
Abstract
Neutron total scattering measurements were conducted on MgTiO3, CaTiO3, SrTiO3, and BaTiO3 to simultaneously investigate the local and average structure of these materials. The local structures of MgTiO3, CaTiO3, and SrTiO3 were well modelled using the refined average structural models: trigonal R\(\bar{3}\), orthorhombic Pbnm, and cubic Pm\(\bar{3}\)m respectively. However the local structure for BaTiO3, at both temperatures where the average structure is orthorhombic Amm2 and tetragonal P4mm, was best described by the rhombohedral R3m model. Only the R3m model was able to account for the observed displacement of titanium in the [111] direction. Furthermore, box-car type refinements were conducted. These refinements show that the coherence length of the rhombohedral distortion is around 10 Å, at larger r-ranges the local distortions become misaligned and average out to Amm2 and P4mm.
Similar content being viewed by others


Introduction
Alkaline earth metal (Group II) titanates of the form ATiO3 (A2+ = Mg, Ca, Sr, and Ba) are both technologically important and increasingly relevant for new material applications. These compounds are generally parent structures for complex materials that are used in a variety of applications. Magnesium titanate (MgTiO3) is a component in low-loss dielectric compositions for microwave applications, with a variety of uses including band-pass filters, communication antennas, direct broadcasting satellite and global positioning systems among others. Although an effective synthetic route has yet to be found for MgTiO3, limiting its current usage1,2. Calcium titanate (CaTiO3) is used in numerous applications, such as: optics, magnetics, electronics, and superconductors3. Most recently CaTiO3 has found renewed interest in hybrid organic-inorganic perovskites for solar cell applications3. Both strontium titanate (SrTiO3) and barium titanate (BaTiO3) are widely used in dielectric and ferroelectric applications4. The remaining group II elements beryllium and radium were not considered due to their toxicity and radioactivity, respectively.
Magnesium titanate crystallizes in the ilmenite structure which has the form \({{\rm{A}}}^{2+}{{\rm{B}}}^{4+}{X}_{3}^{2-}\). In general, the ilmenite structure is a layered derivative of the corundum structure where the cations are ordered into two nonequivalent octahedral A- and B-sites. The A and B sites are ordered along the hexagonal c direction in alternating layers of face shared A-X6 and B-X6 octahedra. The structure of MgTiO3 was first found to be R\(\bar{3}\) by Posnjak and Barth in 19345. A more recent study using neutron diffraction was conducted by Wechsler and Von Dreele in 19896. This neutron diffraction study confirmed the average structure of MgTiO3 to be trigonal R\(\bar{3}\) and found complete ordering of the Mg-O6 and Ti-O6 layers. The structure of MgTiO3 is stable over a wide range of temperatures (25–1025 °C) as no phase transitions have been reported7.
The rest of the materials (CaTiO3, SrTiO3, and BaTiO3) crystallize in the perovskite structure. The general perovskite structure is ABX3, where the A-site cations surrounded by 12 anions in cubo-octahedral coordination and the B-site cations are surrounded by 6 anions in octahedral coordination. The B-O6 octahedra form a network of corner sharing octahedra that surround the larger A-site cubo-octahedra. In the perovskite structure the B-site cations and anions are generally similar in size, whereas the A-site cations are relatively large in comparison. The relative size of the cations and anions can be used to predict the stability and distortions within the perovskite structure according to the Goldschmidt tolerance factor (t):
where rA is the ionic radius of the A-site cation, rB the radius B-site cation, and rO the radius of oxygen. The value of the tolerance factor t can be used to predict the structure, for t < 0.71 the structure is generally not a perovskite, for t = 0.71–0.9 orthorhombic/rhombohedral symmetry is preferred, for t = 0.9–1 cubic is stable, and for t > 1 hexagonal or tetragonal distortions are preferred8. It should be noted that these tolerance factor (t) ranges are guidelines based on observations.
Most materials with the perovskite structure are not cubic, mainly due to cation displacements and octahedral rotations. The archetypical perovskite CaTiO3 is orthorhombic Pbnm (t = 0.97) from room temperature until 1375 K. At 1375 K the structure begins to undergo a phase transition to tetragonal I4/mcm and becomes single-phase tetragonal at 1425 K. The final structural transition from tetragonal to cubic Pm\(\bar{3}\)m occurs at 1525 K9.
At low temperatures the average structure of SrTiO3 is tetragonal I4/mcm, and around 100 K the structure transitions to the room temperature phase cubic Pm\(\bar{3}\)m (t = 1.00)10. It has been reported that a lower temperature (35–55 K) orthorhombic phase exists11. Although, a pair distribution function study of SrTiO3 at 5 and 293 K found only cubic symmetry present for the local structure12.
The average structure of BaTiO3 (t = 1.06) has been reported to undergo several phase transitions. Below 190 K, the structure is rhombohedral R3m. From 190 to 280 K the structure is orthorhombic Amm2, from 280 to 360 K the structure is tetragonal P4mm, and the structure finally becomes cubic Pm\(\bar{3}\)m above 360 K13,14. These reported structures are all related to the undistorted cubic phase and characterized by the direction of the B-site titanium atoms (R3m – [111], Amm2 – [011], P4mm – [001]). In general, these materials undergo displacive phase transitions, where the structure changes as atoms move due to increased thermal energy. Another type of phase transition is an order-disorder transition, where the structure of a material changes due to a change in the degree of positional or orientational ordering/disordering.
In 1968, an order-disorder model was proposed by Comes et al. for BaTiO3 based on the diffuse scattering from single crystals15. In this model, only the rhombohedral phase was considered ordered and the orthorhombic, tetragonal or cubic phases observed were the result of an averaging of titanium atom displacements in disordered rhombohedral directions15. Since then, many studies have been conducted on BaTiO3 using a variety of methods and techniques to explain the complex local and average structures. The methods and summarized results of these studies are listed in Table 1. The most recent study from Senn et al.16 describes the order-disorder behavior observed in BaTiO3 as emanating from local titanium displacements along the <111> directions associated with rhombohedral R3m symmetry. At low temperatures, these rhombohedral displacements are all aligned (along the [111] direction), but as the temperature increases they become disordered (i.e. [111] and [\(\bar{1}11\)]) to a vector average associated with orthorhombic Amm2 symmetry ([011] direction). At higher temperatures, the Ti displacements disorder along four possible directions ([111], [\(\bar{1}11\)], [\(1\bar{1}1\)] and [\(\bar{1}\bar{1}1\)]) to a vector average associated with tetragonal P4mm symmetry ([001] direction). The structure appears cubic Pm\(\bar{3}\)m at higher temperatures when the local rhombohedral displacements are completely disordered relative to one-another (eight possible displacement directions so that the net Ti displacement is localized in the center of the oxygen octahedron).
Additionally, the local structures of nanocrystalline BaxSr1–xTiO3 (x = 1, 0.5, 0) have also been investigated by synchrotron total scattering. For nanocrystalline SrTiO3 the local and average structure were found to be cubic. However for nanocrystalline BaTiO3 the local structure was found to possess tetragonal distortions in the 10–15 Å regime, whereas the average structure of the nanocrystals was best described as cubic17.
In the present work, to provide insight into the local and average structure of these materials, neutron total scattering measurements of bulk ceramics were conducted. The advantage of collecting neutron total scattering is two-fold. Firstly, using total scattering one can study both the average and local structures of a material simultaneously. The process of simultaneous data collection is important, as a single model must be able to explain the apparent discrepancy between both coherent data sets. The second key advantage is intrinsic to neutron scattering, where neutron scattering is not dependent on the elemental number (Z) as is the case for X-ray scattering. In particular, oxygen ions scatter neutrons strongly (oxygen coh b = 5.80 fm) and titanium ions have a negative scattering lengths (titanium coh b = −3.438 fm) which provide better signal and contrast respectively for accurate refinements18. The advantages of neutron total scattering can be seen in the work of Page et al.19 who employed the technique on niobium doped SrTiO3 (SrTi0.875Nb0.125O3) and BaTiO3 (BaTi0.875Nb0.125O3). At room temperature the local structure of niobium doped SrTiO3 remains cubic like the parent structure of SrTiO3. For niobium doped BaTiO3, the local structure appears to possess rhombohedral symmetry19.
The purpose of this work is to provide the community with structural refinements on high resolution neutron total scattering measurements to clearly define both the local and average structure of the accessible group II titanates. In particular, the focus of this work is to present the refined structures of MgTiO3, CaTiO3, and SrTiO3, to serve as a reference point to understand the structural details in BaTiO3. Furthermore, this work seeks to provide a summarization of the literature studies on BaTiO3 to contextualize these nuances. To accomplish these goals, phase pure samples were synthesized, and neutron total scattering data were collected at both 225 and 290 K (particularly to show the phase sequence of BaTiO3) for all compositions.
Results
Magnesium Titanate
As discussed in the introduction, MgTiO3 crystallizes in the ilmenite structure with trigonal R\(\bar{3}\) symmetry at room temperature. In the ilmenite structure both magnesium and titanium are in 6-coordinate octahedral oxygen environments6.
From the initial X-ray diffractograms there was a minor MgTi2O5 phase observed in the MgTiO3 sample (shown in Supplemental Fig. S1). However this phase was not observed in the neutron total scattering data. The results of the diffraction and PDF refinements of MgTiO3 at 290 K with the R\(\bar{3}\) space group in the equivalent hexagonal setting (a = b, α = β = 90°, γ = 120°) are shown in Fig. 1. The tabulated results for the refinement at 290 K and the complete results for the refinement at 225 K are included in the supplemental information. The diffraction refinement converged on a fit criterion, Rwp = 4.146%, with all reflections well modelled. The crystal structure of the refined structure is shown in Fig. 1(b), with magnesium shown as purple, titanium as grey, and oxygen as red spheres. The neutron PDF, shown in Fig. 1(c), was also well fit with the R\(\bar{3}\) space group, Rwp = 10.012%. The titanium- and magnesium-oxygen bonds for the first coordination sphere are labelled in the low-r region of the PDF (Fig. 1(d)). Both titanium and magnesium are displaced in their respective oxygen octahedral, resulting in three short bonds at 1.8656 Å for titanium and at 2.0497 Å for magnesium and three long bonds at 2.0940 Å for titanium and 2.1660 Å for magnesium. Although there are A- and B-site displacements in the MgTiO3 structure, the R\(\bar{3}\) space group is centrosymmetric and thus not a ferroelectric phase.

(a) Rietveld refinement of neutron diffraction, (b) refined model, (c) small-box modeling of neutron PDF, and (d) zoom-in of neutron PDF for MgTiO3 at 290 K with the R\(\bar{3}\) space group. Data (identified by black circles) and refined models (continuous lines) are shown, along with the difference pattern and hkl indices below (diffraction data only).
The negative scattering length of titanium mentioned previously manifests in negative peaks (or wells) for titanium correlations in the PDF. The diffraction and PDF refinements were conducted independently, but converged on the same model with slight discrepancies. Overall, both the long range average and short range local structure refinements clearly support literature reports describing the ilmenite structure for MgTiO3 at these temperatures6.
Calcium titanate
At room temperature CaTiO3 crystallizes in the perovskite structure with Pbnm (or the equivalent Pnma and Pcmn) symmetry20. As discussed in the introduction, the perovskite structure is a network of corner shared B-site octahedra titanium atoms surrounding the 12-coordinate cubo-octahedral A-site calcium ions.
The results of the neutron diffraction, Rwp = 3.11%, and neutron pair distribution function data, Rwp = 8.05%, for CaTiO3 at 290 K are shown in Fig. 2(a–d) respectively. The refinements for CaTiO3 at 225 K are included in the supplementary information. Both independent refinements at 290 K converged on the same Pbnm model where a = 5.38, b = 5.44, and c = 7.64 Å. Titanium is displaced towards an edge of the Ti-O6 octahedra, yielding two bonds at 1.9514, two at 1.9530, and two at 1.9643 Å. The calcium-oxygen cubo-octahedra is more complex in the refined model, with 8 unique bond distances ranging from 2.3379 to 3.2311 Å. The major peaks that result from these bonds are labelled in the low-r region of the PDF (Fig. 2(d)). Similarly to the results for MgTiO3, the average and local structure of CaTiO3 are well described at 290 K with the Pbnm space group as has been reported in the literature20. The Pbnm space group is centrosymmetric and thus CaTiO3 does not display ferroelectric properties.

(a) Rietveld refinement of neutron diffraction, (b) refined model, (c) small-box modeling of neutron PDF, and (d) zoom-in of neutron PDF for CaTiO3 at 290 K with the Pbnm space group. Data (black circle’s) and refined models (continuous lines) are shown, along with the difference pattern and hkl markers below (diffraction data only).
Strontium titanate
Across a large temperature regime SrTiO3 crystallizes in the perovskite structure with cubic Pm\(\bar{3}\)m symmetry11.
At 290 K the diffraction, Rwp = 4.309%, and PDF, Rwp = 6.970%, refinements results are shown in Fig. 3(a–d) respectively. The results of the refinement at 225 K are similar to those at 290 K and are included in the supplementary information. Due to the cubic symmetry of SrTiO3, the diffraction data shows only a few sharp peaks of which the 420 through 220 reflections are labelled. All of the observed reflections were well fit by the model, with no additional reflections observed. There was some observed peak asymmetry which is characteristic of the NOMAD beamline and spallation sources more generally. Similarly, all of the peaks in the PDF data (Fig. 3(c,d)) were well modelled. The titanium-oxygen and strontium-oxygen bonds are labelled in Fig. 3(d). The width of the single Ti-O peak is a result of the thermal disorder present even though all six Ti-O bonds are equivalent with a bond distance of 1.9534 Å. Similarly, strontium is positioned at the center of the Sr-O12 cubo-octahedra, with twelve Sr-O bonds at 2.7625 Å. The refined models for the average and local structure of SrTiO3 are consistent with what has been reported in the literature11.

(a) Rietveld refinement of neutron diffraction, (b) refined model, (c) small-box modeling of neutron PDF, and (d) zoom-in of neutron PDF for SrTiO3 at 290 K with the Pm\(\bar{3}\)m space group. Data (black circles) and refined models (continuous lines) are shown, along with the difference pattern and hkl markers below (diffraction data only).
Barium titanate
The structure of BaTiO3 is much more complicated than the other titanates in its group, although it still crystallizes in the perovskite structure. The reported phase sequence for BaTiO3, from low to high temperature, is R3m to Amm2 to P4mm to Pm\(\bar{3}\)m. In this study data was collected at 225 K in the orthorhombic Amm2 region, and at 290 K in the tetragonal P4mm region14.
The results of the refinements for BaTiO3 at 225 K are shown in Fig. 4. For the Rietveld refinement, as expected, the Amm2 model fit all of the observed reflections well, with Rwp = 3.421%. At first glance the PDF refinement seems equally successful, Rwp = 6.454%, however upon closer inspection it becomes clear that the Amm2 space group does not adequately model the first two Ti-O bonds at ~2 Å (Fig. 4(d)). The orthorhombic Amm2 symmetry, which corresponds to a titanium displacement in the [011] direction, would result in a broad set of three shallow peaks in the PDF data for the Ti-O correlations (2 short bonds, 2 medium bonds, and 2 longer bonds) – which is not observed in the experimental data. In the data there are two equivalent negative peaks at ~1.9 and ~2.1 Å.

(a) Rietveld refinement of neutron diffraction, (b) refined model, (c) small-box modeling of neutron PDF, and (d) zoom-in of neutron PDF for BaTiO3 at 225 K with the Amm2 space group. Data (black circles) and refined models (continuous lines) are shown, along with the difference pattern and hkl markers below (diffraction data only).
Similarly, the results for the refinements of BaTiO3 at 290 K in the tetragonal P4mm space group are shown in Fig. 5. The refinement of the diffraction data, Fig. 5(a), yielded a good fit where all of the observed reflections were modeled. The diffraction data showed peak splitting in the (220), (310), (311), (400), and (420) pseudocubic reflections, which are indicative of a tetragonal distortion in the [001] direction. As was the case with the refinement at 225 K, the P4mm model at 290 K does not adequately model the two Ti-O peaks in the PDF data. A [001] tetragonal titanium distortion would result in three Ti-O peaks (1 short bond, 4 medium bonds, 1 long bond), which was not observed.

(a) Rietveld refinement of neutron diffraction, (b) refined model, (c) small-box modeling of neutron PDF, and (d) zoom-in of neutron PDF for BaTiO3 at 290 K with the P4mm space group. Data (black circles) and refined models (continuous lines) are shown, along with the difference pattern and hkl markers below (diffraction data only).
To account for the two Ti-O peaks around 1.9 and 2.1 Å that were poorly fit by both the Amm2 space group at 225 K and P4mm space group at 290 K, further refinements were conducted using each of the reported space groups for BaTiO3 (R3m, Amm2, P4mm, Pm\(\bar{3}\)m). The first two Ti-O correlations could only be modelled with the R3m space group. The results of the low-r region refinement at 290 K are shown in Fig. 6(a). The refinement at 225 K yielded an analogous fit and the tabulated results at both 225 K and 290 K are presented in the supplementary information. The bond structure and theoretical neutron PDFs of the reported average structures of BaTiO3 are shown in Fig. 6(b). The refined structure is shown visually in Fig. 6(c). The double peaks observed are characteristic of a rhombohedral distortion in the [111] direction, as the titanium atoms are displaced toward the face of the Ti-O6 octahedra. The face-displaced titanium octahedra results in 3 short bonds (~1.9 Å) and 3 longer bonds (~2.1 Å).

Small-box modeling of local structure (a) for BaTiO3 at 290 K using the R3m space group. Data (black circles) and refined model (continuous line) are shown with the difference pattern below. Correlation labels are provided for clarity within 1 unit cell. Calculated PDF (b) for Pm\(\bar{3}\)m, P4mm, Amm2, and R3m models with bond distances shown as tick lines. (c) Refined local structure R3m model.
In the literature, there are two distinct methods that have been used to model local deviations from an average structure with small box models. The first method, henceforth referred to as the variable range refinement method, involves including an adjustable range of data (x-range) for each refinement from a fixed starting point (typically ~1 Å). For example, Smith et al. investigated BaTiO3 nanoparticles and refined the PDF data set from 1 to 28 Å, with varying refinement lengths (ranging from 1–8, 1–12, 1–16… 1–28 Å, etc.)21. The second method, known as the box-car method, involves taking fixed-length boxes and performing sequential refinements from low to high r-ranges. For example, in the study of BaTiO3-Bi(Zn1/2Ti1/2)O3 by Usher et al., a fixed refinement range of 10 Å was used to refine the structure from 1 to 80 Å (over proscribed ranges of 1–10, 5–15, … 70–80 Å, etc.)22. Both methods were utilized in this study to provide a more complete analysis.
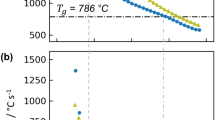
The results of the variable range refinements and box-car refinements are shown in Figs. 7 and 8 for BaTiO3 at 225 and 290 K, respectively. The calculated fit criterion (Rwp) values for each refinement are plotted as a function of the refinement range for the variable range refinement and as a function of the high-r edge of the box (i.e. x = 15 for the 5–10 Å box) for the box-car method. For each method insets are provided to show an example of a characteristic refinement.

Results (Rwp) of the small-box analysis using the (a) variable range refinement method (refinement range shown as x-axis, i.e. x = 5 corresponds to 1–5 refinement, x = 10 to 1–10, … x = 50 to 1–50 Å, etc.) and (b) box-car refinement method (r-range shown on the x-axis corresponds to the high-r edge of box, i.e. x = 10 corresponds to the 0–10 Å box, x = 15 to the 5–15 Å box, …, x = 50, the 40–50 Å box, etc.) for neutron PDF data of BaTiO3 at 225 K using the R3m (violet squares) and Amm2 (red circles) space groups. The insets for each method show an example of one refinement distance or box.

Results (Rwp) of the small-box analysis using the (a) variable range refinement method and (b) box-car refinement method for neutron PDF data of BaTiO3 at 290 K using the R3m (violet squares) and P4mm (green triangles) space groups. The insets for each method show an example of one refinement distance or box.
From the variable range refinement method, at both temperatures the R3m fit yields the lowest Rwp for the 1–5 Å and 1–10 Å refinements where the local Ti-O distortions dominate the fit quality. At larger length scales the Amm2 (for 225 K) and P4mm (for 290 K) average structures yield the best fits for as the penalty from the poorly fit local Ti-O peaks are overcome by a better overall fit to the high-r data. The comparatively larger Rwp values in the first box from 1 to 5 Å for each data set are due to the larger influence of low-r noise on the refinement.
For the box-car refinements at 225 K, the rhombohedral R3m space group fits the data better until the 20–30 Å box where the Amm2 space group yields a lower Rwp. However the next two refinement boxes, 25–35 and 30–40 Å, are best fit by the R3m structure with anomalously low Rwp values. At higher r-ranges the Amm2 average structure again yields the lower Rwp. These results are unclear and the back-switching of the fit criterion is likely due to the decreased intensity of the PDF signal at high r-ranges. Further information on the 20–30, 25–35, and 30–40 Å box-car refinements are presented in the supplementary information.
The box-car method refinements at 290 K are more clear where the local R3m structure fits the data better for the first 1–10 Å box that incorporates the local Ti-O6 distortion. At higher r-ranges the P4mm structure yields the lower Rwp as expected from the average structure refinements. From these results, the coherence length of the local rhombohedral distortions can be estimated to be around ~10 Å at 290 K. At larger distances, the distortions are misaligned relative to one another resulting in the P4mm average structure yielding the superior fit. This observed phenomenon is formalized as the order-disorder model for BaTiO3. An example of the order-disorder model is well visualized in Fig. 3 within the work by Senn et al.16.
Discussion
For MgTiO3, CaTiO3, and SrTiO3 the refinements average and local structures were internally consistent at each temperature. However the average structures for BaTiO3 (Amm2 at 225 and P4mm at 290 K) show distinct features with a different symmetry than the symmetry observed at the local structure. There are clear rhombohedral distortions present in the PDF data at both temperatures which are consistent with the order-disorder model for BaTiO3. The local titanium octahedra distortions are only observed in the first Ti-O correlations around 2 Å. Furthermore, these distortions average out above 10 Å in the PDF data and are not observable in the neutron diffraction data.
The contrast in local structure versus long range average structure observed in BaTiO3 has profound impacts on the physical properties of the material. The other compounds in this study, MgTiO3, CaTiO3, SrTiO3, show a clear correlation between the local and average structure and all exhibit a linear dielectric response. In contrast, in BaTiO3 the unique structural features detailed in this work are linked to its strong non-linear characteristics which are observed in physical phenomena in which there is a correlation between the local structure and the long-range structure. One example includes the occurrence of ferroelectricity in BaTiO3, in which the local dipole moments interact to form a spontaneous polarization. Another consequence of the variable local structural distortions in BaTiO3 is the resultant flat potential energy landscape that leads to the high polarizability and ease of switching of the Ti4+ ion23. Macroscopically, this is observed in BaTiO3 ceramics as large relative permittivities, large polarizations, and large field induced electromechanical strains. This is also clearly relevant to other systems where these contrasting structural length scales influence the material response such as in relaxor ferroelectrics24. Thus, materials systems in which there is a disconnect between the long-range average structure and local structural arrangements are of both scientific and technological interest.
In this work, we present the average and local structure of group II titanates, ATiO3 (A2+ = Mg, Ca, Sr, Ba). For all materials except BaTiO3, there are no observed differences between the local and average structures. It was shown that MgTiO3 crystallizes into the ilmenite structure with trigonal R\(\bar{3}\) symmetry. Both CaTiO3 and SrTiO3 crystallize in the perovskite structure with orthorhombic Pbnm and cubic Pm\(\bar{3}\)m symmetry, respectively. The average structure of BaTiO3 at 225 K is orthorhombic Amm2 and tetragonal P4mm at 290 K. However, the local structure at both temperatures is best described with small box modeling as rhombohedral R3m when the refinements were confined to data below ~10 Å, showing that the coherence length of the distortions is limited to only one to two unit cells in the structure. At larger length scales, the PDF data statistically fits the average structural model better, as the vector sum of the local titanium displacement appears to be in the [011] direction at 225 K and the [001] direction at 290 K corresponding to the orthorhombic Amm2 and tetragonal P4mm average structures, respectively.
Methods
Polycrystalline samples of MgTiO3, CaTiO3, SrTiO3, and BaTiO3 were prepared via standard solid-state synthesis. For each compound, stoichiometric amounts of the component oxide and carbonate reagents were mixed. The reagents MgO (Alfa Aesar 99.95%), CaCO3 (Alfa Aesar 99.997%), SrCO3 (Alfa Aesar 99.994%), BaCO3 (Strem Chemicals Inc. 99.9%), and TiO2 (Sigma Aldrich 99.997%) were used. The carbonate powders (CaCO3, SrCO3, and BaCO3) were heated at 110 °C for 24 h before being weighed. After the initial mixing of powder reagents the mixtures were ground well with a mortar and pestle. The homogenized samples were then ball milled for 4 h in a planetary micromill (Pullverisette 7 Classic Line, Fritsch). For the milling process, each sample mixture was placed in a sealed container with 20 mL of ethanol and eight 10 mm yttria-stabilized zirconia (YSZ) balls. The milled solutions were dried in air at room temperature. Then the dried powders were calcined in alumina crucibles at 800 °C for 24 h, 900 °C for 6 h, 700 °C for 4 h, and 1100 °C for 4 h for MgTiO3, CaTiO3, SrTiO3, and BaTiO3 respectively, with a 4 h step at 450 °C (to allow for degassing of carbonates). The calcined powders were ground well and pressed, uniaxially in a 10-mm die (Carver Inc.) at 2 tons for 2 mins, into pellets. The sample pellets were placed in closed alumina crucibles and sintered at 1200 °C for 12 h, 1350 °C for 12 h, 1100 °C for 12 h, and 1250 °C for 6 h for MgTiO3, CaTiO3, SrTiO3, and BaTiO3 respectively. A heating and cooling rate of 5 °C/min was used for the calcination and sintering steps for all samples. The sintered pellets were ground well and loaded into X-ray diffraction sample holders. X-ray diffraction data was collected using a benchtop X-ray diffractometer (Miniflex 600, Rigaku) to determine the phase purity.
To collect neutron total scattering measurements, samples were loaded in 2 mm diameter quartz capillaries (Hampton) and sent to the Nanoscale Ordered Materials Diffractometer (NOMAD)25 beam-line at the Spallation Neutron Source (SNS)26 at Oak Ridge National Labs (ORNL). Data were collected for each sample at 225 and 290 K with a collection time of 30 mins and a heating ramp rate of 60 K/min.
To determine the average structure, Pawley fitting27 was performed on the collected neutron total scattering data using the Topas Academic (Version 6) software package28,29. The starting models for each refinement were taken from the literature6,11,20,30. For each Pawley fit the background terms, unit cell parameters, and peak profiles were refined. The background for each refinement was modelled with a 10 term polynomial. The results of the Pawley fits were used as starting models for Rietveld analysis31,32 (also performed with Topas) where the scale factor, atomic positions and atomic displacement parameters were refined.
To investigate the local structure of these materials the total scattering data were transformed to the pair distribution function (PDF), G(r), via a sine Fourier transform33. This transformation was performed with in-house software at ORNL with Qmin of 0.50 Å−1 and Qmax of 30.00 Å−1. Again Topas (Version 6) was used to perform small-box modeling using the refined average structure as starting models. The scale, lattice parameters, atomic displacement parameters, and atomic positions were refined. The delta2, Qdamp, and Qbroad parameters were refined from both a nickel and a silicon standard.
References
Sato, T., Miyamoto, R. & Fukasawa, A. Deviation of Dielectric Properties in Magnesium Titanate Ceramics. Jpn. J. Appl. Phys. 20, 151 (1981).
Deng, Y.-F., Tang, S.-D., Lao, L.-Q. & Zhan, S.-Z. Synthesis of magnesium titanate nanocrystallites from a cheap and water-soluble single source precursor. Inorganica Chim. Acta 363, 827–829 (2010).
Thomas, S. & Thankappan, A. Perovskite Photovoltaics: Basic to Advanced Concepts and Implementation. (Academic Press, 2018).
Acosta, M. et al. BaTiO3 -based piezoelectrics: Fundamentals, current status, and perspectives. Appl. Phys. Rev. 4, 041305 (2017).
Barth, T. F. W. & Posnjak, E. The Crystal Structure of Ilmenite. Z. Für Krist. - Cryst. Mater. Teil A, 271–280 (1934).
Wechsler, B. A. & Von Dreele, R. Structure refinements of Mg2TiO4, MgTiO3 and MgTi2O5 by time-of-flight neutron powder diffraction. Acta Crystallogr. B45, 542–549 (1989).
Henderson, C. M. B., Knight, K. S. & Lennie, A. R. Temperature Dependence of Rutile (TiO2) and Geikielite (MgTiO3) Structures Determined Using Neutron Powder Diffraction. Open Mineral. J. 3, 1–11 (2009).
Goldschmidt, V. M. Die Gesetze der Krystallochemie. Naturwissenschaften 14, 477–485.
Redfern, S. A. T. High-temperature structural phase transitions in perovskite (CaTiO3). 9.
Guennou, M., Bouvier, P., Kreisel, J. & Machon, D. Pressure-temperature phase diagram of SrTiO3 up to 53 GPa. Phys. Rev. B 81, 054115 (2010).
Lytle, F. W. X‐Ray Diffractometry of Low‐Temperature Phase Transformations in Strontium Titanate. J. Appl. Phys. 35, 2212–2215 (1964).
Hui, Q., Tucker, M. G., Dove, M. T., Wells, S. A. & Keen, D. A. Total scattering and reverse Monte Carlo study of the 105 K displacive phase transition in strontium titanate. J. Phys. Condens. Matter 17, S111–S124 (2005).
Evans, H. T. The crystal structure of tetragonal barium titanate. Acta Crystallogr. 4, 377–377 (1951).
Kwei, G. H., Lawson, A. C., Billinge, S. J. L. & Cheong, S. W. Structures of the ferroelectric phases of barium titanate. J. Phys. Chem. 97, 2368–2377 (1993).
Comes, R., Lambert, M. & Guinier, A. The Chain Structure of BaTiO3 and KNbO3. Solid State Commun. 6, 715–719 (1968).
Senn, M. S., Keen, D. A., Lucas, T. C. A., Hriljac, J. A. & Goodwin, A. L. Emergence of Long-Range Order in BaTiO3 from Local Symmetry-Breaking Distortions. Phys. Rev. Lett. 116, (2016).
Petkov, V., Gateshki, M., Niederberger, M. & Ren, Y. Atomic-Scale Structure of Nanocrystalline BaxSr1-xTiO3 (x = 1, 0.5, 0) by X-ray Diffraction and the Atomic Pair Distribution Function Technique. Chem. Mater. 18, 814–821 (2006).
Sears, V. Neutron scattering lengths and cross sections. Neutron News 3, (1992).
Page, K., Kolodiazhnyi, T., Proffen, T., Cheetham, A. K. & Seshadri, R. Local Structural Origins of the Distinct Electronic Properties of Nb-Substituted SrTiO3 and BaTiO3. Phys. Rev. Lett. 101, (2008).
Kay, H. F. & Bailey, P. C. Structure and properties of CaTiO3. Acta Crystallogr. 10, 219–226 (1957).
Smith, M. B. et al. Crystal Structure and the Paraelectric-to-Ferroelectric Phase Transition of Nanoscale BaTiO3. J. Am. Chem. Soc. 130, 6955–6963 (2008).
Usher, T.-M. et al. Local and average structures of BaTiO3-Bi(Zn1/2Ti1/2)O3. J. Appl. Phys. 120, 184102 (2016).
Heitmann, A. A. & Rossetti, G. A. Thermodynamics of Ferroelectric Solid Solutions with Morphotropic Phase Boundaries. J. Am. Ceram. Soc. 97, 1661–1685 (2014).
Levin, I. et al. Nanoscale Polar Heterogeneities and Branching Bi-Displacement Directions in K0.5Bi0.5TiO3. Chem. Mater. 31, 2450–2458 (2019).
Neuefeind, J., Feygenson, M., Carruth, J., Hoffmann, R. & Chipley, K. The Nanoscale Ordered Materials Diffractometer NOMAD at the Spallation Neutron Source SNS. Nucl. Instrum. Methods Phys. Res. B 287, 68–75 (2012).
Mason, T. et al. The Spallation Neutron Source in Oak Ridge: A powerful tool for materials research. Phys. B 385–386, 955–960 (2006).
Pawley, G. S. Unit-cell refinement from powder diffraction scans. J. Appl. Crystallogr. 14, 357–361 (1981).
Coelho, A. Whole-profile structure solution from powder diffraction data using simulated annealing. J. Appl. Crystallogr. 33, 899–908 (2000).
Coelho, A. T. O. P. A. S. and TOPAS-Academic: an optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Crystallogr. 51, 210–218 (2018).
Xiao, C. J., Jin, C. Q. & Wang, X. H. Crystal structure of dense nanocrystalline BaTiO3 ceramics. Mater. Chem. Phys. 111, 209–212 (2008).
Rietveld, H. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Crystallogr. 2, 65–71 (1969).
Rietveld, H. M. The Rietveld method. Phys. Scr. 89, (2014).
Egami, T. & Billinge, S. Underneath of Bragg Peaks: Structural Analysis of Complex Materials. vol. 16 (Pergamon, 2012).
Kwei, G. H., Billinge, S. J. L., Cheong, S.-W. & Saxton, J. G. Pair-distribution functions of ferroelectric perovskites: Direct observation of structural ground states. Ferroelectrics 164, 57–73 (1995).
Ravel, B., Stern, E. A., Vedrinskii, R. I. & Kraizman, V. Local structure and the phase transitions of BaTiO3. Ferroelectrics 206, 407–430 (1998).
Zalar, B., Laguta, V. V. & Blinc, R. NMR Evidence for the Coexistence of Order-Disorder and Displacive Components in Barium Titanate. Phys. Rev. Lett. 90, (2003).
Levin, I., Krayzman, V. & Woicik, J. C. Local structure in perovskite (Ba, Sr)TiO3: Reverse Monte Carlo refinements from multiple measurement techniques. Phys. Rev. B 89, (2014).
Acknowledgements
This project was supported by the National Science Foundation (NSF DMR-1832803). This research used resources of the Spallation Neutron Source at the Oak Ridge National Labs (IPTS 21569). The authors would like to thank Dr. Jue Liu for his assistance with the neutron total scattering measurements, and Dr. Igor Levin for his insightful discussions. PHYC is the Bert and Emelyn Christensen professor of OSU and gratefully acknowledges financial support from the Vicki & Patrick F. Stone family. Michelle Dolgos would like to thank the Canada First Research Excellence Fund for financial support.
Author information
Authors and Affiliations
Contributions
C.M.C. initiated the project and synthesized the materials with A.T.F. The material samples were sent to ORNL for neutron total scattering measurements. C.M.C. and M.Y. analyzed and processed the total scattering data. M.R.D., D.P.C. and P.H.-Y.C. supervised the project. All authors contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Culbertson, C.M., Flak, A.T., Yatskin, M. et al. Neutron Total Scattering Studies of Group II Titanates (ATiO3, A2+ = Mg, Ca, Sr, Ba). Sci Rep 10, 3729 (2020). https://doi.org/10.1038/s41598-020-60475-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60475-8
This article is cited by
-
Anharmonic electron-phonon coupling in ultrasoft and locally disordered perovskites
npj Computational Materials (2023)
-
Thermal expansivity of geikielite and ilmenite utilizing in-situ synchrotron X-ray diffraction at high temperature
Acta Geochimica (2023)
-
Direct observation of elemental fluctuation and oxygen octahedral distortion-dependent charge distribution in high entropy oxides
Nature Communications (2022)
-
Study of structural correlations with temperature dependent dielectric response and ferroelectric behavior for (Sr, Mn) co-doped BaTiO3
Journal of Materials Science: Materials in Electronics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
