
Abstract
Understanding the ecological processes that shape spatial genetic patterns of population structure is critical for understanding evolutionary dynamics and defining significant evolutionary and management units in the deep sea. Here, the role of environmental factors (topographic, physico-chemical and biological) in shaping the population genetic structure of four deep-sea habitat-forming species (one sponge - Poecillastra laminaris, three corals - Goniocorella dumosa, Madrepora oculata, Solenosmilia variabilis) was investigated using seascape genetics. Genetic data (nuclear and mitochondrial sequences and microsatellite multilocus genotypes) and environmental variables were employed to build individual-based and population-level models. The results indicated that environmental factors affected genetic variation differently amongst the species, as well as at different geographic scales. For individual-based analyses, different environmental variables explained genetic variation in P. laminaris (dissolved oxygen), G. dumosa (dynamic topography), M. oculata (sea surface temperature and surface water primary productivity), and S. variabilis (tidal current speed). At the population level, factors related to current and food source explained the regional genetic structure in all four species, whilst at the geomorphic features level, factors related to food source and topography were most important. Environmental variation in these parameters may be acting as barriers to gene flow at different scales. This study highlights the utility of seascape genetic studies to better understand the processes shaping the genetic structure of organisms, and to identify environmental factors that can be used to locate sites for the protection of deep-sea Vulnerable Marine Ecosystems.
Similar content being viewed by others

Introduction
How spatially variable environmental and habitat features influence evolutionary processes and population genetic connectivity in the deep sea is poorly understood1,2,3. The deep-sea environment experiences increasing pressure, decreasing pH, and generally decreasing temperature, with increasing depth4. Such single factor or multi-factorial gradients may strongly influence dispersal, settlement and recruitment patterns of deep-sea taxa5,6,7. Additionally, features such as bottom currents8, surface currents9 and bathymetry may play important roles in shaping the patterns of genetic connectivity amongst populations in many deep-sea species5,10,11, with the result that multiple environmental factors may act as barriers to or promoters of gene flow in the deep sea8,9,12 and thereby strongly influence population genetic structure at different spatial and even temporal scales. However, the complexity of the deep-sea physico-chemical environment and the logistical difficulties of sampling (both biological specimens and environmental data) such a large biome have limited our ability to understand multispecies patterns of population genetic structure and how these are influenced by environmental variation3,13,14,15.
Deep-sea vulnerable marine ecosystems (VMEs) are easily disturbed by anthropogenic activities, are very slow to recover, or may never recover, and are physically or functionally fragile16. Coral reefs and sponge gardens are two VMEs in the deep sea16, indicator taxa of which include scleractinian coral species that build reef frameworks and large and/or abundant demo- or hexactinellid sponges that provide physical habitat for other organisms17. Although there are no studies that have quantified the relationships between environmental factors and the maintenance of genetic structure in the deep sea, there are several studies from other research areas that provide information on the importance of some environmental factors for coral reefs and sponge gardens. Generally, factors such as depth, temperature, salinity, particulate organic carbon (POC), and aragonite (for corals) and silica (for sponges) concentrations have been recognised as influencing the formation of deep-sea coral and sponge habitats18,19,20,21,22,23,24,25. In addition, topography and sediment supply are also regarded as important factors influencing the distribution of coral and sponge habitat-forming deep-sea species26,27. At small spatial scales, internal tidal mixing and pH variation may have significant effects on coral (and presumably sponge) larvae and their ability to connect different populations28,29. However, the relative influence (weak versus strong) and the mechanism of action by which these environmental variables moderate the interaction between habitat and population genetic structure and connectivity are largely unknown.
The discipline of seascape genetics is extended from landscape genetics30,31 and attempts to explicitly integrate spatial ecological information with population genetic data to characterise marine environmental factors that contribute to genetic connectivity and/or population genetic isolation32,33. Selkoe et al. summarised seascape genetics studies over the last 10 years and found that temperature, oceanography (e.g., currents) and geography (e.g., distance) showed equal prevalence of influence on spatial genetic patterns of shallow water species34. In addition, the review revealed that > 20 other factors also influence connectivity (gene flow) at distinct spatio-temporal scales. This latter point highlights the complexity of seascape genetics, in particular for the deep sea for which far less is known than in shallow/coastal waters, and there is high uncertainty about species-specific responses at different scales in time and in space. Thus, whilst seascape genetics holds much promise for delivering a new understanding of how environmental variation influences genetic variation (e.g., seascape genetics can help to identify significant spatial differentiation even when the genetic signal is weak35,36) it is important to appreciate that seascape genetics is in its infancy and we are still learning about its limitations and possibilities. This is particularly the case in the deep sea, where there is an information vacuum for the largest single biome on the planet13.
Within the New Zealand Exclusive Economic Zone (EEZ), many species are endemic and most have distributions that encompass a range of environments, and are therefore amenable to quantification of the effect of environmental variation on population genetic variation and genetic connectivity37,38. Species-specific genetic variation in VME indicator taxa in the New Zealand region may be caused by a number of different factors, including historical processes (phylogeography), connectivity (high versus low gene flow within and amongst populations), and environmental variation (contemporary selection pressure)3,14. Whilst there is, at present, no indication that the patterns of species-specific genetic structuring are attributable to local or regional adaptation, the recognition of its existence as a pre-existing condition for seascape genetics analyses is important. Typically, genetic differentiation is described using neutral markers33,39, but with new marker types and new analytical procedures there is increasing focus on non-neutral markers (genes or non-coding regions closely linked to genes), at least some of which may be of particular value in determining the cause and functional mechanism of pronounced patterns of genetic structure that may arise as a consequence of selection15,34,37,40,41.
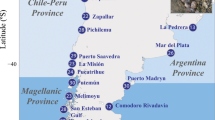
Because of its highly variable seafloor topography and bathymetry and its complex patterns of oceanic dynamics, New Zealand presents an ideal opportunity to test the effect of multi-factorial environmental influences on the genetic variation of deep-sea species. Such testing may be carried out at different spatial scales that are relevant to species’ distributions across the New Zealand region. Firstly, at the largest spatial extent, global assessment of the biogeography of the deep sea based on depth, temperature, salinity, dissolved oxygen, particulate organic carbon (POC), hydrographic and bathymetric parameters42 indicates that the New Zealand region is composed of two provinces, a northern (BY6) and a southern (BY10) province with a boundary at latitude ~45°S. Secondly, superimposed on top of this biogeographic structure are three major currents and their associated fronts around New Zealand - the Tasman Front, the Subtropical Front and the Subantarctic Front43. These currents and frontal systems differentiate water masses in the north and the south of the region, and also influence the direction of northern and southern currents across the Chatham Rise (to the east of New Zealand) that may reduce or perhaps even block gene flow across this feature. Corresponding to these potential hydrographic boundaries, three distinct regions have been recognised3 - above 42°S is the north region, between 42–45°S is the central region that contains the Chatham Rise, and below 45°S is the south region. Thirdly, the seafloor of the New Zealand EEZ is distinguished by different geomorphic features (such as seamounts, plateaux and ridges) that are characterised by different topographies and hydrodynamics that may influence genetic differentiation44,45. As such, populations on major geomorphic features (e.g., the Kermadec Ridge, the Chatham Rise, the Campbell Plateau – see Fig. 1) may be genetically isolated one from another, and this situation may therefore give rise to species-specific patterns of spatially explicit population genetic structure3,14. For example, Poecillastra laminaris (demosponge) and Goniocorella dumosa, Madrepora oculata and Solenosmilia variabilis (stony corals) are all common VME indicator taxa that are widely distributed throughout the New Zealand EEZ (Fig. 1). Previous studies have shown that different patterns of genetic structure are observed amongst these four species despite large overlaps in their distributions. Significant genetic differentiation was detected at the bioprovince, regional and geomorphic features scales in both P. laminaris and S. variabilis, at the regional and geomorphic features scales in G. dumosa, but only at the regional scale in M. oculata3,14. These different patterns of genetic structure may be related, at least in part, to species-specific reproductive strategies1,6, but a major limitation of research of this kind is that we know very little about the mode of reproduction or the larval dispersal ranges of most deep-sea species1. Whilst no studies have determined the reproductive strategy or mode of larval dispersal of the sponge, P. laminaris, deep-sea sponges are expected to produce lecithotrophic larvae with limited dispersal capacity14. The three coral species are expected to have very similar modes of reproduction and of larval dispersal46,47,48,49, are expected to be seasonal gonochoric spawners (although S. variabilis may be a continuous spawner7) with relatively short planktonic dispersal phases of perhaps a few days or weeks7,50. There is an indication that larval dispersal potential may be positively correlated with oocyte size amongst Goniocorella dumosa, Madrepora oculata and Solenosmilia variabilis3.

Map showing the distribution of samples amongst lower bathyal biogeographic provinces (yellow dashed line is the boundary between the northern and southern provinces), regions (yellow and red dashed lines indicate the boundaries for the north-central-south regions), and geomorphic features (named features) used for the analysis of genetic population structure for three species of deep-sea stony corals (blue, yellow and red dots) and one species of demosponge (green dots).
To explore how environmental variation may affect patterns of genetic structure, in the present study, environmental variables and genetic data were employed to quantify the relationships between environmental and genetic variation of these four VME indicator taxa at three different spatial scales. Three main hypotheses were tested: (1) that species-specific genetic variation is independent of environmental variation at the level of individuals within the New Zealand region, and for populations at different spatial scales, (2) that factors such as variation in silicate (for the sponge) and calcite (for the corals) explained significant variation in species-specific genetic variation, and (3) that multispecies genetic variation is associated with a common suite of environmental variables, that may include factors such as dissolved oxygen, some aspect of water movement (delivering suspended particle food and removing excretion products), and water temperature. The results provide information about which environmental variables contribute most to the genetic structure of populations of the four species, how connectivity amongst VMEs is influenced by environmental variation, if environmental variation plays a consistent role across multiple taxa in explaining population genetic structure, and how such information may be used to inform appropriate management measures, which in the present context, are likely to involve the establishment of a new offshore network of marine protected areas.
Results
Bivariate correlations revealed that several of the 26 topographic, physico-chemical and biological variables were significantly correlated (Table S1). Notably, pairwise combinations of nitrate, phosphate, aragonite and calcite concentrations were all highly correlated (r2 > 0.95), as were bottom temperature and temperature-pressure residual (r2 > 0.95). Therefore, aragonite was selected to represent nitrate, phosphate and calcite, and bottom temperature was selected to represent temperature-pressure residual in the modelling analyses. This selection procedure resulted in 22 variables for the sponge environmental data set (Table 1), and 21 variables for the coral environmental data set (Table 2).
Model building for individual-based variation
For all 4 species using the different genetic markers a range of models was successfully built. However, because BIC modelling was less successful at building models (i.e. microsatellites in G. dumosa) than AIC modelling, only AIC-based models were employed to identify environmental variables that explained genetic variation.
GLM analyses – sponge
The best model fit (p = 0.003) for individual-based P. laminaris explaining variation in the COI sequence data contained 11 of the 22 variables, the best model (p < 0.001) for Cytb variation contained 9 variables, and the best model fit for variation in the microsatellite data contained 6 and 11 variables for all (p = 0.031) and neutral (p = 0.232) loci, respectively (Table 1). Based on adjusted r2 values and their associated p-values, the models for the mtDNA sequence variation were a better fit than the models for all loci or neutral loci microsatellite variation (Table 1). Dissolved oxygen was the only variable that occurred in all four models, although it was only statistically significant in two of them (Table 1). Three environmental variables occurred in three of four models, but none were statistically significant on all three occurrences (Table 1). Scatterplots indicated that dissolved oxygen concentration was positively correlated with genetic differentiation for all genetic marker types (p < 0.05) (Figure S1).
GLM analyses - corals
In total, eleven models were developed and tested for individual-based genetic variation of the three corals (Table 2). Although most multiple r2 values of the models were low ( < 0.500), seven of 11 models were statistically significant (p < 0.05). For all 11 models, no one variable was significant in explaining genetic differentiation amongst individuals of all three corals. However, seven of the 21 variables were included in five models, and one variable, sea surface temperature, was included in six models.
For G. dumosa, two of four models were statistically significant and both involved the microsatellite loci (Table 2). The best model fit for the D-loop sequence data included 15 variables (of which six were statistically significant, p < 0.05), but the whole model was not significant (p = 0.603). For the ITS sequence data the best model fit contained 8 variables (of which only 3 were statistically significant, p < 0.05), whilst for microsatellite variation the best model fit contained 3 and 7 variables for all and neutral loci, respectively. Amongst all 4 models, dynamic topography was always included and contributed significantly in three, whilst sea surface temperature gradient) was also a significant contributor to all models except for that based on the all loci microsatellite data (Table 2). Scatterplots indicated that dynamic topography was positively correlated with genetic differentiation for all markers except the D-loop (p < 0.05) (Figure S2).
For M. oculata, two of three models were statistically significant (Table 2). For the ITS sequence data, the best model fit contained 12 of the 21 environmental variables, of which 9 variables were significant (p < 0.05) (Table 2). The best model fits for the microsatellite variation included nine variables (all significant) for all loci and seven (all significant) for the neutral loci. Sea surface temperature and surface water primary productivity were present in all models (Table 2). Scatterplots revealed that surface water primary productivity was positively correlated with genetic differentiation, whereas sea surface temperature was negatively correlated with genetic differentiation in two of three models (Figure S3).
For S. variabilis, three of four models were statistically significant (Table 2). For D-loop, the best model fit contained five variables (three were significant), and a five-variable model (three were significant) also provided the best fit (but not statistically significant) for variation of the ITS sequence. The models for microsatellite variation included four variables (all significant) for both the all loci and neutral loci datasets. There were no variables that were shared amongst all four models, but all models except ITS included tidal current speed. A positive relationship was observed between tidal current speed and genetic distance, even in the non-significant ITS model (Figure S4).
Hierarchical spatial testing
North-Central-South regional populations
Marginal tests resulting from DistLM analysis of population genetic variation at the regional scale showed that the genetic structure of the four species was related to different environmental factors (Table S2). After removal of the variables that did not explain genetic variation, through sequential testing, only one variable was retained in each model. Physico-chemical variables, silicate, bottom current speed, and bottom water temperature) explained variation in the all microsatellites, neutral microsatellites, COI sequences and Cytb sequences of P. laminaris, respectively (Table S2). Bottom water temperature for P. laminaris and M. oculata and dynamic topography for M. oculata and S. variabilis had a relative separation amongst genetically-structured regional populations (Figure S5).
For G. dumosa, the biological factor surface water primary productivity explained regional genetic structure (r2 > 0.669) in both the all and neutral microsatellite loci data sets. For M. oculata, dynamic topography and bottom water temperature contributed to genetic differentiation in the all and neutral loci data sets, respectively, whilst salinity was the best interpreter for the genetic structure revealed from ITS sequences. For S. variabilis, the variable dynamic topography and dissolved oxygen explained variation in the all loci data set, whereas dynamic topography explained most variation in the neutral loci data set (Table S2).
Geomorphic feature populations
Marginal tests resulting from DistLM analyses showed that genetic variation of the sponge P. laminaris exhibited a significant relationship with aragonite, bottom current speed, water density, dynamic topography, seamount, sea surface temperature. When considered alone, seamount explained 46.0% of the variability and water density 33.9% in the all loci data set, whilst seamount 49. 9% and bottom current speed 33.6% in the neutral loci data set. In the sequences data set, aragonite and sea surface temperature explained 39.9% and 35.7% in COI, and water density and dynamic topography were 41.6% and 29.9% in Cytb (Table S3). The dbRDA results illustrated that the populations from the Campbell Plateau and the Kermadec Ridge were genetically different from all other populations, and that water density and seamount were the main contributors to this pattern in the two data sets (Fig. 2A–D). The bottom current speed was in the best explained model in both BEST analyses of COI and Cytb, whilst salinity and seamount were the common variables in all and neutral microsatellites (Table 3).

dbRDA plots of genetic variation for COI (A), CytB (B), all microsatellites (D–F) and neutral microsatellites (C,E,F) of Goniocorella dumosa (E,F), Solenosmilia variabilis (G,H) and Poecillastra laminaris (A–D). Key to geomorphic feature abbreviated names: NE Slope = NE continental slope, Campbell = Campbell Plateau, Challenger = Challenger Plateau, Chatham = Chatham Rise, Hikurangi = Hikurangi Margin, Kermadec = Kermadec Ridge, Louisville = Louisville Seamount Chain, Macquarie = Macquarie Ridge and Tasman Basin.
For G. dumosa, the DistLM analysis from the all loci and neutral loci data sets showed that bottom current speed and seamount were the main factors that explained microsatellite variation: they explained 76.9% of the all microsatellites genetic variation and 78.7% of the neutral loci genetic variation (Table S3). The dbRDA plot showed that populations were separated from each other by the combination of bottom current speed and seamount (Fig. 2E,F). Additionally, aragonite was in the best explained model in BEST analyses of all microsatellites, whilst it was sea surface temperature in neutral microsatellites (Table 3).
Microsatellite variation of all loci of S. variabilis was partially explained by standard deviation of slope, seamount, bottom water temperature, slope percentage, dynamic topography, and tidal current speed. These six variables explained 92.1% of the observed genetic variability, with the highest contribution being made by seamount (Table S3). In the dbRDA plot, the Louisville Seamount Chain population was separated by standard deviation of slope from the other populations. In the results of the neutral microsatellite loci analysis, standard deviation of slope, seamount, bottom water temperature, sea surface temperature, slope percentage and surface water primary productivity were the factors selected in the DistLM analysis, and these 6 variables accounted for 92.2% of the genetic variation. Amongst these variables, standard deviation of slope was the most significant variable in the DistLM analysis, and it explained 25.9% of the microsatellite genetic variation (Fig. 2). The dbRDA plots revealed that the 8 geomorphic feature populations were genetically different from one another, and that this separation was influenced by the combination of these 6 environmental variables. Furthermore, the Louisville Seamount Chain population was also distinct from the other populations due to the influence of standard deviation of slope (Fig. 2G,H). The BEST analysis of the all and neutral microsatellite data sets agreed that bpi-broad, bottom current speed and bottom water temperature were the variables that explained the genetic variation.
In summary, based on the frequency of occurrences in different models, bottom current speed and bottom water temperature were the most frequently included variables in the models of all examined species.
Discussion
Seascape genetics as a discipline has advanced dramatically over the last decade as environmental data sets have increased in size and coverage (both spatial and temporal), and as new molecular markers and analytical tools have been developed (reviewed by32,33,34). However, such work has been exclusively focused on coastal and/or shallow water) species and systems, sometimes from the perspective of environmental or evolutionary biology, and sometimes with a management (e.g., fisheries, conservation) focus (e.g.,38,51,52,53,54,55). This study is amongst the first to apply the approach to deep-sea species and environments. It proved successful at uncovering relationships between genetic structure and environmental factors, although also revealed some issues that will need to be addressed in future applications of the method.
Limitations of the present study
Contrasting patterns of population genetic structure have been reported for the sponge and the three coral taxa throughout their distributional ranges in the New Zealand region3,14. Here, multi-factorial environmental variability has been examined to help explain the taxon-specific genetic variation. Small sample size is, however, a very big challenge that deep-sea researchers have to face2,56, and is a consideration in the interpretation of results from the present study. Because our sampling was based on availability of archived material collected by different research voyages over the years, rather than from a directed sampling programme, the distributions of the specimens analysed here had a spatial bias, with the highest density being from the Chatham Rise, associated with opportunistic collections during fisheries research and benthic biodiversity surveys. This bias may contribute to a failure to reveal seascape genetic patterns at a geomorphic feature level because large samples sizes for the Chatham Rise region may obscure a signal from small sample sizes from other regions. Moreover, because this is the first test of the role of environmental variability explaining genetic variability for deep-sea species, published information from previous studies for comparative purposes was absent. Nonetheless, despite small population sample sizes in some cases (minimum n is 4) and a lack of comparative information about the relationships between environmental factors and genetic differentiation, the results generated here contribute to a more comprehensive understanding about environmental and genetic interactions at both small and large spatial scales.
Different results between genetic markers
The selected environmental variables varied amongst the different models for each species according to the different genetic markers. For DNA sequence markers, mitochondrial markers had a better performance than nuclear markers, as judged by higher r2 values in GLM. One possible reason for this result is that the mitochondrial genome is more sensitive than the nuclear genome to environmental variability, perhaps because almost all of the mitogenome is coding whereas most of the nuclear genome is non-coding. Microsatellites, which had a better performance in population genetic studies, were less sensitive to environmental factors, as might be expected for markers that are assumed to be selectively neutral. Whether there is a direct selection-based association between genetic and environmental variation, or whether the environmental variables simply act as barriers to gene flow (a more neutral rather than selection-based explanation) remains to be determined.
Of note is the fact that a single environmental variable may be included in all models for a single species, but for one marker the variable’s contribution was positive and for another marker within the same species the variable’s contribution was negative (see Tables 1 and 2). Determining how a variable may have both a positive and negative contribution to genetic variation within the same species but for different markers will require further investigation. It may, however, be explained by direct selection pressure on one marker (e.g., the mitogenome) and an absence of selection pressure on another marker type (e.g., the nuclear genome). Given the linked nature of regions (genes) on the circular mitogenome, associations (either positive or negative) may be expected to be the same for two or more regions of mitochondrial DNA. The results here do not however, always support this expectation, indicating that different regions of the mitogenome may be responding differentially to the environmental variation, either as settled adults or as dispersive larvae.
Environmental effects and genetic variation
Topographic factors (such as bathymetric variation), physico-chemical factors (such as temperature, current speed, oxygen, aragonite saturation state, and nutrients), and biological factors (such as surface water Chl-a concentration) were examined directly in the present study. Such variables have previously been identified as environmental drivers for controlling the large-scale distribution of deep-sea corals and sponges9,20,57,58,59,60. In addition, variables such as hydrodynamics (in various forms), temperature (in various forms), bathymetry, and Chl-a concentration are often reported as being significant explanators of genetic variation in a range of marine taxa, including macroalgae, invertebrates and vertebrates (e.g.34,38,52,53,55,).
At the North-Central-South regional scale, the main variables driving the genetic variation amongst coral populations were bottom water temperature, dissolved oxygen, dynamic topography, salinity and surface water primary productivity. Dynamic topography, which is a measure of relative sea surface current velocity, reflects large scale current movements61 and may therefore also reflect the dispersal of coral larvae. Significant contributions of dynamic topography to explain genetic variation in M. oculata and S. variabilis supported the hypothesis in a previous study that large-scale currents associated with the Tropical and Subtropical fronts shaped the regional genetic structure3. For G. dumosa, the regional structure was influenced by surface water primary productivity revealed in both all and neutral microsatellites, which is related to the amount of food available to suspension-feeding corals62. The importance of this variable is also probably related to the influence of currents and fronts on genetic connectivity between the Central region and the North and South regions. That is, G. dumasa was more common in relatively shallower water at 400–500 m (compared to M. oculata and S. variabilis) on the Chatham Rise of the Central region, which is an area of particularly high sea surface primary productivity coincident with the subtropical front (ref for coincidence of high prod and STF). Furthermore, the variables associated with bottom currents and fronts, such as bottom water temperature, dissolved oxygen and salinity, also support the contention that environmental factors associated with the Chatham Rise are responsible for patterns of genetic variation across the three regions3. In the sponge, P. laminaris, bottom current speed, bottom water temperature and silicate explained more than half of the regional variability in different models. The contribution of silicate to the regional structure was revealed by all microsatellites, whilst bottom current speed and bottom water temperature explained the regional structure detected by COI, Cytb and neutral microsatellites. This result might indicate that the genome of P. laminaris is under some sort of (unknown) selection pressure for silicate, and that the bottom current speed is acting as a genetic barrier by affecting larval settlement and/or the available food source. However, this suggestion will require further evidence to test it. Overall, bottom current was an important factor that contributed to the formation of genetic structures for deep-sea corals and a sponge around New Zealand, and is a variable that is likely to influence genetic variation on multiple spatial scales.
At the geomorphic feature level, the factors that affected genetic connectivity patterns varied across the different species, but seamount was included in the all and neutral microsatellite data sets of all examined species. This indicates that the topography, and associated hydrodynamics, of different geomorphic features may affect the genetic structure of these species - for example, providing an explanation for genetic isolation of populations from Kermadec Ridge, Louisville Seamount Chain and Macquarie Ridge, where topographically-forced oceanographic features such as Taylor columns on the seamounts or along ridge currents could potentially influence larval retention on these features63. Previous studies of corals have detected topographically associated genetic patterns: genetic differentiation was significantly different between slope and ridges in Desmophyllum dianthus5, and in the coral genus Narella11. However, neither of these studies discussed the relationship between genetic differentiation and topography: further studies are required to verify this relationship. Bottom current speed was another factor that shaped genetic structure in G. dumosa, and the terrain variables (e.g., standard deviation of slope and slope percentage) were the most important factors that influenced the genetic variation of populations at the geomorphic features scale, in both the all and neutral microsatellite data sets of S. variabilis. For P. laminaris, water density (also referred to as sigma-theta) was the crucial explanator of population genetic variation at the geomorphic features scale. This variable reflects the temperature, salinity, and pressure of the water above the geomorphic features, and as such is probably operating as a single proxy variable for the influence of these water mass characteristics on genetic variation. A possible interpretation for all of the aforementioned variables acting as barriers to population connectivity relates to them reflecting the particular environment suitable for larval settlement and development amongst different geomorphic features. That is, the population genetic variation may be driven by different conditions amongst geomorphic features that challenged the larvae from other populations to survive, colonise and/or reproduce once established as an adult, such that little or no genetic contribution amongst local gene pools induced genetic differentiation between local populations. However, to our knowledge, there are no studies available for deep-sea species to provide evidence to support this suggestion.
At the individual level, the variables that elucidated the patterns of genetic variation differed not only amongst species but also by genetic marker. The important variables identified at this level were similar to those identified by habitat suitability modelling58,61,64,65, but the importance of each variable in the present study was different from those identified by these previous studies. Sea surface temperature gradient was significant as an explanator of genetic variation in G. dumosa and M. oculata and sea surface temperature was also significant in most models for these species. Sea surface temperature and sea surface temperature gradient are both related to primary productivity, and the latter to the location of frontal systems66. Primary productivity and frontal systems are proxies for food availability67,68, and the genetic variation patterns of these two species might therefore be driven by food source availability even at small spatial scales. In S. variabilis, although no common variables were found in all four models, tidal current speed was included in three models but not in ITS. Tidal current speed is related to the distribution of sedentary suspension-feeding organisms, such as corals and sponges, that rely on currents to deliver food28,58. Overall, at small scales, the genetic variations were explained by food-related variables. Food is closely related to an organism’s growth, fecundity, larval dispersal and settlement69,70,71,72,73,74, and thus food availability indirectly effects the genetic contribution made by one population to another population (by fecundity) and/or the genetic connectivity between populations (by larval dispersal and settlement). However, the mechanism by which food resources manipulate genetic connectivity patterns is unclear, and still needs investigation. Interestingly, dynamic topography was an important contribution to explaining the individual-based genetic variation of G. dumosa in all 4 models but not at large scales (regional and geomorphic feature level) for this species (it was important at these scales for the other two coral species). As noted above, dynamic topography is related to large-scale current dynamics and can influence large-scale patterns of genetic connectivity via larval dispersal. However, this variable has also been shown previously to be related to the distribution of suspension-feeding organisms through influencing food source availability and variability28,58, which provides more support for the explanation that food availability might explain the local genetic structure observed in G. dumosa.
For the sponge, P. laminaris, dissolved oxygen was the only variable that was included in all four models. Dissolved oxygen is directly related to metabolic rate and low concentrations of dissolved oxygen are expected to adversely affect sponge physiological performance75. In addition to dissolved oxygen, POC was also an important variable for P. laminaris in the GLM analysis. A significant relationship between POC and genetic structure was also detected in a shallow-sea reef sponge (Stylissa carteri)76. A previous study has found that POC is a very important food source for sponges77, further suggesting that food source availability might be the reason for the genetic differentiation observed in these deep-sea organisms even at small spatial scales. However, in contrast to the three corals, sea surface temperature and sea surface temperature gradient were not main contributors to the local genetic variations in P. laminaris. Similarly, sea surface temperature did not contribute to the habitat suitability model for predicting the distribution of the sponge Geodia78. This result possibly indicates that forces that influence the availability of the food sources and that shape the genetic structure may vary between deep-sea corals and sponges.
Generally, the results at the different spatial scales of this study suggest that bottom currents, food resource and terrain variability shape the genetic structure of both corals and sponges. A previous study has found that deep-sea corals and sponges have similar environmental demands79, and this might help to explain the similar genetic differentiations that were observed across these species. However, the variables that influenced the patterns of genetic structure at the different scales were species-specific, a finding that has been reported previously in the limited number of shallow water multi-species studies (reviewed by34,36). Nevertheless, while our seascape genetics approach has revealed which environmental variables are potentially important, how these different variables modified the species-specific patterns of genetic connectivity is still unclear. Additionally, high correlation coefficients leave as yet unresolved the question of whether species-specific genetic variation is correlated with a single environmental variable or multiple variables. Investigations of genotypic, phenotypic and environmental variability, as well as of gene-environment interactions are required to resolve these questions.
Implications for management
Marine environmental managers are increasingly incorporating genetic information into management planning80,81,82, and the results of seascape genetic modelling can help inform management options that conserve genetic structure and connectivity. To maintain population-level genetic diversity and effective population size, it is essential to understand the patterns of genetic connectivity that exist amongst deep-sea populations. However, open spatial population structures, logistical sampling difficulties and unknown barriers to gene flow present challenges to connectivity research. To overcome these challenges, seascape genetics research may open another window to improve understanding of linkages between genetics and the spatial ecology of populations83.
The genetic separation of some deep-sea coral and sponge populations into regions or locations that are characterised by particular temperature or food availability regimes illustrates the importance of protecting these VME indicator taxa via spatial management options that include sites (e.g. marine protected areas) that represent these different environmental regimes. For example, these different environmental regimes could be represented by a spatial classification of variables (e.g.,84,85) identified by this study as being correlated with genetic variation, which could then be used, along with other data inputs that represent other forms of biological variation in a region, to select the sites of the marine protected areas (e.g.,2,86). Additionally, the identification of variables that help to explain coral and sponge genetic variation indicates that networks of protected areas should include representatives of different geomorphic features, and conservation priorities could be allocated to sites that contain particularly complex topography (e.g., seamounts61,63).
As seascape genetics moves beyond the search for patterns amongst populations of a single species to a more rigorous approach involving hypothesis testing, individual-based and population-level assessments, greater awareness of spatial and temporal coverage, and comparisons across multiple species, it is expected that new insights into the environmental factors influencing gene flow will be forthcoming, and that such information will be of value in evolutionary studies and management (e.g.,31,87,88,89).
Materials and methods
Sample collection
Individuals of the demosponge, Poecillastra laminaris, and the stony corals, Goniocorella dumosa, Madrepora oculata and Solenosmilia variabilis, were obtained from the New Zealand National Institute of Water and Atmospheric Research (NIWA) invertebrate collection (NIC). Material had been collected from multiple sites throughout the South Pacific region over multiple years dating back to the 1960s3,14. Here, we recognise 12 distinct geomorphic features from which samples had been collected (Table S4). The majority of specimens were from seamount and slope habitats (122–1,805 m water depth). Most material was stored in ethanol, but a small number of samples (mostly sponges) had been air-dried. Only one fragment per species was subsampled from each collection sample (defined as a single sampling event at a single location) to avoid colonies produced by asexual reproduction and/or fragments of the same colony produced by damage caused by sampling gear. To permit testing of spatially explicit data, all specimens were allocated into three hierarchical geographic groups to define the populations for analysis: northern and southern bioprovinces (separated at ~45°S, following42), north-central-south regions (separated at ~42°S and ~45°S, following3,14) and geomorphic features (12 distinct features - NE continental slope, Bollons Seamount, Bounty Plateau, Bounty Trough, Campbell Plateau, Challenger Plateau, Chatham Rise, Hikurangi Margin, Kermadec Ridge, Louisville Seamount Chain, Macquarie Ridge and Tasman Basin) (Fig. 1). Spatial coverage was dictated by availability of archived material in the NIC. The minimum sample size for a population was set at four to achieve a balance between the validation of results and extracting maximum information content from the specimens. Further details of the ecology and life-histories of the four VME indicator taxa are provided by Zeng et al.3,14.
Genetic variation
Two different approaches, sequencing (nucleotide diversity) and genotyping (microsatellite allelic frequencies), were employed to detect species-specific genetic variation. The methodological details of development and identification of appropriate markers and the sequencing and genotyping are described in previous studies3,14. Briefly, mitochondrial DNA regions (COI and Cytb) were sequenced for the sponge, and mitochondrial (D-loop) and nuclear (ITS) loci were utilised for the corals. In total, 10, 20, 24 and 27 microsatellite loci were genotyped for P. laminaris, G. dumosa, M. oculata and S. variabilis, respectively. Because neutral markers can provide unbiased estimates of time since reproductive isolation and the amount of genetic drift90, the neutral microsatellite loci identified by LOSITAN91 and Micro-checker92 were employed for all analyses, where appropriate (Table 4). Following previous studies41,93 we also employed all loci, regardless of neutrality, in all analyses where appropriate, because non-neutral loci may be informative about population genetic structure and therefore useful for management purposes. In total, 6, 8, 6 and 12 neutral microsatellite loci were identified for P. laminaris, G. dumosa, M. oculata and S. variabilis, respectively. High rates of apparently non-neutral loci are typical of many marine invertebrates, including deep-sea corals and sponges, and may arise for different reasons, including null alleles, coloniality, inbreeding, selection, the Wahlund effect and poor state of sample preservation2,3,7,14. Previous work has shown that for population genetic analyses, the two data sets (all loci versus neutral loci only) perform very similarly, but with some small and perhaps important differences3,14. The work described here uses the genetic data sets from the population genetics research described by Zeng et al.3,14, but analyses these in the novel context of environmental variation to test for associations between genetic and environmental variation (pairwise estimates of ΦST and FST are presented in Table S5).
Environmental variables
Data for 26 environmental variables − 6 topographic, 17 physico-chemical and 3 biological variables - were obtained from various sources (Table 5). Data for environmental variables were obtained for the same geographic location from which specimens had been sampled. These environmental variables have been shown to potentially influence the distribution of coral and sponge species (see references in Introduction and61). The variable calcite was excluded from the analysis of sponge data, whilst silicate was excluded from the analysis of coral data due to their expected lack of biological relevance. Missing values in the environmental data set (about 4.58% missing values) were imputed by Primer-e (v7) using the expectation-maximization algorithm.
Highly correlated environmental variables were identified and eliminated from subsequent analysis to reduce problems that arise for the interpretation of models that include a large number of collinear predictors94. Pearson correlation coefficients between all pairs of environmental variables were calculated for those data collected at the sponge and coral sample locations using the R project (v3.2.2,95). To overcome colinearity, one of the predicator variables was eliminated from the data set when bivariate correlations were > 0.9537.
Modelling analyses
To test the hypothesis that multi-factorial environmental variation does not significantly explain species-specific genetic variation, three statistical methodologies were employed:
-
(i)
a generalized linear model (GLM), which is a multiple regression analysis between a number of environmental variables and a dependent variable (mean pairwise genetic distances between individuals of any one species). The individual pairwise distances from DNA sequences were generated using MEGA (v6), and a pairwise individual-by-individual genotypic distance matrix was calculated from the microsatellite data using the Codom-Genotypic distance option within GenAlEx96. This approach is a modification (based on pairwise individual genetic distance) of the approach using mean pairwise genetic distance between populations employed by38 and37. GLM analyses were performed separately for each species using a stepwise model, and the best model fit was chosen based on the lowest Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC) value models. All analyses were conducted using the package MASS in R. Scatterplots of the best-fit model were generated using the package ggplot2 in R.
-
(ii)
a distance-based linear model (DistLM) routine in PERMANOVA+ 97 that tests for associations between genetic and environmental variation37. DistLM was used to perform an ordination of fitted values from a given model and is constrained to find linear combinations of predictor variables (environmental data) that explain the greatest variation in the data cloud (population-specific haplotypic or allele frequencies). The best-fitting relationship was chosen for the final regression models by comparing the adjusted r2 selection value. Relationships between environmental parameters were initially examined by analysing each predictor separately (marginal tests), and then sequentially using the adjusted r2 selection procedure. Similarity matrices in DistLM analyses were built using Bray-Curtis resemblance matrices of haplotypic/allelic frequencies. For each species, haplotypic frequency matrices were calculated for each population in Excel, and allelic frequency matrices were calculated using GenAlEx. The p-values for individual predictor variables were obtained using 9,999 permutations. Distance-based redundancy analysis (dbRDA) plots were also generated using PERMANOVA+ to visualise the results once the best DistLM model of each species was obtained.
-
(iii)
a biological environmental stepwise analysis (BEST) that tests for the relationship between resemblance matrices of dependent (species-specific and population-specific haplotypic/allelic frequencies) and predictor (environmental) variables38. The BVSTEP analysis in Primer-e was utilised to search for the best model fit between genetic and environmental variables. The best-fitting relationship was chosen for the final regression models by comparing adjusted r2 selection criterion. A Bray–Curtis resemblance matrix was employed for the haplotypic/allelic frequency variables and a Euclidean distance resemblance matrix was employed for the environmental variables. To test for species-specific correlations between the two matrices, the BIOENV subroutine of the BEST routine was implemented using the non-parametric Spearman correlation coefficient method (rS) to test all combinations of factors.
These three analytical approaches operate in slightly different ways by testing different associations between the species-specific and site-specific genetic and environmental data. GLM is based on genetic variation at the individual, not population level. Whilst DistLM and BEST are based on population-specific haplotypic/allelic frequencies per locus, the former is a permutational model building analysis whereas the latter is a non-parametric test of all possible models, from the 26 single variable models to the one 26-factor model. All three analyses are species-specific and employ the same population-specific environmental data sets.
Rather than testing all possible data set combinations (i.e., all species × all genetic markers × all environmental variables) we only carried out seascape genetics analyses where significant spatial population genetic differentiation has already been identified. This is based on previous studies of deep-sea VME indicator coral3 and sponge14 population genetic variation. However, the dbRDA and BEST tests do not allow for testing of difference between only two groups of data, which means that testing of differences at the Northern-Southern provinces level could not be carried out. Instead, we focus on analyses of regional and geomorphic population genetic variation. In total, we conducted 15 analyses of individual-based genetic diversity for all four species and their respective markers, plus we conducted a further 12 tests of population-level genetic diversity at the scale of regions and geomorphic features, across all four species and their respective markers (Table 4).
Data availability
Data in study are available in the supplementary files.
References
Hilário, A. et al. Estimating dispersal distance in the deep sea: Challenges and applications to marine reserves. Front. Mar. Sci. 2, 6 (2015).
Baco, A. R. et al. A synthesis of genetic connectivity in deep-sea fauna and implications for marine reserve design. Mol. Ecol. 25, 3276–3298 (2016).
Zeng, C., Rowden, A. A., Clark, M. R. & Gardner, J. P. A. Population genetic structure and connectivity of deep-sea stony corals (Order Scleractinia) in the New Zealand region: Implications for the conservation and management of vulnerable marine ecosystems. Evol. Appl. 10, 1040–1054 (2017).
Amils, R., Ellis-Evans, C., Hinghofer-Szalkay, H., Rothschild, L. J. & Mancinelli, R. L. Life in extreme environments. Nature 409, 1092–1101 (2001).
Miller, K. J., Williams, A., Rowden, A. A., Knowles, C. & Dunshea, G. Conflicting estimates of connectivity among deep-sea coral populations. Mar. Ecol. 31, 144–157 (2010).
Bors, E. K., Rowden, A. A., Maas, E. W., Clark, M. R. & Shank, T. M. Patterns of deep-sea genetic connectivity in the New Zealand region: Implications for management of benthic ecosystems. PLoS One 7, e49474 (2012).
Miller, K.J. & Gunasekera, R. A comparison of genetic connectivity in two deep sea corals to examine whether seamounts are isolated islands or stepping stones for dispersal. Sci. Rep. 7, (2017).
Chevaldonné, P., Joilivet, D., Vangriesheim, A. & Desbruyéres, D. Hydrothermal-vent alvinellid polychaete dispersal in the eastern Pacific. 1. Influence of vent site distribution, bottom currents, and biological patterns. Limnol. Oceanogr. 42, 67–80 (1997).
Arellano, S. M., Van Gaest, A. L., Johnson, S. B., Vrijenhoek, R. C. & Young, C. M. Larvae from deep-sea methane seeps disperse in surface waters. Proc. R. Soc. B 281, 20133276 (2014).
Miller, K., Rowden, A., Williams, A. & Häussermann, V. Out of their depth? Isolated deep populations of the cosmopolitan coral Desmophyllum dianthus may be highly vulnerable to environmental change. PLoS One 6, e19004 (2011).
Baco, A. R. & Cairns, S. D. Comparing molecular variation to morphological species designations in the deep-sea coral narella reveals new insights into seamount coral ranges. PLoS One 7, e45555 (2012).
Catarino, D. et al. The Pillars of Hercules as a bathymetric barrier to gene flow promoting isolation in a global deep-sea shark (Centroscymnus coelolepis). Mol. Ecol. 24, 6061–6079 (2015).
Ramirez-Llodra, E. et al. Man and the last great wilderness: Human impact on the deep sea. PLoS One 6, e22588 (2011).
Zeng, C., Clark, M.R., Rowden, A.A. & Gardner, J. P. A. The use of spatially explicit genetic variation data from four deep-sea sponges to inform the protection of Vulnerable Marine Ecosystems. Sci. Rep. (2019).
Taylor, M. L. & Roterman, C. N. Invertebrate population genetics across Earth’s largest habitat: The deep-sea floor. Mol. Ecol. 26, 4872–4896 (2017).
FAO. International guidelines for the management of deep-sea fisheries in the high seas. https://doi.org/10.1007/s13398-014-0173-7.2 (2009).
Aguilar, R., Perry, A. L. & López, J. Conservation and management of vulnerable marine benthic ecosystems. in Marine animal forests: The ecology of benthic biodiversity hotspots (eds. Rossi, S., Bramanti, L., Gori, A. & Covadonga, O.) 1–43 (Springer International Publishing, 2017). https://doi.org/10.1007/978-3-319-17001-5_34-1.
Adkins, J. F., Boyle, E. A., Curry, W. B. & Lutringer, A. Stable isotopes in deep-sea corals and a new mechanism for ‘vital effects’. Geochim. Cosmochim. Acta 67, 1129–1143 (2003).
Kiriakoulakis, K. et al. Lipids and nitrogen isotopes of two deep-water corals from the North-East Atlantic: Initial results and implications for their nutrition. in Cold-water Corals and Ecosystems 715–729, https://doi.org/10.1007/3-540-27673-4_37 (2005).
Davies, A. J., Wisshak, M., Orr, J. C. & Murray Roberts, J. Predicting suitable habitat for the cold-water coral Lophelia pertusa (Scleractinia). Deep. Res. Part I Oceanogr. Res. Pap. 55, 1048–1062 (2008).
Dullo, W. C., Flögel, S. & Rüggeberg, A. Cold-water coral growth in relation to the hydrography of the Celtic and Nordic European continental margin. Mar. Ecol. Prog. Ser. 371, 165–176 (2008).
Dodds, L. A., Black, K. D., Orr, H. & Roberts, J. M. Lipid biomarkers reveal geographical differences in food supply to the cold-water coral Lophelia pertusa (Scleractinia). Mar. Ecol. Prog. Ser. 397, 113–124 (2009).
Guinan, J., Grehan, A. J., Dolan, M. F. J. & Brown, C. Quantifying relationships between video observations of cold-water coral cover and seafloor features in rockall trough, west of Ireland. Mar. Ecol. Prog. Ser. 375, 125–138 (2009).
Roberts, J. M. et al. Mingulay reef complex: An interdisciplinary study of cold-water coral habitat, hydrography and biodiversity. Mar. Ecol. Prog. Ser. 397, 139–151 (2009).
Case, D. H., Robinson, L. F., Auro, M. E. & Gagnon, A. C. Environmental and biological controls on Mg and Li in deep-sea scleractinian corals. Earth Planet. Sci. Lett. 300, 215–225 (2010).
Dolan, M. F. J., Grehan, A. J., Guinan, J. C. & Brown, C. Modelling the local distribution of cold-water corals in relation to bathymetric variables: Adding spatial context to deep-sea video data. Deep. Res. Part I Oceanogr. Res. Pap. 55, 1564–1579 (2008).
Brooke, S. D., Holmes, M. W. & Young, C. M. Sediment tolerance of two different morphotypes of the deep-sea coral Lophelia pertusa from the Gulf of Mexico. Mar. Ecol. Prog. Ser. 390, 137–144 (2009).
Frederiksen, R., Jensen, A. & Westerberg, H. The distribution of the scleractinian coral Lophelia pertusa around the Faroe Islands and the relation to internal tidal mixing. Sarsia 77, 157–171 (1992).
McCulloch, M. et al. Resilience of cold-water scleractinian corals to ocean acidification: Boron isotopic systematics of pH and saturation state up-regulation. Geochim. Cosmochim. Acta 87, 21–34 (2012).
Manel, S., Schwartz, M. K., Luikart, G. & Taberlet, P. Landscape genetics: Combining landscape ecology and population genetics. Trends Ecol. Evol. 18, 189–197 (2003).
Richardson, J. L., Brady, S. P., Wang, I. J. & Spear, S. F. Navigating the pitfalls and promise of landscape genetics. Mol. Ecol. 25, 849–863 (2016).
Selkoe, K. A., Henzler, C. M. & Gaines, S. D. Seascape genetics and the spatial ecology of marine populations. Fish Fish. 9, 363–377 (2008).
Riginos, C. & Liggins, L. Seascape genetics: Populations, individuals, and genes marooned and adrift. Geogr. Compass 7, 197–216 (2013).
Selkoe, K. A. et al. A decade of seascape genetics: Contributions to basic and applied marine connectivity. Mar. Ecol. Prog. Ser. 554, 1–19 (2016).
Crandall, E. D., Treml, E. A. & Barber, P. H. Coalescent and biophysical models of stepping-stone gene flow in neritid snails. Mol. Ecol. 21, 5579–5598 (2012).
Liggins, L., Treml, E. A., Possingham, H. P. & Riginos, C. Seascape features, rather than dispersal traits, predict spatial genetic patterns in co-distributed reef fishes. J. Biogeogr. 43, 256–267 (2016).
Silva, C. N. S. & Gardner, J. P. A. Identifying environmental factors associated with the genetic structure of the New Zealand scallop: Linking seascape genetics and ecophysiological tolerance. ICES J. Mar. Sci. 73, 1925–1934 (2016).
Wei, K., Wood, A. R. & Gardner, J. P. A. Seascape genetics of the New Zealand greenshell mussel: Sea surface temperature explains macrogeographic scale genetic variation. Mar. Ecol. Prog. Ser. 477, 107–121 (2013).
Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 154, 1219–1228 (1997).
Holderegger, R., Kamm, U. & Gugerli, F. Adaptive vs. neutral genetic diversity: Implications for landscape genetics. Landsc. Ecol. 21, 797–807 (2006).
Wei, K., Wood, A. R. & Gardner, J. P. A. Population genetic variation in the New Zealand greenshell mussel: Locus-dependent conflicting signals of weak structure and high gene flow balanced against pronounced structure and high self-recruitment. Mar. Biol. 160, 931–949 (2013).
Watling, L., Guinotte, J., Clark, M. R. & Smith, C. R. A proposed biogeography of the deep ocean floor. Prog. Oceanogr. 111, 91–112 (2013).
Chiswell, S. M., Bostock, H. C., Sutton, P. J. & Williams, M. J. Physical oceanography of the deep seas around New Zealand: A review. New Zeal. J. Mar. Freshw. Res. 49, 1–32 (2015).
Bucklin, A., Wilson, R. R. & Smith, K. L. Genetic differentiation of seamount and basin populations of the deep-sea amphipod Eurythenes gryllus. Deep Sea Res. Part A, Oceanogr. Res. Pap. 34, 1795–1810 (1987).
Yang, C. H. et al. Connectivity of the squat lobsters Shinkaia crosnieri (Crustacea: Decapoda: Galatheidae) between cold seep and hydrothermal vent habitats. Bull. Mar. Sci. 32, 257 (2015).
Waller, R. G. & Tyler, P. A. The reproductive biology of two deep-water, reef-building scleractinians from the NE Atlantic Ocean. Coral Reefs 24, 514–522 (2005).
Waller, R. G. Deep-water Scleractinia (Cnidaria: Anthozoa): Current knowledge of reproductive processes. Cold-water Corals Ecosyst. 691–700, https://doi.org/10.1007/3-540-27673-4_35 (2005).
Waller, R. G., Tyler, P. A. & Gage, J. D. Sexual reproduction in three hermaphroditic deep-sea Caryophyllia species (Anthozoa: Scleractinia) from the NE Atlantic Ocean. Coral Reefs 24, 594–602 (2005).
Pires, D. O., Silva, J. C. & Bastos, N. D. Reproduction of deep-sea reef-building corals from the southwestern. Atlantic. Deep. Res. Part II Top. Stud. Oceanogr. 99, 51–63 (2014).
Burgess, S. N. & Babcock, R. C. In Cold-water Corals and Ecosystems. (eds Freiwald, A. & Roberts, J. M.) 701–713 (Springer-verlag 2015).
Galindo, H. M. et al. Seascape genetics along a steep cline: Using genetic patterns to test predictions of marine larval dispersal. Mol. Ecol. https://doi.org/10.1111/j.1365-294X.2010.04694.x (2010).
Nanninga, G. B., Saenz-Agudelo, P., Manica, A. & Berumen, M. L. Environmental gradients predict the genetic population structure of a coral reef fish in the Red Sea. Mol. Ecol. https://doi.org/10.1111/mec.12623 (2014).
Saha, A. et al. Seascape genetics of saithe (Pollachius virens) across the North Atlantic using single nucleotide polymorphisms. ICES J. Mar. Sci. 72, 2732–2741 (2015).
Silva, C. N. S. & Gardner, J. P. A. Emerging patterns of genetic variation in the New Zealand endemic scallop Pecten novaezelandiae. Mol. Ecol. 24, 5379–5393 (2015).
Durrant, H. M. S., Barrett, N. S., Edgar, G. J., Coleman, M. A. & Burridge, C. P. Seascape habitat patchiness and hydrodynamics explain genetic structuring of kelp populations. Mar. Ecol. Prog. Ser. 587, 81–92 (2018).
Boschen, R. E., Rowden, A. A., Clark, M. R. & Gardner, J. P. A. Limitations in the use of archived vent mussel samples to assess genetic connectivity among seafloor massive sulfide deposits: A case study with implications for environmental management. Front. Mar. Sci. 2, 1–14 (2015).
Bryan, T. L. & Metaxas, A. Distribution of deep-water corals along the North American continental margins: Relationships with environmental factors. Deep. Res. Part I Oceanogr. Res. Pap. 53, 1865–1879 (2006).
Tittensor, D. P. et al. Predicting global habitat suitability for stony corals on seamounts. J. Biogeogr. 36, 1111–1128 (2009).
Woodby, D., Carlile, D. & Hulbert, L. Predictive modeling of coral distribution in the central Aleutian Islands, USA. Mar. Ecol. Prog. Ser. 397, 227–240 (2009).
Fisher, C. R. et al. Footprint of deepwater horizon blowout impact to deep-water coral communities. Proc. Natl. Acad. Sci. USA 111, 11744–9 (2014).
Tracey, D. M., Rowden, A. A., Mackay, K. A. & Compton, T. Habitat-forming cold-water corals show affinity for seamounts in the New Zealand region. Mar. Ecol. Prog. Ser. 430, 1–22 (2011).
Thiem, Ø., Ravagnan, E., Fosså, J. H. & Berntsen, J. Food supply mechanisms for cold-water corals along a continental shelf edge. J. Mar. Syst. https://doi.org/10.1016/j.jmarsys.2005.12.004 (2006).
White, M., Bashmachnikov, I., Arístegui, J. & Martins, A. Physical processes and seamount productivity. in Seamounts: Ecology, fisheries & conservation (eds. Pitcher, T. J. et al.) 10.1002/9780470691953.ch4. (Blackwell Publishing Ltd, 2008).
Guinotte, J. M. et al. Will human-induced changes in seawater chemistry alter the distribution of deep-sea scleractinian corals? Front. Ecol. Environ. 4, 141–146 (2006).
Davies, A. J. & Guinotte, J. M. Global habitat suitability for framework-forming cold-water corals. PLoS One 6, e18483 (2011).
Hurst, R., Renwick, J., Uddstrom, M., Kennan, S. & Rickard, G. J. Climate and ocean trends of potential relevance to fisheries in the New Zealand region.New Zealand Aquatic Environment and Biodiversity Report No. 90. (2012).
Abelson, A., Miloh, T. & Loya, Y. Flow patterns induced by substrata and body morphologies of benthic organisms, and their roles in determining availability of food particles. Limnol. Oceanogr. 38, 1116–1124 (1993).
Kaiser, M. J. et al. Marine ecology: Processes, systems and impacts. Marine Ecology vol. 0 (Oxford University Press, 2011).
Thorson, G. Reproductive and larval ecology of marine bottom invertebrates. Biol. Rev. 25, 1–45 (1950).
Buss, L. W. & Jackson, J. B. C. Planktonic food availability and suspension-feeder abundance: Evidence of in situ depletion. J. Exp. Mar. Bio. Ecol. 49, 151–161 (1981).
Bertness, M. D. & Gaines, S. D. Larval dispersal and local adaptation in acorn barnacles. Evolution (N. Y). 47, 316–320 (1993).
Qian, P. Y. Effect of food quantity on growth and reproductive characteristics of Capitella sp (Annelida: Polychaeta). Invertebr. Reprod. Dev. 26, 175–185 (1994).
Pechenik, J. A., Estrella, M. S. & Hammer, K. Food limitation stimulates metamorphosis of competent larvae and alters postmetamorphic growth rate in the marine prosobranch gastropod Crepidula fornicata. Mar. Biol. 127, 267–275 (1996).
Fouzai, N., Opdal, A., Jørgensen, C. & Fiksen, Ø. Effects of temperature and food availability on larval cod survival: A model for behaviour in vertical gradients. Mar. Ecol. Prog. Ser. 529, 199–212 (2015).
Seibel, B. A. & Drazen, J. C. The rate of metabolism in marine animals: Environmental constraints, ecological demands and energetic opportunities. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 362, 2061–78 (2007).
Giles, E. C., Saenz-Agudelo, P., Hussey, N. E., Ravasi, T. & Berumen, M. L. Exploring seascape genetics and kinship in the reef sponge Stylissa carteri in the Red Sea. Ecol. Evol. 5, 2487–2502 (2015).
Hadas, E., Shpigel, M. & Ilan, M. Particulate organic matter as a food source for a coral reef sponge. J. Exp. Biol. 212, 3643–3650 (2009).
Knudby, A., Kenchington, E. & Murillo, F. J. Modeling the distribution of Geodia sponges and sponge grounds in the Northwest Atlantic. PLoS One 8, e82306 (2013).
Rooper, C. N., Zimmermann, M., Prescott, M. M. & Hermann, A. J. Predictive models of coral and sponge distribution, abundance and diversity in bottom trawl surveys of the Aleutian Islands, Alaska. Mar. Ecol. Prog. Ser. 503, 157–176 (2014).
Waples, R. S. & Do, C. ldne: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Resour. 8, 753–6 (2008).
McCook, L. J. et al. Management under uncertainty: Guide-lines for incorporating connectivity into the protection of coral reefs. Coral Reefs 28, 353–366 (2009).
Haig, S. M. et al. The conservation genetics juggling act: Integrating genetics and ecology, science and policy. Evol. Appl. 9, 181–195 (2016).
Selkoe, K. A. et al. The DNA of coral reef biodiversity: Predicting and protecting genetic diversity of reef assemblages. Proc. R. Soc. B Biol. Sci. 283, 20160354 (2016).
Anderson, O. F. et al. Field validation of habitat suitability models for vulnerable marine ecosystems in the South Pacific Ocean: Implications for the use of broad-scale models in fisheries management. Ocean Coast. Manag. 120, 110–126 (2016).
Anderson, O. F. et al. Habitat suitability models for predicting the occurrence of vulnerable marine ecosystems in the seas around New Zealand. Deep. Res. Part I Oceanogr. Res. Pap. 115, 265–292 (2016).
Boschen, R. E. et al. A primer for use of genetic tools in selecting and testing the suitability of set-aside sites protected from deep-sea seafloor massive sulfide mining activities. Ocean Coast. Manag. 122, 37–48 (2016).
Prunier, J. G. et al. Optimizing the trade-off between spatial and genetic sampling efforts in patchy populations: Towards a better assessment of functional connectivity using an individual-based sampling scheme. Mol. Ecol. 22, 5516–5530 (2013).
Hall, L. A. & Beissinger, S. R. A practical toolbox for design and analysis of landscape genetics studies. Landsc. Ecol. 29, 1487–1504 (2014).
Luximon, N., Petit, E. J. & Broquet, T. Performance of individual vs. group sampling for inferring dispersal under isolation-by-distance. Mol. Ecol. Resour. 14, 745–752 (2014).
Gebremedhin, B. et al. Frontiers in identifying conservation units: From neutral markers to adaptive genetic variation. Anim. Conserv. 12, 107–109 (2009).
Antao, T., Lopes, A., Lopes, R. J., Beja-Pereira, A. & Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9, 1–5 (2008).
van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M. & Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538 (2004).
Gagnaire, P. A. et al. Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evol. Appl. 8, 769–786 (2015).
Graham, M. H. Confronting multicollinearity in ecological multiple regression. Ecology 84, 2809–2815 (2003).
Team, R. C. R. A Language and Environment for Statistical Computing. Vienna, Austria (2019).
Peakall, R. & Smouse, P. E. GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539 (2012).
Anderson, M. J., Gorley, R. N. & Clarke, K. R. PERMANOVA+ for PRIMER: Guide to Software and Statistical Methods. (primer-E Limited, 2008).
Acknowledgements
Sponge material was collected by NIWA and supplied for genetic work by the NIWA Invertebrate Collection (NIC); we are particularly grateful to Sadie Mills and Kareen Schnabel for their diligent assistance with sample location and processing. All values of environmental variables were generated by Mike Williams (NIWA).The work was funded by the New Zealand Ministry of Business, Innovation and Employment 712 as part of the NIWA-led project “Predicting the occurrence of vulnerable marine ecosystems for planning spatial management in the South Pacific region” [CO1X1229], NIWA under the Coasts and Oceans Research Programme 2 Marine Biological Resources: Discovery and definition of the marine biota of New Zealand [SCIs, 2014/2015, 2015/2016 & 2017/2018], and Startup Foundation for Advanced Scholars, Hunan Agricultural University.
Author information
Authors and Affiliations
Contributions
All authors contributed to research design; M.R.C. and A.A.R. provided logistical support for specimen collection; C.Z. completed all molecular experiments and analysis; all authors contributed to the writing of the manuscript and the interpretation of the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, C., Rowden, A.A., Clark, M.R. et al. Species-specific genetic variation in response to deep-sea environmental variation amongst Vulnerable Marine Ecosystem indicator taxa. Sci Rep 10, 2844 (2020). https://doi.org/10.1038/s41598-020-59210-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59210-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
