
Abstract
Salmonella enterica serovar Enteritidis is a major cause of foodborne disease in Uruguay since 1995. We used a genomic approach to study a set of isolates from different sources and years. Whole genome phylogeny showed that most of the strains are distributed in two major lineages (E1 and E2), both belonging to MLST sequence type 11 the major ST among serovar Enteritidis. Strikingly, E2 isolates are over-represented in periods of outbreak abundance in Uruguay, while E1 span all epidemic periods. Both lineages circulate in neighbor countries at the same timescale as in Uruguay, and are present in minor numbers in distant countries. We identified allelic variants associated with each lineage. Three genes, ycdX, pduD and hsdM, have distinctive variants in E1 that may result in defective products. Another four genes (ybiO, yiaN, aas, aceA) present variants specific for the E2 lineage. Overall this work shows that S. enterica serovar Enteritidis strains circulating in Uruguay have the same phylogenetic profile than strains circulating in the region, as well as in more distant countries. Based on these results we hypothesize that the E2 lineage, which is more prevalent during epidemics, exhibits a combination of allelic variants that could be associated with its epidemic ability.
Similar content being viewed by others


Introduction
Salmonella is a major cause of human foodborne disease worldwide. A singular epidemiological feature of human salmonellosis is that one particular serovar can become prevalent over the others, but the prevalent serovar may change over time. Previous to the 1980’s, Salmonella enterica serovar Typhimurium was the most commonly isolated serovar worldwide, but then at the beginning of the 1990’s S. enterica serovar Enteritidis emerged as the most common cause of human salmonellosis, first in Europe and then in many other countries arround the world1,2,3,4,5,6. The reasons for this serovar shift are still not fully understood.
Several studies have addressed the phylogenetic diversity within S. enterica serovar Enteritidis, and suggested that diversification events could be related to its epidemiological features7,8,9,10. The work of Allard et al. first reported the use of whole genome sequencing (WGS) and single nucleotide polymorphisms (SNP) analysis to address molecular epidemiology of a set of isolates previously indistinguishable by other techniques. Deng et al. suggested that serovar Enteritidis diversified from a few major lineages spread worldwide, and associated some lineages with geographic and epidemiological characteristics. Feasey et al. found a strong correlation between prophage content and accessory genome features with the establishment of a new epidemiological course. In our studies, we found that the acquisition of a new prophage was probably determinant for the onset of a particular lineage8. The vast majority of S. enterica serovar Enteritidis isolates worldwide belong to the eBurst Group 4 (EBG4). Achtman et al. and more recently our group described that among EBG4, multi locus sequence type (MLST) ST11 is the major ST and constitutes a polyphyletic lineage from which several other minor lineages emerged8,11,12.
In Uruguay, until 1994 S. enterica serovar Enteritidis was only sporadically isolated, but in 1995 an important outbreak of human salmonellosis associated to this serovar occurred. Since then it became the most prevalent serovar in the country despite some periodical declining1. Five different epidemiological periods can be distinguished, as related to the prevalence of serovar Enteritidis in the country: a pre-epidemic period, the first epidemic (1995–2004), a declining (2005–2008), a second epidemic period (2009–2013) and further declining afterward (see Fig. 1(a)).

(a) Distribution of S. enterica serovars received at the National Salmonella Center over the time (1975 to 2017). The 5 epidemiological periods of isolation are indicated with numbers. In white: non epidemic periods (1, 3 and 5). In orange: epidemic periods (2 and 4). (b) Phylogenetic tree showing the two clusters among Uruguayan S. enterica serovar Enteritidis genomes (E1 in blue, E2 in red and other strains in black). Highlight over the strain designation indicate the period of isolation as explained above. The genome of reference (P125109) is highlighted in green and marked with an asterisk. The isolates are named using a number followed by an underscore and the last two digits of the year of isolation. Scale bar units represents changes per variant site.
In previous studies, we found that the two oldest pre-epidemic isolates (31/88 and 8/89) belong to a unique genetic type by RAPD, PFGE, and MLST and have impaired behavior in different models of infection1,8,13. In spite of these observations, we could not find a correlation between genetic features and different pathogenic or epidemiological abilities among the isolates. In the present study, we analyze the genomic sequences of 61 S. enterica serovar Enteritidis strains isolated in Uruguay between 1988 and 2017 from different sources by comparative genomics and SNP based phylogenetic analysis. We extended the study to include strains from Argentina and Brazil. An analysis of the genomic differences between the lineages that have been circulating since the beginning of the S. enterica serovar Enteritidis epidemic period is presented.
Methods
Bacterial strains, culture and DNA extraction
A total of 61 S. enterica serovar Enteritidis strains isolated in Uruguay from different sources and periods were selected from the collection of the National Salmonella Center (Instituto de Higiene, Universidad de la República). All strains were previously characterized by our group using different genetic and phenotypic methods13,14. Strain names, source and date of isolation are shown in Supplementary Table 1. The strains were recovered from freeze stocks (kept at −80 °C in LB broth with 16% glycerol) by culture in LB agar (Sigma-Miller). Liquid cultures used for DNA extraction were performed in LB broth at 37 °C with shaking. DNA extraction and purification were performed using commercial purification kits (DNeasy Blood & Tissue Kit, Qiagen).
Genome sequencing and assembly
Genome sequencing was performed using Illumina technology, and the sequences were deposited in the NCBI database. The total number of produced paired-end reads for each strain ranged from 0.66 × 106 to 7.9 × 106, with a read size of 100, 277 and 76 for platforms Hiseq. 2000, MySeq v3, and Illumina Genome Analyzer II, respectively. Generated reads were trimmed using sickle (available at github.com/najoshi/sickle), with a phred score threshold of 30 (Q30). Quality was finally checked using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). For platform and sequencing details see Supplementary Table 1. De novo assembly was performed with Spades (version 3.6.1)15 using a pre-assembly approach with Velvet (version 1.2.10)16. The first assembly was done using Velvet with default parameters. The final assembly was performed in Spades using a range of k-mer sizes between 29 and the maximum read size minus one. Velvet-generated contigs were used as a reference with the “--untrusted-contigs” option. On average 97.7% of the generated reads were mapped and the mean observed nucleotide coverage ranged from 27 to 254×. N50 contig size has an average size of 447.312. Assembly stats are summarized in Supplementary Table 1. All the Uruguayan genomes are available in the NCBI BioProject database with accession numbers PRJEB2130 and PRJEB4649 (Supplementary Table 1).
Comparative genomics
The genome of phage type 4 strain P125109, (GenBank ID AM933172.1, NCBI refseq accession NC_011294.1), was used as reference when needed. All the genomic sequences were annotated using RASTtk17 and verified with EnteroBase annotation pipeline. Comparative SNP analysis was performed using NUCmer (NUCleotide MUMmer version 3.1) and local blast (version 2.2.30)18. NUCmer was run with the parameters “-maxmatch” and the minimum length of a single exact match set to 12 (-l parameter). SNPs were obtained using the show-snps module with the –C option, so excluding all SNPs from repeats.
Phylogeny figures were generated using iTOL19. Figures showing the alignment between variants of genes were generated through BLASTn alignments using EasyFig20, exported to svg images and edited using Inkscape software (inkscape.org).
For phylogenetic analysis all fastq files were submitted to EnteroBase which automatically performs a pipeline of assembly, annotation and some in silico typing schemes including Achtman 7 gene MLST and ribosomal MLST (rMLST) (http://enterobase.warwick.ac.uk)12. EnteroBase tools were used to perform a whole genome SNP phylogeny, using the genome of strain P125109 as reference. The EnteroBase algorithm performs de novo assembly from reads and uses LAST software to call the SNPs from the assembled genomes, filters out SNPs from repetitive regions and low quality variants, and builds a SNP matrix which is used as input to perform a RAxML maximum likelihood phylogeny21. For the SNP tree we considered only the sites present in 100% of the genomes.
Genomes from strains isolated in Argentina and Brazil that were included in phylogenetic analysis are shown in Supplementary Table 2 and were selected from the available genomes in EnteroBase.
Results and Discussion
Comparative genomics
First we compared the 61 Uruguayan isolates using SNP analysis with the P125109 genome as reference. This analysis showed that the two oldest isolates have more than 600 SNPs of difference compared to the reference genome (607 and 652 for 31/88 and 08/89 respectively), whereas all other 59 isolates have less than two hundred SNPs (from 60 to 166) (Supplementary Table 1). On the other hand, all Uruguayan genomes share 17 SNPs of difference with P125109 genome.
A phylogenetic analysis based on SNPs revealed the existence of two clades that we named E1 and E2 (Fig. 1(b)) that comprise 57 out of the 61 strains. These 57 strains belong to the major S. enterica serovar Enteritidis MLST sequence type 11 (ST-11) and have been co-circulating in the country since 1994. The remaining four isolates (31/88, 8/89, 77/02 and 89/02) did not group with any of the two clades (Fig. 1(b)). We previously reported that the two oldest isolates (31/88 and 8/89) belong to the MLST sequence type 1974 (ST1974) and that ST1974 originated from ST11 after the acquisition of a characteristic prophage8. The other two isolates, 77/02 and 89/02, belong to ST11 as the majority of the strains.
The E1 clade comprises 21 strains that span all five epidemiological periods, and were isolated from food, animal or human infections. Instead, E2 clade is represented by 36 isolates of which 34 were isolated in the periods associated to epidemics (Fig. 1(a,b)). Hence, it is reasonable to hypothesize that E2 may constitute a lineage that was responsible for the S. enterica serovar Enteritidis epidemics in the country.
Previously we reported a phylogenetic analysis of 203 strains, representing intra-serovar diversity among S. enterica serovar Enteritidis EBG4 isolates worldwide, that included 6 of the 61 Uruguayan isolates used in the present study8. As we found now, half of these 6 strains are E1 and half are E2 (E1: 53/94, 206/99 and 214/02; E2: 8/02, 251/01 and 253/01), and each of these two groups is closely related with strains from Europe and North America. This relation is showed in Supplementary Fig. S1, containing the previously reported phylogeny with a zoom on the clade formed by these strains. This suggests that E1 and E2 may have a broad circulation pattern.
In order to further support this assumption we performed a new phylogenetic analysis, this time with focus in strains isolated in the region, including 12 from Argentina, 122 from Brazil and the same 61 genomes from Uruguay (Fig. 2(a)). Interestingly we found that all these strains clustered in a highly similar way of what we found among Uruguayan isolates, i.e. E1 cluster now contains strains from Argentina and Brazil whereas E2 cluster contains strains from Brazil (103 in E1, and 62 in E2). Given the low number of genomes from Argentina available in EnteroBase, we can not exclude the possibility that lineage E2 was circulating in this country. Only 30 strains from all three countries grouped outside E1 and E2, of which 24 were isolated before 1994 (Fig. 2(a), Supplementary Table 2). All in all our results strongly suggest that E1 and E2 strains were introduced in the region arround 1994 and have been circulating ever since in Uruguay, Brazil, Argentina, Europe and North America, supporting the idea that both constitute lineages of the serovar Enteritidis.

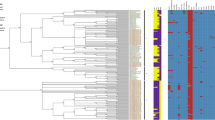
(a) Phylogenetic tree of the 195 genomes including 61 from Uruguay (white leaves), 12 from Argentina (light blue leaves) and 122 from Brazil (yellow leaves). Branches of E1 lineage are colored in blue. Branches of E2 lineage are indicated in red. Lanes at right represent with different colors the allelic variants for ycdX, pduD, hsdM, ybiO, yiaN, aas and aceA respectively. See colour legends inside the (b). Scale bar units represents changes per variant site. (b) Gene alignments for the different allelic variants of ycdX, pduD, hsdM, ybiO, yiaN, aas and aceA respectively. Genes are depicted as annotated in the genome containing each variant and aligned to the reference P125109. The scale of the alignment in base pairs (bp) is indicated by horizontal bars. SNPs associated to each variant respect to the reference genome are detailed in Table 1. SNPs causing non synonymous variants are marked as X → Y (in single letter aminoacid code) relative to its position in the gene. Synonymous SNPs are only marked relative to its position in the gene (syn SNP).
We then performed a comparative genomics analysis aimed to identify genetic differences associated with lineages E1 and E2, including 165 genomes: 57 from Uruguay, 11 from Argentina and 97 from Brazil (Fig. 2(a)).
Allelic variants among E1 and E2 lineages
As we did not find differences in accessory genome between E1 and E2 lineages, we looked for SNP differences among the 165 selected strains. This analysis showed that the main differences between E1 and E2 are located in three genes: ycdX, pduD and hsdM. These genes are interrupted in E1 genomes when compared to their counterparts in the E2 genomes. In addition, we found allelic variants in other four genes, ybiO, yiaN, aas and aceA, that are specific for each lineage (Fig. 2(a,b); Table 1).
All the 103 E1 strains (11 from Argentina, 71 from Brazil and 21 from Uruguay) show a 4 base pair insertion located in gene ycdX (SEN1912) as compared to the reference and to all E2 strains (Table 1). This insertion introduces a premature stop codon, which may produce a pseudo-gene or a truncated version of the protein (Fig. 2(b) and Table 1). The ycdX gene encodes an hydrolase that belongs to the superfamily of PHP proteins, that contain a NH2-terminal php domain (polymerase histidinol phosphatase). This domain is characteristic of the DNA polymerase III alpha subunit of bacteria and is involved in the proofreading function22. It has been suggested that YcdX is involved in the DNA repair pathways in E. coli and could act as a nuclease or a phosphatase in these pathways23. Recently, Wang et al. found two allelic variants for this gene (non synonymous SNP) between S. enterica serovar Enteritidis isolates with markedly different abilities to survive in egg white24. Inoue et al. showed that mutation in ycdY (the adjacent gene in the ycdWXYZ operon) caused repression of swarming in E. coli25.
All the strains from the E2 lineage have the same allelic variant for pduD gene (SEN2039) while 102 out of the 103 strains from E1 lineage show a base substitution that introduce a stop codon, similar to the one observed for ycdX (Table 1). The remaining strain (i.e. 11/07 from Uruguay) completely lacks the pduD gene (Fig. 2(b) and Table 1). pduD encodes for a propanediol dehydratase, part of the pdu operon which encodes all the required proteins for the 1,2-propanediol catabolism. This operon that was acquired by lateral gene transfer in Salmonella26,27 is disrupted in different serovars that cause invasive extra-intestinal infections in human such as Typhi and Paratyphi A amongst others28. Faber et al. reported that the functionality of this operon can be important to compete with intestinal microbiota, as 1,2 propanediol is a metabolic product in the anaerobic gut environment that can be used by Salmonella when coupled to the tetrathionate anaerobic respiration29. It is proposed that ability to use 1,2 propanediol allow the expansion of the pathogen population in the gut increasing the excretion in feces, which is an important factor for epidemic dissemination29.
Furthermore, all E2 strains have the same allelic variant for the hsdM gene (SEN4290) but among the E1 strains there are several variants for this gene (Fig. 2(b) and Table 1). Most of the E1 strains contain variants of hsdM that would produce either a truncated version (4 out of the 6 variants) or a non synonymous substitution for the protein. Only three strains in the E1 (including the Uruguayan 44/07) have the same variant as E2, suggesting that only in E1 strains hsdM is prone to accumulate mutations (Fig. 2(b)). The hsdM gene encodes a DNA methylase involved in type I restriction-modification system (RM) in enterobacteria30,31. Previous reports linked restriction modification systems type 2 with pathogenicity in Salmonella due to epigenetic effects on the virulence gene expression32. Silva et al. showed that mutations in the RM genes produce an impaired phenotype in the mouse model of invasive salmonellosis33. Type 1 RM systems were also described as involved in pathogenicity of Yersinia pseudotuberculosis34.
Given that E2 but not E1 have intact copies of ycdX, pduD and hsdM, and considering that these genes have been previously implicated in Salmonella pathogenicity, it can be taken to suggest that these three genes may be relevant for the epidemic ability of the E2 strains. We also looked for the allelic variants in the global phylogeny representing the intra-serovar diversity (Supplementary Fig. 1 and ref. 8) and found that the predominant allele is the E2 allele in all cases, whereas the disrupted variants are specific for E1 (data not shown).
As mentioned above, another four genes ybiO, yiaN, aas and aceA were found with minor sequence differences between both lineages. These four genes present a single variant in E2 strains whereas E1 are mostly identical to P125109 reference strain.
All E2 strains present a particular allelic variant of the ybiO gene (SEN0772) due to a substitution that produce a Gly(601)→Arg in the protein (Fig. 2(a,b), Table 1). The ybiO gene encode for a putative mechanosensitive channel protein involved in osmotic adaptation that is activated by hypo-osmotic shock in E. coli35. It was previously reported that there are two allelic variants for this gene among S. enterica serovar Enteritidis isolates that have differing ability to survive in egg white24. For yiaN (SEN3494) (L-dehydroascorbate transporter large permease subunit, a putative membrane protein) all E2 genomes present the same allele that differs in Gly(163)→Ala respect to E1. Similarly, all E2 share the same variant for aas (2-acylglycerophosphoethanolamine acyltransferase/acyl-ACP synthetase, SEN2853) and aceA (isocitrate lyase, SEN3966) genes. In E1, most of the strains have an aas gene identical to the reference, and a few strains present either a non synonymous substitution (5 strains) or a synonymous substitution (1 strain) (Fig. 2(a), Table 1). Similarly, the aceA gene is identical to the reference in all but one of the E1 genomes.
In summary, we found that the E2 alleles for ybiO, yiaN, aas and aceA are specific for this lineage.
Conclusions
Two different lineages have been circulating in Uruguay after the start of the epidemic period. These same two lineages also circulated in Brazil in similar periods of time as well as in Europe and North America. Our results are in agreement with those recently published by Campioni et al. that report the existence of a shift in the genomic profile of S. enterica serovar Enteritidis circulating in South America after 199436. Based on all these results, we hypothesize that around 1994 phage type 4 strains (likely from E1 and E2 lineages) were introduced in the region and successfully spread causing epidemics. These introduced strains belong to one of the sub-clades described by Feasey as part of the global epidemic S. enterica serovar Enteritidis clade10.
Further, as reported here, at least in Uruguay, the E2 lineage is clearly associated with epidemics. This lineage presents a combination of allelic variants that could be associated with its epidemic ability. The fact that genes ycdX, pduD and hsdM are disrupted in E1 strains that come from different regions and periods of isolation is suggestive of a biological relevance for conserving this disruption. The disrupted version of these genes is a genetic marker for the E1 lineage, and we propose that it might be related to impaired epidemic ability. On the other hand, the allelic variants of another four genes (ybiO, yiaN, aas, aceA) are specific markers for the E2 lineage, and could be useful for tracking the presence of this lineage in further epidemic events.
Data availability
Change history
13 May 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Betancor, L. et al. Prevalence of Salmonella enterica in poultry and eggs in Uruguay during an epidemic due to Salmonella enterica serovar Enteritidis. J. Clin. Microbiol. 48, 2413–23 (2010).
Cogan, T. A. & Humphrey, T. J. The rise and fall of Salmonella Enteritidis in the UK. J. Appl. Microbiol. 94, 114–119 (2004).
Herikstad, H., Motarjemi, Y. & Tauxe, R. V. Salmonella surveillance: A global survey of public health serotyping. Epidemiol. Infect. 129, 1–8 (2002).
Rodrigue, D. et al. Outbreaks of Salmonella enteritidis Infections in the United States, 1985–1991. J. Infect. Dis. 169, 547–552 (2011).
Poppe, C. In Salmonella enterica serovar Enteritidis in human and animals (eds. Saeed, A. M., Gast, R. K., Potter, M. E. & Wall, P. G.) 3–18 (Iowa State University Press, 1999).
Schroeder, C. M. et al. Estimate of illnesses from Salmonella Enteritidis in eggs, United States, 2000. Emerg. Infect. Dis. 11, 113–115 (2005).
Allard, M. W. et al. On the Evolutionary History, Population Genetics and Diversity among Isolates of Salmonella Enteritidis PFGE Pattern JEGX01.0004. PLoS One 8 (2013).
D’Alessandro, B. et al. A novel prophage identified in strains from Salmonella enterica serovar Enteritidis is a phylogenetic signature of the lineage ST-1974. Microb. Genomics 4 (2018).
Deng, X. et al. Genomic epidemiology of Salmonella enterica serotype Enteritidis based on population structure of prevalent lineages. Emerg. Infect. Dis. 20, 1481–1489 (2014).
Feasey, N. A. et al. Distinct Salmonella Enteritidis lineages associated with enterocolitis in high-income settings and invasive disease in low-income settings. Nat. Genet. 48, 1211–1217 (2016).
Achtman, M. et al. Multilocus Sequence Typing as a Replacement for Serotyping in Salmonella enterica. PLoS Pathog. 8, e1002776 (2012).
Alikhan, N.-F., Zhou, Z., Sergeant, M. J. & Achtman, M. A genomic overview of the population structure of Salmonella. PLOS Genet. 14, e1007261 (2018).
Yim, L. et al. Differential Phenotypic Diversity among Epidemic-Spanning Salmonella enterica Serovar Enteritidis Isolates from Humans or Animals. Appl. Environ. Microbiol. 76, 6812–6820 (2010).
Betancor, L. et al. Genomic and phenotypic variation in epidemic-spanning Salmonella enterica serovar Enteritidis isolates. BMC Microbiol. 9, 237 (2009).
Bankevich, A. et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 19, 455–477 (2012).
Zerbino, D. R. & Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Brettin, T. et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5, 8365 (2015).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016).
Sullivan, M. J., Petty, N. K. & Beatson, S. A. Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010 (2011).
Zhou, Z. et al. Pan-genome Analysis of Ancient and Modern Salmonella enterica Demonstrates Genomic Stability of the Invasive Para C Lineage for Millennia. Curr. Biol. 28, 2420–2428.e10 (2018).
Wieczorek, A. & McHenry, C. S. The NH 2 -terminal php Domain of the α Subunit of the Escherichia coli Replicase Binds the ϵ Proofreading Subunit. J. Biol. Chem. 281, 12561–12567 (2006).
Teplyakov, A. et al. Crystal structure of theEscherichia coli YcdX protein reveals a trinuclear zinc active site. Proteins Struct. Funct. Genet. 51, 315–318 (2003).
Wang, Y. et al. Comparative Genomic Analysis and Characterization of Two Salmonella enterica Serovar Enteritidis Isolates From Poultry With Notably Different Survival Abilities in Egg Whites. Front. Microbiol. 9, 1–14 (2018).
Inoue, T. et al. Genome-Wide Screening of Genes Required for Swarming Motility in Escherichia coli K-12. J. Bacteriol. 189, 950–957 (2007).
Bobik, T. A., Xu, Y., Jeter, R. M., Otto, K. E. & Roth, J. R. Propanediol utilization genes (pdu) of Salmonella typhimurium: three genes for the propanediol dehydratase. J. Bacteriol. 179, 6633–9 (1997).
Bobik, T. A., Havemann, G. D., Busch, R. J., Williams, D. S. & Aldrich, H. C. The propanediol utilization (pdu) operon of Salmonella enterica serovar Typhimurium LT2 includes genes necessary for formation of polyhedral organelles involved in coenzyme B(12)-dependent 1, 2-propanediol degradation. J. Bacteriol. 181, 5967–75 (1999).
Nuccio, S.-P. & Bäumler, A. J. Comparative Analysis of Salmonella Genomes Identifies a Metabolic Network for Escalating Growth in the Inflamed Gut. MBio 5, e00929–14 (2014).
Faber, F. et al. Respiration of Microbiota-Derived 1,2-propanediol Drives Salmonella Expansion during Colitis. PLOS Pathog. 13, e1006129 (2017).
Sharp, P. M., Kelleher, J. E., Daniel, A. S., Cowan, G. M. & Murray, N. E. Roles of selection and recombination in the evolution of type I restriction-modification systems in enterobacteria. Proc. Natl. Acad. Sci. 89, 9836 LP–9840 (1992).
Taylor, J. E., Swiderska, A. & Geoff Kneale, G. A rapid purification procedure for the HsdM protein of EcoR124I and biophysical characterization of the purified protein. Protein Expr. Purif. 87, 136–140 (2013).
Heusipp, G., Falker, S. & Alexandershmidt, M. DNA adenine methylation and bacterial pathogenesis. Int. J. Med. Microbiol. 297, 1–7 (2007).
Silva, C. A. et al. Infection of Mice by Salmonella enterica Serovar Enteritidis Involves Additional Genes That Are Absent in the Genome of Serovar Typhimurium. Infect. Immun. 80, 839–849 (2012).
Pouillot, F., Fayolle, C. & Carniel, E. A putative DNA adenine methyltransferase is involved in Yersinia pseudotuberculosis pathogenicity. Microbiology 153, 2426–2434 (2007).
Edwards, M. D. et al. Characterization of three novel mechanosensitive channel activities in Escherichia coli. Channels 6, 272–281 (2012).
Campioni, F. et al. Changing of the Genomic Pattern of Salmonella Enteritidis Strains Isolated in Brazil Over a 48 year-period revealed by Whole Genome SNP Analyses. Sci. Rep. 8, 10478 (2018).
Acknowledgements
We thanks to Arací Martínez for her technical assistance with the collection of isolates. We thanks to Professor Gordon Dougan and Professor Nicholas Thomson for their collaboration with the sequencing of genomes. This work was supported by the Universitary Research Council (Comisión Sectorial de Investigación Científica, CSIC), Universidad de la República, Uruguay (Research groups program PI Laura Betancor and Lucía Yim).
Author information
Authors and Affiliations
Contributions
B.D., A.I., L.B., V.P.E. and F.G. performed all the bioinformatic analysis. L.B., L.Y. and D.P. participate on strain collection, conservation and sequencing. B.D., L.B., A.I., L.Y. and J.A.C. participate in the interpretation of results, discussion and conclusions. B.D. and L.B. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
D’Alessandro, B., Pérez Escanda, V., Balestrazzi, L. et al. Comparative genomics of Salmonella enterica serovar Enteritidis ST-11 isolated in Uruguay reveals lineages associated with particular epidemiological traits. Sci Rep 10, 3638 (2020). https://doi.org/10.1038/s41598-020-60502-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60502-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
