity, metal ion and nucleotide binding activity. In the case of IR, transcripts with ribonuclease activity, nucleic acid and ATP binding activity were in greater number. The largest number of transcripts belonged to nucleic acid binding, ribonuclease and polymerase activity for genotype TH. Among the cellular components, transcripts related to membrane, plastid, chloroplast and integral component of membrane were largest in AR whereas transcripts related to mitochondria, plastid and membrane bound vesicle were largest in IR. Transcripts related to mitochondria, plastid and membrane bound vesicle were largest in TH.

Top ten GO terms in each of the three categories, molecular function, cellular component and biological process for the three rice genotypes AR, IR and TH. The top GO terms for molecular function (blue), cellular component (green) and biological process (red) categorised for genotypes AR (A), IR (B) and TH (C).
The functionally annotated genes were mapped for the three genotypes to check whether any particular pathway related genes were differentially regulated in tolerant and susceptible genotypes (Fig. 4). It was observed that the susceptible genotype (IR) showed upregulation of enzymes like phosphoglycerate kinase involved in exergonic process (02g0169300). In the case of IR, an ABC transporter (01g0732500) was also upregulated. The acidic soil adapted TH showed an increase in transcript encoding citrate synthase (02g0194100). The genotype AR on other hand showed no differential expression for the transcripts for the two enzymes and downregulation for enolase (06g0136600); phosphofructokinase (05g0524400) and fructose bisphosphate (01g0118000) (Fig. 4A). In TH, transcripts for methionine synthase (012g0624000) and zeaxanthin epoxidase (04g0448900) were also upregulated. Arginine biosynthetic gene (03g0279400) and lignin biosynthetic genes (06g0656500 and 02g0697400) were significantly upregulated in IR (Fig. 4B) but downregulated in AR.

GO enrichment in response to Al stress across three rice genotypes. Significantly, enriched GO categories in the up-regulated genes in tolerant genotype AR (a) and susceptible genotype IR (B) and TH (C) are shown. The genes were analyzed using BiNGO and the biological process terms showing significant enrichment are shown. Node size is proportional to the number of genes in each category and shades represent the significance level (yellow—no significant difference; red −>log2 fold upregulated; green − <log2 fold downregulated; scale, P = 005 to P < 00000005).
We also performed K-means clustering analysis for the three genotypes in response to Al stress condition and generated major clusters. Among the clusters generated, three groups of genes with distinct gene expression patterns (Fig. 5) were identified. Group I genes were up-regulated under Al stress. Group II genes were down-regulated under normal conditions. Group III genes were down-regulated under Al stress.

Heat map representation and K-means clustering of expression profiles of genes differentially expressed under Al stress conditions in AR (A), IR (B) and TH (C). The ion torrent RNA-seq data were re-analyzed, and the FPKM values were log2 transformed and heat map generated using MeV v4.11 software. Clustering was performed on log2 fold change for each gene under Al stress conditions when compared with control condition across the three genotypes. Genes exhibiting a similar pattern of expression under Al stress conditions cluster together. Red and green colours denote control (no Al3+) and treatment (0.54 mM Al3+), respectively. Bar at the bottom represents log2 fold values.
Al toxicity response in rice genotypes with respect to known genes/QTLs
A set of 54 already reported rice genes (Supplementary Table S4) playing different molecular and physiological roles in Al toxicity were cross-referred in the significantly expressed dataset produced for the three rice genotypes in this study. Surprisingly, DEGs from the tolerant genotype AR show no overlap (Fig. 6A). In fact, across genotypes, none of the upregulated DEGs show any overlap with reported genes for Al toxicity. In TH, two significantly downregulated transcripts show an overlap with an unknown protein and an expansin (Fig. 6A, Supplementary Table S5). A set of nine QTLs lying on chromosomes 1, 2, 6, 9 and 12 of rice have been previously identified using hydroponics based screening13 (Supplementary Table S6). The DEGs from tolerant genotype AR do not overlap with these reported QTLs. However, TH DEGs overlap with QTL AltTRG1.1, AltPRG1.1, AltPRG6.1, AltLRG9.1, AltTRG1.1 and AltTRG12.1. IR DEGs overlap with QTL AltTRG1.1, AltPRG1.1 and AltPRG6.1. Among the 7 genomic loci/segments chosen, none of them harbored even one up regulated gene. Down regulated genes were found in chromosomes (chr #) 1, 4, 5 and 7. Chromosome 1 harbored the maximum number of down regulated genes (13 genes). ART1 is a transcription factor which is constitutively expressed and its expression is not affected by Al. It regulates a set of 31 genes in rice. We checked the status of these 31 genes in DEGs of the three genotypes. It was found that the DEGs found in the three genotypes are not regulated by ART1 (data not shown).

Mapping statistics of DEGs against already reported (A) genes and (B) QTLs for Al toxicity tolerance in rice. Total number of reported genes mapped- Number; Upregulated DEGs- Up_Regulated, Downregulated DEGs; Down_Regulated; Upregulated DEGs mapped to a reported rice QTL- Up_QTL_mapped; Downregulated DEGs mapped to reported rice QTL- Down_QTL_mapped.
Significantly expressed DEGs were also mapped to check overlap with reported rice QTLs. In genotype AR, three downregulated DEGs map to 2 QTLs for plant hopper resistance (chr # 2) and ultra violet light resistance (chr # 1) (Fig. 6B, Supplementary Table S5). Genotype IR showed overlap with QTLs for plant hopper resistance (chr # 2), ultra violet light resistance (chr # 1 and 4) and spikelet number (chr # 2 and 9).Genotype TH showed maximum overlap (a total of 16 QTLs) including QTLs for spikelet density (chr # 3), spikelet number (chr # 2 and 9), spikelet sterility (chr # 5), tiller number (chr # 3 and 4), seed dormancy (chr # 1, 2, 3 and 8), plant height (chr # 1 and 4), leaf length (chr # 5 and 10) and root number (chr # 3 and 5).
Sequencing 3061 bp across Nrat1 revealed a total of six nucleotide variations (Fig. 7A). Five nucleotide substitutions were observed in the intronic region. IR contains one SNP in the exonic region and two SNP in the intronic region. TH was found to carry the complete reference haplotype. Sequencing data showed that the exonic region of both azucena (AZ) and AR possess reference type allele but nucleotide variation in the intronic region was observed. AR has a SNP at position 729 in the genomic region while AZ has three nucleotide variations at positions 769, 781 and 3435. AZ had one SNP in common with IR at position 621 in the genomic region. One putative functional polymorphism specific to the Al sensitive genotype (IR) at position 3157 in the genomic region was identified. Change in the nucleotide level did not lead to any changes at the protein level in any of the genotypes. Though the SNP lie in the conserved region of the protein, it does not affect the conserved domain (Fig. 7c). All the tolerant and susceptible genotypes translate same protein for Nrat1. Four transversions and two transitions were observed for this gene. The nucleotide diversity was calculated for each genotype for genomic and coding sequence (CDS) regions (Supplemental Table S7). A genomic region of 2590 bp region covering 470 bp of the 5′ untranslated region (UTR), the whole CDS region and 574 bp of the 3′ UTR region was sequenced across 4 genotypes for glycine-rich A3 (Fig. 7B). A total of fifteen nucleotide variations were observed. Eight nucleotide substitutions were observed in the 5′ UTR region while three nucleotide substitutions were observed in the exonic region and four in the 3′ UTR. AR contains one nucleotide substitution in the exonic region which is common between TH and IR. It also contains five nucleotide substitutions in the 5′ UTR and two in the 3′ UTR region. AZ possess a complete reference type haplotype except for one nucleotide variation observed in the 3′ UTR region at position 2271 bp (Fig. 7D). This nucleotide substitution in the 3′ UTR region was found to be common in all the four genotypes. Both IR and TH contain 3 common nucleotide substitutions in the exonic region as well as in the 5′ UTR region of the gene. At the protein level, three synonymous substitutions were observed. Nucleotide substitution unique to AR was observed at position 2504 bp in the 3′ UTR region. Similarly, SNP in the 3′ UTR region at position 2423 bp was observed only in IR. A total of twelve transitions and three transversions were observed in the genomic region. The nucleotide diversity was calculated for the genomic region and the CDS region for each genotype (Supplemental Table S7).

Single nucleotide polymorphism (SNP) and sequence alignment across NRAT 1 (A,C) and glycine-rich A3 (B,D) genes for five rice genotypes. Dark grey and white colours indicate reference and novel allele, respectively. Sequence alignment of NRAT 1 (C) and glycine-rich A3 for five rice genotypes where numbers 1, 2, 3, 4 and 5 indicate reference genotype, AR, AZ, TH and IR, respectively. The encircle portions indicate SNPs unique to AR.
Expression pattern of differentially regulated top 40 genes in three genotypes
A set of 40 transcripts highly expressed (log2 fold) in tolerant genotype AR were cross examined in the other two genotypes for expression levels (Table 5). Most of the DEGs were differentially regulated between the tolerant and susceptible genotypes. The highly expressed DEGs can be classified into; (a) signal transduction/regulatory components like transcripts encoding zinc finger protein (12g0581900) auxin induced protein (02g0445600), calcineurin binding protein (03g0685700); micro RNA gene (miR424a gene (AY730701.1), genes involved in chromatin modelling (02g02290) and RNA binding regulatory protein (12g01010); (b) cell wall associated transcripts like EP_003637074 and EXC01915 and cell wall hydrolase (AP014959). Hydrolase was significantly downregulated in AR (as compared with IR). (C) Genes for enzymes or accessory factors like peroxidase (11g0112200), isoflavone reductase (10g01044), peptidase (06g0625400), etc. are expressed in contrasting manner between AR, IR and TH. Transcripts encoding accessory factors for enzymes like LYR motif containing proteins (01g0342800) and UV repressible protein (02g0240100) are down regulated in AR but upregulated in TH and IR; (d) transcripts for components of translation machinery like 40S ribosomal protein S-15-like (03g0798600), 60S ribosomal protein L28-1 (05g0541900) and 18S rRNA sequence and (e) transcripts of unknown function (EPS70027, EXB92316, XP_007154367, 10g0102900, XP_006854095, 10g0154566, AP014960, 03g0160900, AK064232.1) were also differentially expressed.
Discussion
The major constrains to obtaining potential yields are various abiotic and biotic stresses encountered by plants. An estimated 39% and 52% of land suited to rice production in South and Southeast Asia, respectively is affected by severe soil problems30. These various soil problems are due to toxicity or deficiency of macro- and/or micro-nutrients. Therefore, a better understanding of molecular basis of uptake, transport, homeostasis and assimilation of these nutrients followed by application holds potential for gaining increases in crop productivity. In the case of acid soils, Al toxicity is the primary limitation to plant growth31. Avoidance and/or tolerance to Al is by exclusion and detoxification, respectively31. Till date various molecular and physiological strategies to combat Al toxicity from different higher plants have been reported like exclusion of Al3+ using malate transporter4, reactive oxygen species32, ART14, modification of DNA33, etc. It is also known that most genes induced by Al are involved in various stress responses including phosphorus deficiency34. Rice is the most Al-tolerant of the cereal crop35 species. Inhibition of root growth, deposition of callose at the root apex along with cellular damage is the major Al toxicity symptom observed in plants4,36. Degree of toxicity varies depending on the genotype, Al concentration and duration of exposure37. Role of multiple mechanisms with underlying numerous genes has been suggested for high level of Al resistance in rice6. Therefore, it is important to understand Al toxicity response mechanism in diverse rice genotypes. Acidic soils adapted rice genotypes are therefore, one such genetically diverse pool, which can be targeted for better understanding of this complex mechanism. In our study, root transcriptome of diverse rice genotypes was targeted for better molecular understanding. The root growth inhibition in plants in Al3+ toxicity may not offer enough information about the level of tolerance38. It has also been reported that root biomass can be taken as an indirect trait for selection of tolerant genotypes. Similar visual toxic symptoms of Al3+ treated roots as observed by us have been reported earlier39. It has been proposed that cell wall of root cells is the site of both Al3+ toxicity and Al3+ exclusion. Most of the Al3+ absorbed by roots is localized to the apoplast37. Al3+ probably binds to the pectin matrix, replacing Ca2+ and makes the cell wall rigid, thereby affecting cell elongation37.
The data generated in the current study has revealed that none of DEGs identified from tolerant upland rice genotype overlap with the previous reported QTLs for Al toxicity tolerance13. However, TH DEGs overlap with QTLs AltTRG1.1, AltPRG1.1, AltPRG6.1, AltLRG9.1, AltTRG1.1 and AltTRG12.1. IR DEGs overlap with QTLs AltTRG1.1, AltPRG1.1 and AltPRG6.1 with chromosome 1 showing maximum number of down-regulated genes in this susceptible genotype.
In the case of Al tolerance, using a set of 36,901 SNPs on a diverse set of 383 genotypes along with two immortal populations, three regions overlapping with genes ART 1, STAR 2 and NRAT 1 have been identified13. Further work on NRAT 135 using 24 rice genotypes led to identification of alleles associated with Al tolerance. AR carries a distinct haplotype for NRAT 1 (as well as glycine-rich A3), but the functional relevance, if any needs to be further investigated. ART1 is a transcription factor which is constitutively expressed and its expression is not affected by aluminum. It regulates a set of 31 genes in rice. We checked the status of these 31 genes in DEGs of the three genotypes. It was found that the DEGs found in the three genotypes are not regulated by ART1. It has been shown for an Al-tolerant cultivar Koshihikari, that other mechanisms apart from ART1-regulated mechanism do exist40. Root proteome studies on Al tolerant rice has revealed modulating the available energy may be one way of combating Al toxicity41. In high Al accumulating species like buckwheat, it has been shown that most of the ART1-regulated gene homologs were not differentially regulated in response to high Al levels42. ART2, which is a homolog of ART1 has also been reported to regulate Al toxicity tolerance albeit with a different set of genes43. These genes were also not differentially regulated in AR genotype. An ABC transporter was significantly upregulated in IR. Previously, ABC transporters located on tonoplast have been implicated in Al detoxification20. It would be of interest to check the role of this ABC transporter in Al toxicity response.
One of the transcripts that were highly upregulated in response to Al in AR encodes S15- like ribosomal protein. Members of ribosomal S15 proteins in Arabidopsis viz. RPS15aA, -D and -F are expressed abundantly under temperature and metal stress44. It has been suggested that stress may result in altering composition of ribosomes and biased translation45. Another transcript that was differentially expressed codes for large subunit of ribulose bisphosphate. It is reported that stresses like salt46, drought, ozone, high temperature and cadmium47 lead to accumulation of large chain of ribulose bisphosphate in rice and Arabidopsis cells, respectively. This is a major leaf protein involved in photosynthesis and photorespiration.
Increased and better signal transduction in response to Al stress can be one of the strategies of tolerance (Fig. 8). The various components of signal transduction and regulatory machinery like zinc finger protein, calcieurin binding protein, cell wall associated transcripts are among the highly upregulated DEGs in tolerant genotype AR. It has been reported that cell wall associated kinases play a vital role in signaling mechanisms in response to heavy metal toxicity48. It is to be seen whether the cell wall associated proteins coded by transcripts upregulated in AR have kinase activity. AR DEGs reflect a better and efficient response to Al stress (as indicated by the GO terms). Also, based on pathway analysis of functionally annotated genes, it appears that AR conserves energy by downregulating key genes (fructose bisphosphate, enolase and phosphofructokinase) of glycolysis pathway and maintaining transcript levels of two key exergonic step metabolizing genes (citrate synthase and phosglycerate kinase) under Al stress (Fig. 4A). Thickening of rice roots due to callose deposition is an energy dependent process wherein sugar is diverted for deposition along cell walls6,36,37. Previously, it has been reported that metal stress leads to significant alterations in proteins involved in energy metabolism41,49. The number of downregulated transcripts is much higher in Al toxicity conditions. Another transcript which was significantly different between tolerant AR and susceptible IR encodes LYR motif containing protein. These motifs are characteristic of Fe-S proteins, which in turn are involved in electron transport, photosynthesis and DNA repair. Previously, it has been shown that rice genotypes carrying SUB1A gene survive flooding stress by suppressing shoot elongation and conserving energy50. This is achieved by higher level induction of genes, Slender Rice-1 (SLR1) and SLR Like-1 (SLRL1), involved in ethylene signalling pathway50.

Schematic representation of the probable mechanism of susceptibility, tolerance and adaptation based on Al root transcriptome data for genotypes IR, AR and TH, respectively ↑–upregulation and ↓– downregulation.
Our work provides insight into a novel mechanism of Al toxicity tolerance in indica type rice, AR. The comparison of Al3+ accumulation, suggests that susceptible genotype IR accumulates significant amounts of Al in its roots whereas AR and TH do not. Transcriptome data generated on roots identified a total of 511, 804 and 912 DEGs in genotypes AR, IR and TH, respectively. The tolerance in AR appears to be as a result of novel mechanism as none of the reported Al toxicity genes or QTLs overlap with significant DEGs. Transcriptome reveals aluminum toxicity tolerant rice adapts to high aluminum through expression of genes involved in cell signaling and energy conservation whereas susceptible genotype responds though energy intensive processes. Sequencing of NRAT1 and glycine-rich protein A3 revealed distinct haplotype for indica type AR. The newly identified components of Al tolerance will be helpful in development of molecular breeding strategies for development of Al toxicity tolerant rice varieties.
Materials and method
Plant material and Al toxicity treatment
Three rice genotypes (ARR 09 (AR); IR1552 (IR), and theruvii (TH)) were selected for generating NGS data for seedling stage Al toxicity tolerance. Seeds (30–40) of the selected genotypes were germinated on petri plates containing moist filter paper and kept in the green house. The 5-day old seedlings of each genotype were transferred to full strength Magnavaca’s solution containing 0.54 mM AlCl3 to create Al toxic hydroponic condition51. A separate set of seedlings was grown as control in Magvaca’s solution without AlCl3. After 5 days of treatment, phenotypic traits like fresh root weight and shoot weight were measured. The samples were divided in half: one half was cryopreserved for RNA extraction and transcriptome analysis. The other half was dried; dry weight measured and estimation of total Al carried out using Atomic Absorption Spectrometer (Perkin Elmer Analyst 200). Three independent replicates were used per genotype.
Hematoxylin staining
The roots of 10-day old seedlings grown in presence or absence of Al, were used for hematoxylin staining [0.2% hematoxylin (Himedia) and 0.02% potassium iodide, w/v] following standard procedure52. Free hand cross sections for the root were made and studied under the microscope (Leica DM 750) at 10X and 40X.
RNA extraction and generation of raw reads
All the six samples were sent to the commercial service provider, Xcelris Genomics Ltd, India for sequencing. The extraction of total RNA, RNA quality and quantity analysis, library generation and identification of quality reads was performed following previously reported methodology53. The mapping of the reads was done with spliced read mapper TopHat version 1.4.1 using Nipponbare (MSU release 6.1) genome as reference. The collection of mapped paired end reads and read counts were done using Picard 1.63 and HTSeq version 0.5.3, respectively53 (Supplemental Fig. S1).
Quality check, data assembly and transcriptome generation
The assembly of QC passed sequence reads was performed to identify putative transcripts that were detected in each pair of rice genotypes (control and treatment) using two MIRA and TRINITY assemblers independently. The transcripts were divided into various sizes to find heterogeneity and degree of fragmentation. Typically, transcripts >100 bp in length were considered for downstream analysis (for ion torrent data).
Differential gene expression and annotation of transcriptome
The differential analysis was done using DESeq v 2.054 and annotation using Blast2go55 for all the six samples. Nonredundant protein database of National Center for Biotechnology Information (NCBI) was used for BLASTx56 (E value ≤0.00001; annotation cutoff = 55; GO weight = 5). The transcriptome for the three genotypes was analyzed for a) DEGs and b) transcripts which were specific to the Al concentration (treatment (0.54 mM Al3+)).
Confirmation of random DEGs by qRT-PCR
To validate the results from the transcriptome experiment, 15 randomly selected DEGs were analyzed using qRT-PCR. Total RNA was extracted from roots using Spectrum plant total RNA kit (SIGMA) according to the manufacturer’s protocol. The first strand cDNA synthesis was carried out and rice ubiquitin gene54 was used as reference gene for normalization as previously described57. Three replicates were performed.
Gene ontology, functional annotation and identification of pathways enriched in Al toxicity
The statistically significant genes were identified and classified using standard methods as previously described53. For functional annotation of the genes, all the pathway related genes sequence involved in rice were downloaded and blast of all pathway genes (as database) and transcripts (as query) from all samples was done. Mapped LOC ID (Pathway involved gene id) was converted into RAP ID and each RAP ID (Pathway involved gene id) was mapped to our transcript expression data and linked with pathway name. This data was used as input in programme cytoscape for image generation (≥2 (upregulate, red colour, ≤ −2 downregulated, green colour).
Mapping of known/reported loci/QTLs
Sequences for all genomic positions were extracted and BLAST on up- and down-regulated transcript from all samples (as query) against genomic sequence (as database) was performed.
Sequencing across reported genes for Al toxicity tolerance
Sequencing across the two reported genes, NRAT 1 and glycine-rich protein A313 was done following standard methods3. All the four genotypes (including Azucena (AZ), an already reported tolerant aus genotype was included for sequencing)13 produced single amplicon with the gene-specific primers using high fidelity Taq polymerase (Sigma-Aldrich, USA) when analyzed by agarose gel electrophoresis. The PCR products were sent for sequencing using forward and reverse primers to Amnion Biosciences Pvt. Ltd, India. Sequences obtained were aligned using BioEdit 7.2.5. Nipponbare was used as a reference genome for sequence alignment, putative SNPs identified based on sequence homology and nucleotide diversity (π) calculated.
Data availability
The RNAseq data reported are available in NCBI sequence read archive under the accession number SUB986310 (PRJNA287493). The data sets supporting the results of this article are included within the article.
References
Uexküll, H. R. & Mutert, E. Global extent, development and economic impact of acid soils. Plant Soil 171, 1–15 (1995).
Kumar, M. et al. Variable lime requirement based on differences in organic matter content of iso-acidic soils. Indian J. Hill Farming 25, 26–30 (2012).
Yumnam, J. S., Rai, M. & Tyagi, W. Allele mining across two low-P tolerant genes PSTOL1 and PupK20-2 reveals novel haplotypes in rice genotypes adapted to acidic soils. Plant Genet Resour-C 15, 221–229 (2017).
Abate, E., Hussien, S., Laing, M. & Mengistu, F. Aluminium toxicity tolerance in cereals: Mechanisms, genetic control and breeding methods. F. Afr. J. Agric. Res. 8, 711–722 (2013).
Ma, J. F., Chen, Z. C. & Shen, R. F. Molecular mechanisms of Al tolerance in gramineous plants. Plant Soil 381, 1–12 (2014).
Kochian, L. V., Pineros-Miguel, A., Liu, J. & Magalhaes, J. V. Plant adaptation to acid soils: The molecular basis for crop aluminum resistance. Annu. Rev. Plant Biol. 66, 571–598 (2015).
Delhaize, E., Ma, J. F. & Ryan, P. R. Transcriptional regulation of aluminium tolerance genes. Trends Plant Sci. 17, 341–348 (2012).
Nguyen, V. T. et al. Molecular mapping of genes conferring aluminum tolerance in rice (Oryza sativa L.). Theor. Appl. Genet. 102, 1002–1010 (2001).
Nguyen, V. T. et al. Mapping of genes controlling aluminum tolerance in rice: comparison of different genetic backgrounds. Mol. Genet. Genomics 267, 772–780 (2002).
Nguyen, B. D. et al. Identification and mapping of the QTL for aluminum tolerance introgressed from the new source, Oryza rufipogon Griff. into indica rice (Oryza sativa L.). Theor. Appl. Genet. 106, 583–593 (2003).
Wu, P. et al. QTLs and epistasis for aluminum tolerance in rice (Oryza sativa L.) at different seedling stages. Theor. Appl. Genet. 100, 1295–1303 (2000).
Spindel, J. et al. Bridging the genotyping gap: using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor. Appl. Genet. 126, 2699–2716 (2013).
Famoso, A. N. et al. Genetic architecture of aluminum tolerance in rice (Oryza sativa) determined through genome-wide association analysis and QTL mapping. PLoS Genet. 7, 1–16 (2011).
Zhang, P., Zhong, K., Tong, H., Shahid, M. & Li, J. Association mapping for aluminium tolerance in a core collection of rice landraces. Front. Plant Sci. 7, 1415 (2016).
Heuer, S. et al. Improving phosphorus use efficiency – A complex trait with emerging opportunities. Plant J. 90, 868–885 (2017).
Liu, J., Piñeros, M. A. & Kochian, L. V. The role of aluminum sensing and signaling in plant aluminum resistance. J. Integr. Plant Biol. 56, 221–230 (2014).
Delhaize, E. et al. Engineering high-level aluminum tolerance in barley with the ALMT1 gene. Proc. Natl. Acad. Sci. USA 101, 15249–15254 (2004).
Yokosho, K., Yamaji, N., Ueno, D., Mitani, N. & Ma, J. F. OsFRDL1 is a citrate transporter required for efficient translocation of iron in rice. Plant Physiol. 149, 297–305 (2009).
Xia, J., Yamaji, N., Kasai, T. & Ma, J. Plasma membrane-localized transporter for aluminum in rice. Proc. Natl. Acad. Sci. USA 107, 18381 (2010).
Huang, C. F., Yamaji, N., Chen, Z. & Ma, J. F. A tonoplast-localized half-size ABC transporter is required for internal detoxification of aluminum in rice. Plant J. 69, 857–867 (2012).
Huang, C. F. et al. A bacterial-type ABC transporter is involved in aluminium tolerance in rice. Plant Cell 21, 655–667 (2009).
Ryan, P. R. et al. The identification of aluminium-resistance genes provides opportunities for enhancing crop production on acid soils. J. Exp. Bot. 62, 9–20 (2011).
Yamaji, N. et al. A zinc finger transcription factor ART1 regulates multiple genes implicated in aluminum tolerance in rice. Plant Cell 21, 3339–3349 (2009).
Iuchi, S. et al. Zinc finger protein STOP1 is critical for proton tolerance in Arabidopsis and coregulates a key gene in aluminum tolerance. Proc. Natl. Acad. Sci. USA 104, 9900–9905 (2007).
Sawaki, Y. et al. STOP1 regulates multiple genes that protect Arabidopsis from proton and aluminum toxicities. Plant Physiol. 150, 281–294 (2009).
Tsutsui, T., Yamaji, N. & Ma, J. F. Identification of a cis-acting element of ART1, a C2H2-type zinc-finger transcription factor for aluminum tolerance in rice. Plant Physiol. 156, 925–931 (2011).
Chen, Z. C., Yamaji, N., Motoyama, R., Nagamura, Y. & Ma, J. F. Up-regulation of a magnesium transporter gene OsMGT1 is required for conferring aluminum tolerance in rice. Plant Physiol. 159, 624–1633 (2012).
Xia, J., Yamaji, N. & Ma, J. F. A plasma membrane-localized small peptide is involved in rice Al tolerance. Plant J. 76, 345–355 (2013).
Challam, C., Kharshing, G. A., Yumnam, J. S., Rai, M. & Tyagi, W. Association of qLTG3-1 with germination stage cold tolerance in diverse rice germplasm from the Indian subcontinent. Plant Genet Resour-C. 11, 206–211 (2013).
Haefele, S. M., Nelson, A. & Hijmans, R. J. Soil quality and constraints in global rice production. Geoderma 235–236, 250–259 (2014).
Inostroza-Blancheteau, C. et al. Molecular and physiological strategies to increase aluminum resistance in plants. Mol. Bio. Rep. 39, 2069–2079 (2012).
Bian, M., Zhou, M., Sun, D. & Li, C. Molecular approaches unravel the mechanism of acid tolerance in plants. Crop J. 1, 91–104 (2013).
Eekhout, T., Larsen, P. & Veylder, L. D. Modifications of DNA checkpoints to confer aluminum tolerance. Trends Plant Sci. 22, 102–105 (2017).
Magalhaes, J. V., Pineros, M. A., Maciel, L. S. & Kochian, L. V. Emerging pleiotropic mechanisms underlying aluminum resistance and phosphorus acquisition on acidic soils. Front. Plant Sci. 9, 1420 (2018).
Li, J. L. et al. Natural variation underlies alteration in Nramp aluminium transporter (NRAT 1) expression and function that play role in rice aluminum tolerance. Proc. Natl. Acad. Sci. USA 111, 6503–6508 (2014).
Panda, S. K., Baluska, F. & Masumoto, H. Aluminium stress signalling in plants. Plant Signal. Behav. 4, 592–597 (2009).
Kochian, L. V., Pineros, M. A. & Hoekenga, O. A. The physiology, genetics and molecular biology of plant aluminum resistance and toxicity. Plant Soil 274, 175–195 (2005).
Alvim, M. N., Ramos, F. T., Oliveira, D. C., Isaias, R. M. S. & França, M. G. C. Aluminium localization and toxicity symptoms related to root growth inhibition in rice (Oryza sativa L.) seedlings. J. Biosci. 37, 1079–1088 (2012).
Alvarez, I. et al. Morphological and cellular changes in rice roots (Oryza sativa L.) caused by Al stress. Bot. Stud. 53, 67–73 (2012).
Tsutsui, T. et al. Comparative genome-wide transcriptional analysis of Al-responsive genes reveals novel tolerance mechanisms in rice. PLoS One 7(10), e48197 (2012).
Wang, C. Y., Shen, R. F., Chao Wang, C. & Wang, W. Root protein profile changes induced by Al exposure in two rice cultivars differing in Al tolerance. J. Proteom. 78, 281–293 (2013).
Zhu, H. et al. Genome-wide transcriptomic and phylogenetic analyses reveal distinct aluminum-tolerance mechanisms in the aluminum-accumulating species buckwheat (Fagopyrum tataricum). BMC Plant Biol. 15, 16 (2015).
Che, J., Tsutsui, T., Yokosho, K., Yamaji, N. & Ma, J. F. Functional characterization of an aluminum (Al)-inducible transcription factor, ART2, revealed a different pathway for Al tolerance in rice. New Phytol. 220, 209–218 (2018).
Hulm, J. L., Mcintosh K. B. & Bonham-Smith, P. C. Variation in transcript among the four members of the Arabidopsis thaliana ribosomal protein S15a gene family. Plant Sci. 169, 267–278 (2005).
Wang, J. et al. Expression changes of ribosomal proteins in phosphate- and iron-deficient Arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genomics 14, 783 (2013).
Kim, D-W. et al. A hydroponic rice seedling culture model system for investigating proteome of salt stress in rice leaf. Electrophoresis 26, 4521–4539 (2005).
Sarry, J-E. et al. The early responses of Arabidopsis thaliana cells to cadmium exposure explored by protein and metabolite profiling analyses. Proteomics 6, 2180–2198 (2006).
Le Gall, H. et al. Cell wall metabolism in response to abiotic stress. Plants 4, 112–166 (2015).
Garcia, J. S., Souza, G. H. M. F., Eberlin, M. N., Arruda, M. A. Z. Evaluation of metal-ion stress in sunflower (Helianthus annuus L.) through proteomic changes. Metallomics 1, 107–113 (2009).
Fukao, T. & Bailey-Serres, J. Submergence tolerance conferred by Sub1A is mediated by SLR1 and SLRL1 restriction of gibberellins responses in rice. Proc. Natl. Acad. Sci. U. S. A. 105, 16814–16819 (2008).
Famoso, A. N. et al. Development of a novel aluminum tolerance phenotyping platform used for comparisons of cereal aluminum tolerance and investigations into rice aluminum tolerance mechanisms. Plant Physiol. 53, 1678–1691 (2010).
Roselló, M., Poschenrieder, C., Gunsé, B., Barceló, J. & Llugany, M. Differential activation of genes related to aluminium tolerance in two contrasting rice cultivars. J. Inorg. Biochem. 152, 160–166 (2015).
Tyagi, W. & Rai, M. Root transcriptomes of two acidic soil adapted Indica rice genotypes suggest diverse and complex mechanism of low phosphorus tolerance. Protoplasma 254, 725–736 (2017).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Jain, M. et al. F-Box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol. 143, 1467–1483 (2007).
Acknowledgements
This work is supported by NAIP (National Agriculture Innovative Project) (C30033/415101-036) from Indian Council for Agricultural Research (ICAR), Government of India to W.T.
Author information
Authors and Affiliations
Contributions
W.T. designed and conceptualized the molecular experiment, interpreted the data and wrote the manuscript. J.S.Y. performed the physiological and sequencing experiment and generated samples for transcriptome analysis. D.S. helped in generating root photographs. M.R. conceptualized the experiment and was involved in critical revision of the manuscript and final approval of the version to be published. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tyagi, W., Yumnam, J.S., Sen, D. et al. Root transcriptome reveals efficient cell signaling and energy conservation key to aluminum toxicity tolerance in acidic soil adapted rice genotype. Sci Rep 10, 4580 (2020). https://doi.org/10.1038/s41598-020-61305-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61305-7
This article is cited by
-
Genetic analysis of purple pigmentation in rice seed and vegetative parts — implications on developing high-yielding purple rice (Oryza sativa L.)
Journal of Applied Genetics (2024)
-
Strategies for alleviating aluminum toxicity in soils and plants
Plant and Soil (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
