
Abstract
Equine arteritis virus (EAV) is maintained in the horse populations through persistently infected stallions. The aims of the study were to monitor the spread of EAV among Polish Hucul horses, to analyse the variability of circulating EAVs both between- and within-horses, and to identify allelic variants of the serving stallions EqCXCL16 gene that had been previously shown to strongly correlate with long-term EAV persistence in stallions. Serum samples (n = 221) from 62 horses including 46 mares and 16 stallions were collected on routine basis between December 2010 and May 2013 and tested for EAV antibodies. In addition, semen from 11 stallions was tested for EAV RNA. A full genomic sequence of EAV from selected breeding stallions was determined using next generation sequencing. The proportion of seropositive mares among the tested population increased from 7% to 92% during the study period, while the proportion of seropositive stallions remained similar (64 to 71%). The EAV genomes from different stallions were 94.7% to 99.6% identical to each other. A number (41 to 310) of single nucleotide variants were identified within EAV sequences from infected stallions. Four stallions possessed EqCXCL16S genotype correlated with development of long-term carrier status, three of which were persistent shedders and the shedder status of the remaining one was undetermined. None of the remaining 12 stallions with EqCXCL16R genotype was identified as a persistent shedder.
Similar content being viewed by others

Introduction
Equine viral arteritis (EVA) is one of the economically important diseases of horses and other equids1. The causative agent is equine arteritis virus (EAV), which has been recently reclassified within a species Alphaarterivirus equid in the genus Alphaarterivirus of the family Arteriviridae in the order Nidovirales2. Infection with EAV is often subclinical, but occasionally can lead to clinical disease of various severity after an incubation period of three to 14 days. The underlying cause is vascular injury, which leads to development of clinical signs that include pyrexia, depression, anorexia, dependent oedema, conjunctivitis, petechial haemorrhages on mucosal surfaces and urticaria. Pregnant mares may abort, and foals may develop severe interstitial pneumonia or pneumoenteric syndrome, depending on the age at the time of infection1.
Antibodies against EAV have been detected in equine sera worldwide, with only Japan, Iceland and New Zealand currently considered free from EAV infections3. The first reported outbreak of EVA in Poland occurred at one of the Thoroughbred studs in 1976–19774. Since then, EAV specific antibodies have been detected in a high percentage of horses of different breeds throughout the country5,6. The virus has been implicated in economic losses, as can be illustrated by isolation of EAV from 23% of cases of abortion or neonatal death over a 34-year period in one Polish-based study7. The virus is transmitted between horses through either respiratory or venereal routes. Horizontal transmission occurs via infectious droplets of respiratory secretions from acutely infected horses or via direct contact between infectious and susceptible animals1,8. Although EAV is considered to be easily inactivated outside of the host, it can remain infective for as long as 75 days at 4 °C under laboratory conditions9, and lateral spread via fomites was speculated to occur under natural conditions by some authors10. Especially important for the epizootiology of EAV is venereal route of transmission through semen of a persistently infected stallion to susceptible mares. Between 10 and 70% of the infected stallions become persistent shedders for periods of time ranging from months to years (in some cases for the whole life of the animal)1,8. It has been recently shown that EqCXCL16 gene is correlated with the establishment of a long-term (>1 year) carrier status by EAV infected stallions. Those with at least one copy of the dominant allele (EqCXCL16Sa or EqCXCL16Sb) associated with in-vitro susceptibility of CD3 + T lymphocytes to EAV infection are more likely to become long-term shedders than those with two copies of the recessive allele (EqCXCL16R) linked to the resistant phenotype11.
The genome of EAV is a linear positive-sense single stranded RNA molecule that encodes 10 open reading frames (ORFs). The two most 5′ proximal ORFs (ORF1a and ORF1b) encode two overlapping polyproteins (1a and 1ab), which are further cleaved into 13 non-structural proteins that are essential for virus replication. The remaining eight ORFs (ORF2a, ORF2b, ORF3, ORF4, ORF5, ORF5a, ORF6 and ORF7) encode structural proteins of the virus2. In persistently infected stallions the virus undergoes mutations that accumulate over time, which may lead to the emergence of variants with increased virulence12.
The aims of the current study were: (1) to monitor the spread of EAV within a population of Hucul horses at one of the Polish national studs in the absence of targeted infection control measures; (2) to determine the variability of circulating EAVs, both within- and between EAV-infected Hucul horses; and (3) to determine the allelic variants of the serving stallions’ EqCXCL16 gene.
Results
EAV status of the sampled horses
Overall, 84/221 (38.0%) serum samples were positive for EAV antibody in the course of the study (Table 1). Out of 17 mares introduced to the stud in 2012, 14 tested negative for EAV antibody at both 05/2012 and 12/2012 samplings, while the remaining three had viral neutralisation test (VNT) titres that ranged between 8 (two mares) and 32 (one mare) at 05/2012, and remained the same six months later at the 12/2012 sampling. A sharp increase in the number of EAV seropositive mares ((χ2 = 48.7, p < 0.00001) was observed between 12/2012 and 05/2013 samplings. All 14 seronegative mares that were introduced to the stud in 2012 seroconverted to EAV by May 2013 with titres ranging from 32 to 128. A rising EAV titres were detected in the remaining three seropositive mares, with a four-fold or greater increase in the EAV titre observed for two of these mares. The proportion of EAV seropositive stallions remained similar within the same period ((χ2 = 0.45, P = 0.45).
Out of 37 semen samples from 11 stallions that were tested for EAV RNA, 21 were positive (Table 2). The positive samples were collected from six stallions, including three that tested positive more than once, and were hence classified as permanent shedders (hucPL2, hucPL3 and hucPL5).
Genomic analysis of EAV
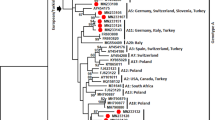
Full genome sequences of EAV were obtained from six semen samples from four infected stallions with an average coverage of 115.78X for EAVhucPL1(05/2013), 146.1X for EAVhucPL2(01/2009), 123.6X for EAVhucPL2(05/2013), 14.7X for EAVhucPL3(01/2009), 290.8X for EAVhucPL3(12/2012) and 26.3X for EAVhucPL4(05/2013). The nucleotide identity between genomic sequences of EAV from different horses varied from 94.7% to 99.6%. Based on the phylogenetic analysis, EAV sequences from the current study were closely related to other Polish EAV sequences from the last two decades (Fig. 1).

Neighbour-joining phylogenetic tree of sequences obtained in the study. The tree was constructed using a 2,895 nucleotide fragment of genomic RNA coding for structural genes of equine arteritis virus (EAV). Sequences acquired in this study are shown within a box. The following additional EAV sequences were included in the tree: PL-06_2011 (JX984459.1), PL-01_2007 (JX984455.1), PL-11_2004 (JX984449.1), F18 (EF492556.1), F23 (EF492561.1), S4216 (GQ903811.1), S3583 (GQ903809.1), S3699 (GQ903796.1), S4417 (GQ903798.1), SR7406 (GQ903866.1), ITA1165 (JN314874.1), 7HANG (JN314866.1), 1HANG (JN314862.1), 1342CAN (JN314858.1), SealeachSWE (JN314888.1), 1128CAN (JN314856.1), 544CAN (JN314854.1), 3142AUS (JN314851.1), LiedenNL (JN314875.1), 490ITA (JN314869.1), 52ITA (JN314867.1), VD7634 (MF598091.1), Wroclaw (JN314876.1), VienneAus (JN314852.1), Bibuna SWZ (JN314889.1), HK116 (EU586274.1), Bucyrus (NC_002532), F5 (EF492543.1), F27 (JN211316.1), CW96 (AY349167.1), CW01 (AY349168.1), GB_Glos (LC000003.1). Country and the date of isolation (when available) are included in the brackets next to each sequence. Numbers represent the average number of nucleotide substitutions between branches.
Single nucleotide variants (SNVs) were analysed in order to determine the intra-host variability of EAV. The total coverage for sequences from EAVhucPL4(05/2013) and EAVhucPL3(01/2009) was considerably lower than the total coverage obtained for viral sequences from the remaining samples, and below a value of 100 recommended for variant detection studies with next generation sequencing (NGS)13. Hence, the NGS data for EAV sequences from hucPL4 was not included in the SNV analysis. Multiple variable sites were identified throughout the viral genomes from each of the remaining three stallions, with the highest number (n = 310) detected in EAVhucPL2(05/2013) and the lowest (n = 41) in EAVhucPL2(01/2009) (Table 3). Distribution of synonymous and non-synonymous SNVs is presented in Fig. 2. For all EAV sequences included in the analyses, the majority of non-synonymous SNVs were located in ORF1a fragment encoding non structural protein (nsp) 2 (Table 4) and in genes encoding viral surface glycoprotein (GP) 2, GP3, GP4 and GP5 (Table 5).

Frequency distribution of single nucleotide variants (SNVs) across an equine arteritis virus (EAV) genome. Synonymous (S) and non-synonymous (N) SNVs across the EAV sequences from EAVhucPL1(05/2013) (A), EAVhucPL2(01/2009) (B), EAVhucPL2 (05/2013) (C), and EAVhucPL3(12/2012) (D). Only SNVs with frequencies ≥10% and ≥5X coverage are shown.
Two samples collected four years apart were available from stallion hucPL2. Based on the analysis of these two samples, the number of SNVs increased from 41 to 310 between 2009 and 2013. Some (n = 12) low-frequency SNVs present in 2009 became predominant in 2013, while others (n = 13) disappeared (Fig. 3).

Frequency distribution of single nucleotide variants (SNVs) across an equine arteritis virus (EAV) genome during persistence in one host. Red dots - SNVs from EAVhucPL2(01/2009), blue dots - SNVs from EAVhucPL2(05/2013). Lines connect SNVs present both in 2009 and 2013, and show changes in their frequencies. Only SNVs with frequencies ≥10% and ≥5X coverage are shown.
EqCXCL16 genotypes of EAV-infected stallions
Among 16 stallions sampled in the study, 12 (75%) were homozygous for EqCXCL16R allele and hence had a genotype associated with resistance of CD3 + T lymphocytes to EAV infection and decreased likelihood of development of long-term carrier status, and four (hucPL2, hucPL3, hucPL4 and hucPL5) were heterozygotes (EqCXCL16R/EqCXCL16Sa) and hence were considered to have a susceptible phenotype correlated with an increased likelihood of establishment of long-term carrier status following EAV infection (Fig. 4).

Allelic variants of EqCXCL16 gene detected among Hucul stallions enrolled in the study. Nucleotide positions 715, 741, 744 and 750 are determinants of CD3 + T lympocyte susceptibility to equine arteritis virus infection in-vitro11 that is correlated with the likelihood of development of EAV carrier satus. Stallions with two copies of recessive EqCXCL16R allele are more likely to clear the virus than stallions with at least one copy of the dominant EqCXCLSa or EqCXCLSb allele, which are more likely to develop persistent EAV infection. Sequences with ambigious bases on positions 715, 741, 744 and 750 were interpreted as representing a heterozygous (susceptible) genotype.
Discussion
Equine arteritis virus, similarly to other RNA viruses, is characterized by a high genetic variability. This results in generation of closely related genetic variants, known as quasispecies, within an individual host14. Until recently, quasispecies were identified by Sanger sequencing of cloned PCR products amplified from either viral isolates or directly from semen of infected horses15. In the current study NGS was used for sequence analysis of EAV. Six full consensus sequences of EAV from four different stallions were obtained, with the sequencing depth that allowed for identification of genetic variants appearing with as low as 10% frequency for four of them. Although multiple variable sites were found in each of the analysed viral sequences, their total number differed between EAVs from the three stallions. The lowest number (n = 41) was found in the 2009 EAV sequence from hucPL2, followed by the 2013 sample from hucPL1 (n = 140). Both numbers represented less than half of the SNVs identified in the EAV sequences obtained from the two remaining samples, including a sample from stallion hucPL2 collected in 2013. It has been previously shown that EAV remains genetically stable after the onset of infection and during early persistence, but its variability increases over time in response to pressures from the host’s immune system16,17. Our results support this conclusion, as hucPL3 and hucPL2 were persistently infected with on-going viral replication for at least six years prior to samplings in 2012/13, while hucPL1 was EAV negative until 2013. In addition, there was a clear increase in the intra-host diversity between EAV sequences from stallion hucPL2 collected four years apart (Table 3, Fig. 3).
The highest numbers of SNVs were identified in genes encoding nsp2, GP2, GP3, GP4 and GP5 of the virus. In general, this was consistent with the results of the previous studies using classical sequencing methods12,15. Similarly, in the most recent study that employed NGS, genes coding for GP3, GP5 and nsp2 showed the highest level of variability with the highest intra-host evolutionary rates recorded for ORF3 and ORF5, coding for GP3 and GP518.
In agreement with the previous data19, non-synonymous SNVs in ORF5 were located predominantly in three variable regions (V) of GP5: V1 encompassing aminoacid (aa) positions 61–121, V2 (aa 141–178), and V3 (aa 202–222). The V1 has been previously shown to contain three out of four major neutralization sites (aa 49, 61, 67–90, and 99–106)20. Each of the analysed viruses showed non-synonymous SNVs in at least one of these four major neutralization sites within GP520. Hence, our data support the previous assumption that variations in ORF5 is driven by the immune selection pressure17.
Non-synonymous SNVs were also found in genes coding for minor EAV glycoproteins (GP2, GP3 and GP4). It has been previously established that those proteins function in trimeric complexes. Hence, there are some structural constraints limiting their variability in regions responsible for binding21. This was consistent with our NGS results, as all non-synonymous SNVs in ORF2b (coding for GP2) were located in two previously described variable regions: V1 (aa 1–33) or V4 (aa 161–227)22. No SNVs were found at positions encoding cysteine (aa 48, 102, 137 and 195) in any of the viruses analysed, consistent with the importance of these residues for binding other minor glycoproteins23. All fixed amino acid changes within GP2 from a persistently infected stallion that was followed over a seven-year period in a previous study were also limited to the same two regions22.
Variability in GP3 was limited to two previously described highly variable sites (aa 3–41 and 98–121)24. It has been speculated that variants that had lost both glycosylation sites within the overlapping sequon (28NNTT31) through a change from asparagine to another amino acid at positions 28 and 29 may be selected during EAV persistence. This in turn could lead to the cleavage of the signal peptide in N-terminal end of GP3, although it is currently unknown what, if any, functional advantage would this create for the virus25,26. Our results do not support these speculations, as variants lacking one of the glycosylation sites (aa 29, Table 5) were found only in a recently infected stallion hucPL1, while asparagine was conserved at both positions in stallions hucPL2 and hucPL3 that have been persistently infected with EAV for years.
No variability was found in any of the glycoslation sites (aa 33, 55, 65 and 90) in predicted GP4 sequences. However, SNVs were found at nt positions 11 or 109 (corresponding to aa positions 4 and 37) in EAV sequences from two stallions. It has been proposed that these two SNVs may be involved in the attenuation of the vaccine strains of EAV27. Interestingly, substitution at aa position 37 of GP4 was found only in EAV from placenta of an infected pregnant mare, but not in EAV from any of the tested carrier stallions28. Therefore, the role of this SNV remains to be established, but it is unlikely that it is associated with carrier status of the host.
The only nsp where multiple SNVs were detected in the current study was nsp2. It has been previously shown that this protein induces humoral antibody responses in both naturally infected and vaccinated horses29. The majority of SNVs identified in EAVhucPL3(12/2012) and EAVhucPL2(05/2013) (two long-term carriers) were located in the part of ORF1a encoding a hypervariable region (aa 388–480)12,30. However, the pattern of SNVs in stallions that were recently infected with EAV at the time of sampling looked different. Most of SNVs in EAVhucPL1(05/2013) were located within the central region of nsp2, and only one SNV was present in nsp2 from EAVhucPL2(01/2009). Hence, it appears that SNVs accumulate within the hypervariable region of nsp2 during persistence, most likely due to selection driven by the immune response.
Next generations sequencing analysis performed in this study allowed for the determination of frequency and location of SNVs, and for the analysis of changes in the make-up of viral quasispecies within one persistently infected stallion over a period of time. Although this provided some insights into the changes in the dominant SNVs present at various time-points post-infection, a larger group of persistently infected stallions would need to be tested to confirm the observed tendencies and to determine whether or not some SNVs are linked to each other. It was not possible to reconstruct full-length sequences of individual viral haplotypes within the quasispecies population of EAV in each sample due to the use of short read sequencing. Application of novel NGS sequencing methods such as linked-read sequencing or long-read sequencing may allow to overcome this difficulty in the future31.
A number of environmental, viral and host factors may affect the outcome of EAV infection and establishment of persistent infection. These include factors such as breed of the horse, viral genotype or management32,33. Stallions in the current study were all of the same breed, managed under similar conditions, and were exposed to closely related variants of EAV. Hence, these factors were unlikely to be responsible for the observed differences in the duration of viral shedding by infected stallions. Results of two recent studies suggested that stallions with specific alleles of EqCXCL16 gene, which are linked to in-vitro susceptibility of CD3 + T lymphocytes to EAV infection, were at increased risk of becoming persistent shedders of the virus11,32. The majority (12/16) of stallions in the current study were homozygous for the resistant (EqCXCL16R) genotype. Unfortunately, data from reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) and serology were available for only some of the stallions at each of the six sampling occasions, which is a limitation of the study. Nonetheless, 3/16 stallions (hucPL2, hucPL3 and hucPL5) tested positive for EAV RNA in the semen for a period of at least two years and as such, could be classified as persistent shedders. All three stallions had an EqCXCL16 genotype associated with in vitro susceptibility to EAV infection and development of long-term carrier status. One stallion (hucPL11) with resistant genotype was positive for EAV RNA at only one sampling occasion and negative at all subsequent samplings, and hence has presumably cleared the infection. For six stallions the shedder status could not be determined as none, or only one, semen sample was tested by RT-qPCR for the presence of the virus. This group included five stallions with a resistant genotype and one (hucPL4) with a susceptible genotype. The remaining six stallions with a resistant genotype did not show any evidence of EAV infection (were serologically negative) at the time(s) they were tested. Altogether, out of the four EAV positive stallions that could be confidently classified as shedders or non-shedders, only one stallion with the resistant EqCXCL16 genotype cleared the virus. The remaining three that became persistent shedders all had susceptible EqCXCL16 genotypes. This is in agreement with the results reported previously where 29/34 (85.3%) stallions with a resistant genotype cleared the virus following EAV infection, whereas 32/43 (74.4%) stallions with the susceptible genotype became persistent shedders11.
Serological analysis performed in this study was the extension of the previous study6 where EAV seropositivity among horses at the same Hucul stud was monitored between 2006 and 2008. In that study, the differences in EAV antibody status between various age groups were described, while in the current study we were interested in the changes in the serological status of horses over a period of time. For the first five samplings the percentage of EAV seropositive horses remained relatively low around 20–30%, compared with over 55% reported previously6. However, between 2012 and 2013 seroconversion was detected in the majority of the tested mares, which resulted in the increase of the EAV seropositive horses to over 90% at the last sampling. Based on the interview with the stud’s staff, at least four of the EAV infected stallions, including three persistently infected ones (hucPL2, hucPL3 and hucPL5), were used for breeding in 2012 and 2013. It is not unusual to use persistently infected stallions for reproduction34 providing that certain precautions are taken including ensuring that the stallions are bread only seropositive mares and such mares are separated for up to three weeks following cover35. Such precautions were not taken when a group of 17 EAV seronegative mares was introduced to the stud in 2012 and bred for the first time during 2012/13 breeding season. All these mares showed serological evidence of EAV infection between December 2012 and May 2013. It is likely that the initial spread of the virus was via venereal route, however subsequent spread probably also involved respiratory route and possibly fomites, as two previously seronegative stallions (hucPL1 and hucPL4) became EAV positive in 2013. The sharp increase in the proportion of EAV seropositive horses from 22% to 84% over a period of six months provides a good example of how quickly the virus can spread within any horse population if appropriate infection control precautions are not maintained.
In conclusion, the results of this study add to our understanding of EAV epidemiology and genomic variability. To our knowledge, this is the first study that involved horses of the primitive Hucul breed. We have shown that intra-host diversity of EAV sequences is concentrated at specific sites of the genome. The impact of these variations on the expression of disease and virus-host interaction needs to be elucidated in future research.
Methods
Horses and sampling
Hucul horses from a national horse stud located in the southern part of Poland were used for the study. Results of our previous study demonstrated the circulation of EAV within this herd6. Coagulated blood samples for serology were collected between December 2010 and May 2013 every six months from both mares and stallions. In total, 221 blood samples from 16 stallions and 46 mares were collected. These included samples from 17 mares that were introduced to the stud shortly before May 2012 sampling.
Semen was collected from seropositive stallions whenever possible and tested for the presence of viral RNA. Altogether, 35 semen samples from 11 stallions were collected. In addition, two archival semen samples from January 2009 from EAV positive horses hucPL2 and hucPL3 were included in the analyses. Eight stallions from the sampled group (hucPL1, hucPL2, hucPL3, hucPL5, hucPL8, hucPL10, hucPL11, and hucPL13) were used for breeding at least once during the period of the study.
As the sampling and testing was done as part of veterinary management of the stud, no additional approval of the Local Ethics Committee was required for these activities (Directive 2010/63/EU).
Serology
Antibodies to EAV were detected using the VNT performed as described in the OIE Manual36. Briefly, serial two-fold dilutions of heat-inactivated (30 min at 56 °C) serum samples were added to each well of the 96-well microtitre plate in duplicate and mixed with 25 μL (100 to 300 tissue culture infectious dose 50% (TCID50)) of the Bucyrus strain of EAV diluted in Eagle’s minimum essential medium (MEM) (Sigma-Aldrich) containing guinea-pig complement at a final concentration of 10%. After incubation for 1 h at 37 °C, 50 μL (approximately 5 × 103 cells) of rabbit kidney 13 (RK13) cells (ATCC CCL-37) were added to each well. The plates were sealed with parafilm and incubated for three to five days at 37 °C in a humidified atmosphere with 5% CO2. Appropriate virus-, cell- and serum controls were included with each test run. The neutralisation titres were expressed as reciprocals of the highest dilution of the serum that inhibited viral cytopathic effect (CPE). A titre of 4 or greater was considered positive.
Viral RNA extraction and EAV-specific RT-qPCR
Total RNA was extracted directly from seminal plasma using QIAamp Viral RNA Mini Kit (Qiagen). Extractions were performed according to the manufacturer’s instruction with the exception that linear polyacrylamid (1 µL/sample) was used as carrier RNA. Reverse transcriptase qPCR was performed using Quantitect Viral Kit (Qiagen) and primers/probe complementary to a highly conserved region within the ORF7 gene of EAV as previously described37. Each reaction consisted of 1x QuantiTect Virus Master Mix, 1x QuantiTect Virus RT Mix, 0.8 µM of each primer, 75 nM of Taq Probe and 5 µL of template RNA in a total volume of 25 µL. The amplification conditions included the reverse transcriptase (RT) step (35 min at 48 °C), followed by the initial denaturation (10 min at 90 °C), and 40 cycles of denaturation (15 sec at 95 °C) and annealing/elongation (1 min at 60 °C). Samples were considered positive if quantification cycle (Cq) values were lower than 38. Extraction controls consisted of a Bucyrus strain of EAV (ATCC VR-796, positive control) and semen from an EAV seronegative stallion (negative control). Amplification controls consisted of RNA extracted from the Bucyrus strain (positive control) and water (non-template control). The RT-qPCR run was considered valid if all controls showed the expected results.
Next-generation sequencing
Six samples from four EAV infected stallions were subjected to NGS. These were labelled as: EAVhucPL1(05/2013), EAVhucPL2(01/2009), EAVhucPL2(05/2013), EAVhucPL3(01/2009), EAVhucPL3(12/2012) and EAVhucPL4(05/2013) where letters were used for identification of the stallion and numbers in bracket represented month and year of sampling. Stallions hucPL2 and hucPL3 were positive for EAV RNA in their semen since at least 20066 whereas stallions hucPL1 and hucPL4 tested EAV positive for the first time in 2013. Prior to sequencing, all samples were concentrated by ultracentrifugation. Briefly, 6 mL of seminal plasma was diluted five times in phosphate buffered saline pH 7.3 to 7.5 (PBS) and centrifuged at 3,000 × g for 10 min. Supernatant was then layered onto a 30% glycerol cushion (5 mL) in ultraclear thinwall polypropylene ultracentrifuge tube (Beckman). Following addition of PBS to the final volume of 35 mL, each sample was centrifuged for 2 h at 236,000 × g at 4 °C in a 70Ti fixed angle rotor (Beckman). Supernatant was removed and the pellet was resuspended in 0.5 mL of PBS. Viral RNA for NGS was extracted from this preparation using the same procedure as described above for RT-qPCR. Next generation sequencing was performed at Genomed SA using Illumina MiSeq Personal Sequencer.
Analysis of NGS data
Quality of 2 × 250 base pairs (bp) Illumina reads was checked with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Overlapping reads were merged using bbmerge v36.6238 and trimmed with Trimmomatic v0.3639 to remove adapters and low quality reads. Next, reads mapping to ribosomal RNA database SILVA v. 12840 were excluded (BWA MEM41 0.7.12 with default parameters), and the remaining reads were assembled de-novo into contigs using SPAdes 3.9.042. Contigs containing EAV genomic sequences were identified in each sample using an online version of the NCBI blastn program (v2.8.0)43, optimized for highly similar sequences (Megablast). The largest contig with an EAV reference sequence as the top hit was selected for futher analysis.
To identify SNVs within viral populations present in each sample, we used BWA MEM41 with default parameters to map trimmed reads to the reference EAV sequences. A consensus viral sequence obtained from the relevant de-novo assembly was used as the reference sequence for stallions PL1 and PL4, while EAV sequences from 2009 samples were used as reference sequences for stallions PL2 and PL3. Variants were called using LoFreq. 2.1.2 with default parameters44. Frequency distribution of SNVs across each viral genome was visualized using R 3.4.145. We included only variant alleles evidenced by at least five reads (alternative allele depth; as defined in46) and present in at least 10% of the reads at a given loci (alternative allele frequency).
Phylogeny
Neighbour-joining phylogenetic tree was constructed based on the alignment of 2,895 nt long fragments of the viral genome encoding structural proteins (positions 9751–12645 in Bucyrus reference strain - NC_002532) using MEGA5.2 software47. Viral sequences obtained in this study, and selected EAV sequences previously detected in Poland (n = 4) and other countries (n = 15) were included in the analysis.
Analysis of EqCXCL16 gene variant of the stallions
Total DNA was extracted from seminal plasma or serum samples (when semen was not available) collected from each of the stallions using QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions. PCR reactions were performed using JumpStart Taq DNA Polymerase (Sigma-Aldrich). Each reaction mix consisted of 0.4 µM of each primer (CXCL16-F and CXCL16-R11), 0.4 µM of dNTP mix, 1 µL of JumpStart Taq DNA Polymerase, and 2 µL of DNA in 1× reaction buffer in a total volume of 25 µL. Specific PCR products (280 bp) were sequenced using the same primers. EqCXCL16 sequences were assigned to a genotype based on the presence of specific nucleotides at positions 715, 741, 744 and 750 following alignment with the reference sequence (XM_001504756) as described previously11. Two susceptibility alleles (T, C, A, A and T, C, G, A) were designated EqCXCL16Sa and EqCXCL16Sb, respectively. The recessive allele associated with resistance to in-vitro EAV infection of and development of long-term carrier status the establishment of persistent EAV infection (EqCXCL16R) was characterized by A, G, T, and G at the specified positions. All analyses were performed using MEGA5.2 software47.
Nucleotide sequence accession numbers
The Polish EAV sequences obtained in this study have been submitted to GenBank under accession numbers MN056351 - EAVhucPL1(05/2013), MN180159 - EAVhucPL2(01/2009) MN104892 - EAVhucPL2(05/2013), MN056352 - EAVhucPL3 (01/2009), MN204505 - EAVhucPL3(12/2012), MN056353 - EAVhucPL4(05/2013).
Ethics approval and consent to participate
An informed approval was sought from stud managers before commencement of sampling. One of the roles of the National Veterinary Research Institute in Pulawy is monitoring of endemic diseases among Polish livestock. The sampling for the current study was performed within the scope defined by this role.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
References
Balasuriya, U. B. R., Go, Y. Y. & MacLachlan, N. J. Equine arteritis virus. Vet. Microbiol. 167, 93–122 (2013).
King, A. M. et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2018). Arch. Virol. 163, 2601–2631 (2018).
McFadden, A. M. et al. Evidence for absence of equine arteritis virus in the horse population of New Zealand. New Zeal. Vet. J. 61, 300–304 (2013).
Golnik, W. & Michalak, T. Causes of equine viral arteritis (arteritis quorum) in Poland. Med. Weter. 35, 605–606 (1978).
Golnik, W. The results of serological examinations of stallions for equine arteritis virus antibodies. Med. Weter. 56, 573–575 (2000).
Rola, J., Larska, M., Rola, J. G., Belák, S. & Autorino, G. L. Epizotiology and phylogeny of equine arteritis virus in hucul horses. Vet. Microbiol. 148, 402–407 (2011).
Bażanów, B. A., Frącka, A. B., Jackulak, N. A., Staroniewicz, Z. M. & Ploch, S. M. A 34-year retrospective study of equine viral abortion in Poland. Pol. J. Vet. Sci. 17, 607–612 (2014).
Holyoak, G. R., Balasuriya, U. B. R., Broaddus, C. C. & Timoney, P. J. Equine viral arteritis: current status and prevention. Theriogenology 70, 403–414 (2008).
de Vries, A. A. F. The Molecular Biology of Equine Arteritis Virus. Doctoral Thesis, University of Utrecht, Utrecht, the Netherlands (1994).
Guthrie, A. J. et al. Lateral transmission of equine arteritis virus among Lipizzaner stallions in South Africa. Equine. Vet. J. 35, 596–600 (2003).
Sarkar, S. et al. Allelic variation in CXCL16 determines CD3+ T lymphocyte susceptibility to equine arteritis virus infection and establishment of long-term carrier state in the stallion. PLoS Genetics 12, e1006467 (2016).
Balasuriya, U. B. R. et al. Genetic characterization of equine arteritis virus during persistent infection of stallions. J. Gen. Virol. 85, 379–390 (2004).
Van den Hoecke, S., Verhelst, J., Vuylsteke, M. & Saelens, X. Analysis of the genetic diversity of influenza A viruses using next-generation DNA sequencing. BMC Genomics 16, 79 (2015).
Andino, R. & Domingo, E. Viral quasispecies. Virology 479, 46–51 (2015).
Miszczak, F. et al. Emergence of novel equine arteritis virus (EAV) variants during persistent infection in the stallion: origin of the 2007 French EAV outbreak was linked to an EAV strain present in the semen of a persistently infected carrier stallion. Virology 423, 165–174 (2012).
Hedges, J. F., Balasuriya, U. B. R., Timoney, P. J., McCollum, W. H. & MacLachlan, N. J. Genetic divergence with emergence of novel phenotypic variants of equ ine arteritis virus during persistent infection of stallions. J. Virol. 73, 3672–3681 (1999).
Liu, L. et al. Analysis of ORFs 2b, 3, 4, and partial ORF5 of sequential isolates of equine arteritis virus shows genetic variation following experimental infection of horses. Vet. Microbiol. 129, 262–268 (2008).
Nam, B. et al. Intra-host Selection Pressure Drives Equine Arteritis Virus Evolution during Persistent Infection in the Stallion Reproductive Tract. J. Virol. JVI-00045 (2019).
Balasuriya, U. B. R., MacLachlan, N. J., De Vries, A. A., Rossitto, P. V. & Rottier, P. J. Identification of a neutralization site in the major envelope glycoprotein (GL) of equine arteritis virus. Virology 207, 518–527 (1995).
Balasuriya, U. B. R. et al. Neutralization determinants of laboratory strains and field isolates of equine arteritis virus: identification of four neutralization sites in the amino-terminal ectodomain of the GL envelope glycoprotein. Virology 232, 114–128 (1997).
Veit, M., Matczuk, A. K., Sinhadri, B. C., Krause, E. & Thaa, B. Membrane proteins of arterivirus particles: structure, topology, processing and function. Virus Res. 194, 16–36 (2014).
Rola, J., Socha, W. & Żmudziński, J. F. Sequence analysis of minor protein genes of equine arteritis virus during persistent infection. Bull. Vet. Inst. Pulawy 59, 179–184 (2015).
Wieringa, R., de Vries, A. A., Post, S. M. & Rottier, P. J. Intra-and intermolecular disulfide bonds of the GP2b glycoprotein of equine arteritis virus: relevance for virus assembly and infectivity. J. Virol. 77, 12996–13004 (2003).
Archambault, D., Laganiere, G., Carman, S. & St-Laurent, G. Comparison of nucleic and amino acid sequences and phylogenetic analysis of open reading frames 3 and 4 of various equine arteritis virus isolates. Vet. Res. 28, 505–516 (1997).
Zhang, M. & Veit, M. Differences in signal peptide processing between GP3 glycoproteins of Arteriviridae. Virology 517, 69–76 (2018).
Matczuk, A. K. & Veit, M. Signal peptide cleavage from GP3 enabled by removal of adjacent glycosylation sites does not impair replication of equine arteritis virus in cell culture, but the hydrophobic C-terminus is essential. Virus. Res. 183, 107–111 (2014).
Zhang, J. et al. Amino acid substitutions in the structural or nonstructural proteins of a vaccine strain of equine arteritis virus are associated with its attenuation. Virology 378, 355–362 (2008).
Balasuriya, U. B. R. et al. Genetic stability of equine arteritis virus during horizontal and vertical transmission in an outbreak of equine viral arteritis. J. Gen. Virol. 80, 1949–1958 (1999).
Go, Y. Y., Snijder, E. J., Timoney, P. J. & Balasuriya, U. B. R. Characterization of equine humoral antibody response to the nonstructural proteins of equine arteritis virus. Clin. Vaccine Immunol. 18, 268–279 (2011).
Snijder, E. J., Kikkert, M. & Fang, Y. Arterivirus molecular biology and pathogenesis. J. Gen. Virol. 94, 2141–2163 (2013).
De Coster, W. & van Broeckhoven, C. Newest methods for detecting structural variations. Trends in biotechnology, https://doi.org/10.1016/j.tibtech.2019.02.003 (2019).
Balasuriya, U. B. R. et al. Host factors that contribute to equine arteritis virus persistence in the stallion: An update. J. Equine Vet. Sci. 43(Suppl), 11–17 (2016).
Balasuriya, U. B., Zhang, J., Go, Y. Y. & MacLachlan, N. J. Experiences with infectious cDNA clones of equine arteritis virus: lessons learned and insights gained. Virology 462, 388–403 (2014).
Balasuriya, U. B. R., Carossino, M. & Timoney, P. J. Equine viral arteritis: A respiratory and reproductive disease of significant economic importance to the equine industry. Equine Vet. Educ. 30, 497–512 (2018).
McCollum, W. H., Timoney, P. J., Roberts, A. W., Willard, J. E. & Carswell, G. D. Response of vaccinated and non-vaccinated mares to artificial insemination with semen from stallions persistently infected with equine arteritis virus. Proceedings of the 5th International Conference on Equine Infectious Diseases. The University Press of Kentucky, Lexington, Ky pp 13–18 (1988).
Anonymous. Equine viral arteritis. (Chapter 2.5.10). In: OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals (2013).
Balasuriya, U. B. R. et al. Detection of equine arteritis virus by real-time TaqMan® reverse transcription-PCR assay. J. Virol. Methods 101, 21–28 (2002).
Bushnell, B. BBTools. https://jgi.doe.gov/data-and-tools/bbtools/ (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41(D1), D590–D596 (2013).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Johnson, M. et al. NCBI BLAST: a better web interface. Nucleic. Acids Res. 36(suppl_2), W5–W9 (2008).
Wilm, A. et al. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Research 40(22), 11189–201 (2012).
R Core Team. R: a language and environment for statistical computing. Foundation for Statistical Computing, Vienna, Austria. https://www.r-project.org (2015).
Hasing, M. E., Hazes, B., Lee, B. E., Preiksaitis, J. K. & Pang, X. L. A next generation sequencing-based method to study the intra-host genetic diversity of norovirus in patients with acute and chronic infection. BMC Genomics 17, 480 (2016).
Tamura, K. et al. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Acknowledgements
The authors wish to acknowledge the help of the stud’s manager and staff in collection of samples. The work described in this manuscript was funded by a grant number 05-1/KNOW2/2015 from Leading National Research Centre (KNOW) Scientific Consortium “Healthy Animal – Safe Food” awarded by the Polish Ministry of Science and Higher Education.
Author information
Authors and Affiliations
Contributions
W.S., J.R. participated in study design, analysis of the results, writing up and editing of the final manuscript, B.J. assisted in sample collection, participated in editing of the final manuscript, P.S. participated in data analysis and editing of the final manuscript, M.D. participated in writing up and editing of final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Socha, W., Sztromwasser, P., Dunowska, M. et al. Spread of equine arteritis virus among Hucul horses with different EqCXCL16 genotypes and analysis of viral quasispecies from semen of selected stallions. Sci Rep 10, 2909 (2020). https://doi.org/10.1038/s41598-020-59870-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59870-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
