
Abstract
The study’s objectives were to examine the effects of electrofusion on rabbit somatic cell nuclear transfer (SCNT) embryos, and to test melatonin as a protective agent against electrofusion damage to SCNT embryos. The levels of reactive oxygen species (ROS), the epigenetic state (H3K9me3), and the content of endoplasmic reticulum (ER) stress-associated transcripts (IRE-1 and CHOP) were measured. Melatonin was added during the preimplantation development period. The total blastocyst cell numbers were counted, and the fragmentation rate and apoptotic index were determined and used to assess embryonic development. Electrofusion increased (1) ROS levels at the 1-, 2-, 4-, and 8-cell stages; (2) H3K9me3 levels at the 2-, 4-, and 8-cell stage; and (3) the expression of IRE-1 and CHOP at the 8-cell, 16-cell, morula, and blastocyst stages. The treatment of SCNT embryos with melatonin significantly reduced the level of ROS and H3K9me3, and the expression levels of IRE-1 and CHOP. This treatment also significantly reduced the fragmentation rate and apoptotic index of blastocysts and increased their total cell number. In conclusion, the electrofusion of rabbit SCNT embryos induced oxidative stress, disturbed the epigenetic state, and caused ER stress, while melatonin reduced this damage. Our findings are of signal importance for improving the efficiency of SCNT and for optimizing the application of electrical stimulation in other biomedical areas.
Similar content being viewed by others

Introduction
Somatic cell nuclear transfer (SCNT) is important in generating genetically modified animals for improving breeding stock, modelling human diseases, and as a method of treatment with therapeutic potential1. Since the first cloned sheep Dolly was generated by SCNT in 1997, dozens of mammals have been successfully cloned over the past two decades2. However, SCNT technology still suffers from low efficiency, and this limits its application in agriculture and biomedicine3. Some factors, such as the cell cycle of oocytes and donor cells, the damage to reconstructed embryos, inferior culture medium, and incomplete reprogramming, have been identified as important factors affecting normal SCNT embryonic development1,4,5. The method used to fuse the donor cells and recipient oocytes is particularly important in determining the outcome of SCNT. There are several fusion methods. One is chemical fusion, which is generally toxic to embryos and thus rarely used6,7. Another is virus fusion using inactivated hemagglutinating virus of Japan (HVJ), however, the potential risk of HVJ to the embryo development remains unclear; the biological safety of this virus is questionable and the fusion rate is not especially high. Electrofusion is a method that has been widely used with a good success rate8,9. One benefit of using electrofusion is that the physical parameters of the technique can be fixed accurately and are easily reproducible. This can yield a higher fusion rate relative to other fusion10. The mechanism of electrofusion centers on the liquid flow model. When the plasmalemmas of donor and recipient cells contact each other under electrofusion, they generate micropores and the rearrangement of phospholipid molecules results in cell fusion11. Although electrostimulation is effective in many embryonic manipulations, the potential adverse effects on embryonic development are not well understood.
Previous studies reported that electrostimulation induced ROS in the embryos of rats, mice and pigs but only a few studies focused on SCNT embryos12,13. SCNT embryos can manifest higher ROS levels, and this can cause damage to mitochondria, cell membranes, and DNA, and abnormal gene expression5,14,15. Also, abnormal epigenetic reprogramming occurs in a large proportion of cloned embryos, and this impedes improvement in SCNT efficiency1,9. Electrostimulation also regulates the epigenetic state in somatic cells; however, there are few reports on the effect of electrostimulation on the epigenetic state of embryos16,17. In this study, we optimized the electrofusion procedure, and studied its effects on SCNT embryos, oxidative stress, epigenetic state and ER stress. Also, the developmental competence of SCNT embryos was assessed in terms of their fragmentation rate, apoptotic index, blastocyst rate, and total cell number in blastocysts.
Results
Rabbit SCNT embryos after electrofusion had higher cleavage and blastocyst rates
The reconstructed embryos and the cleavage embryos were observed and analyzed. There was no significant difference in reconstruction rate among the non-electrofusion group, the electrofusion group at 1.6 kV/cm, and the electrofusion group at 3.2 kV/cm, but the cleavage and blastocyst rates in the non-electrofusion group were significantly lower than in the electrofusion groups at 1.6 kV/cm and 3.2 kV/cm (P < 0.05) (Table 1). There was no significant difference in cleavage and blastocyst rates between the groups undergoing electrofusion at 1.6 kV/cm and 3.2 kV/cm (Table 1).
Electrofusion induced oxidative stress in rabbit SCNT embryo
To measure the levels of ROS in SCNT embryos at the 1-, 2-, 4-, 8-, and 16-cell, morula and blastocyst stages, dichlorodihydrofluorescein diacetate (DCFH-DA) was used (Fig. 1A). The results showed that ROS levels in electrically stimulated SCNT embryos at the 1-, 2-, 4-, and 8-cell stages were significantly higher than those in the embryos in the non-electrofusion group (P < 0.05) (Fig. 1B). The level of ROS in the 3.2 kV/cm SCNT group was higher than that in the 1.6 kV/cm SCNT group (P < 0.05) (Fig. 1B). There were no significant differences of ROS levels in embryos at 16-cell, morula, and blastocyst stages among all groups (Fig. 1B).

Effects of electrofusion on rabbit SCNT embryos. (A) A representative fluorescent image of rabbit SCNT embryos stained with dichlorodihydrofluorescein diacetate (DCFH-DA) assay for ROS production (green). The quantitation of ROS (B) in rabbit SCNT embryos at the indicated stages in non-electrofusion, and electrofusion at 1.6 kV/cm and 3.2 kV/cm, respectively. (C) A representative image of rabbit SCNT embryos stained with H3K9me3 (green) and DAPI for nuclei (blue). Relative levels of H3K9me3 (D) in rabbit SCNT embryos at the indicated stage in non-electrofusion, and electrofusion at 1.6 kV/cm and at 3.2 kV/cm, respectively. The relative expression levels of IRE1 (E) and CHOP (F) in rabbit SCNT embryos at the indicated stages in non-electrofusion, and electrofusion at 1.6 kV/cm and at 3.2 kV/cm, respectively. Different superscripts at the same stage in each group above the bars denote statistically significant differences (P < 0.05).
Electrofusion increased H3K9me3 levels in rabbit SCNT embryos
To detect the epigenetic states of SCNT embryos at the 1-, 2-, 4-, 8-, and 16-cell, morula, and blastocyst stages, the level of histone H3 lysine 9 trimethylation (H3K9me3) was assessed by immunofluorescence (Fig. 1C). The results showed that the levels of H3K9me3 in the electrically stimulated SCNT groups were significantly higher than those in the non-electrofusion group at the 2-, 4- and 8-cell stages (P < 0.05). The levels of H3K9me3 in the 3.2 kV/cm groups were significantly higher than those in the 1.6 kV/cm group at the 2-, 4- and 8-cell stages (P < 0.05) (Fig. 1D).
Electrofusion-induced ER stress in rabbit SCNT embryos
The levels of ER stress-associated transcripts, including IRE-1 and CHOP, were measured at the 1-, 2-, 4-, 8-, and 16-cell, morula, and blastocyst stages using real-time PCR (Fig. 1E,F). In the results, there were no significant differences of the expressions of IRE-1 and CHOP in the 1-, 2-, and 4-cell stage embryos among the 3.2 kV/cm SCNT group, 1.6 kV/cm SCNT group and non-electrofusion group. Compared with those in the non-electrofusion group, the expression of IRE-1 and CHOP was markedly increased in the 3.2 kV/cm SCNT group and the 1.6 kV/cm SCNT group at the 8- and 16-cell, morula, and blastocyst stages (P < 0.05). Additionally, the expressions of IRE-1 and CHOP in the 1.6 kV/cm SCNT group were significantly lower than that in the 3.2 kV/cm SCNT group (P < 0.05).
Melatonin reduced ROS and ameliorated the abnormal epigenetic state and ER stress in rabbit SCNT embryos
To determine the optimal melatonin concentration, SCNT embryos were treated with different concentrations of melatonin throughout the preimplantation development period. The results showed that the blastocyst rate in the 10−6 M melatonin group was significantly higher than that in other groups (P < 0.05) (Table 2). Compared with the group without melatonin, the group with 10−6 M melatonin had significantly reduced ROS throughout the preimplantation stage (P < 0.05) (Fig. 2A), and decreased H3K9me3 levels at the 2-, 4-, 8-cell, morula, and blastocyst stages in the SCNT group (P < 0.05) (Fig. 2B). The treatment with melatonin also decreased the expression of IRE-1 (P < 0.05) (Fig. 2C) and CHOP (P < 0.05) (Fig. 2D) at the 8- and 16-cell, morula, and blastocyst stages.

Effects of melatonin on SCNT embryos. (A) The quantitation of ROS in rabbit SCNT embryos treated with melatonin at 0 and 10−6 M at the indicated stage. (B) The effect of electrofusion on H3K9me3 in SCNT embryos treated with melatonin at 0 and 10−6 M at the indicated stage. The relative expression levels of IRE1 (C) and CHOP (D) in rabbit SCNT embryos treated with melatonin at 0 and 10−6 M at the indicated stage. (*P < 0.05).
Melatonin protects against electrofusion-induced damage in rabbit SCNT embryos
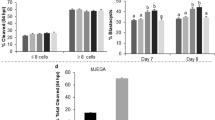
The embryos at 1.6 kV/cm electrical stimulation were cultured with or without melatonin (10−6 M). The fragmentation rate, the total cell number of blastocysts and the apoptotic index of blastocysts were analyzed. The results showed that the treatment with melatonin significantly reduced the fragmentation rate (P < 0.05) (Fig. 3A,B), increased the total cell numbers (P < 0.05) (Fig. 3C,D), and decreased the apoptotic index of blastocysts (P < 0.05) (Fig. 3C,E).

Effects of melatonin on SCNT blastocyst development. (A) A bright-field image of four embryos showing normal cleavage or fragmentation. (B) The percentage of fragmented embryos after treatment with melatonin at 0 and 10−6 M. (C) Blastomeres stained by TUNEL assay for apoptosis (green) and DAPI for nuclei (blue). The total number of cells per blastocyst (D) with 10−6 M melatonin treatment were significantly increased compared to the untreated group. The apoptotic index (E) of the group treated with 10−6 M melatonin was significantly reduced compared to the untreated group (*P < 0.05). The number of embryos per group is indicated by ‘n’.
Discussion
Up to now, electrofusion was the most commonly used method for most SCNT studies8,9,18,19. Regarding the piezo microinjection method, researchers were concerned about the risk of mercury contamination during the injection using micropipettes20. The mercury vapor and mercury compounds are highly toxic, so the piezo method is very risky to operators and embryos21,22. And also, the electric or chemical activations were required immediately after the establishment of embryos23. Here, we determined that the cleavage and blastocyst rates in the non-electrofusion group were significantly lower than in the electrofusion groups, indicating that the electrofusion method was better than the piezo method in terms of activating SCNT embryos. Considering its safety, applicability and better activation, we opted to use the electrofusion procedure and studied its effects on SCNT embryos, oxidative stress, epigenetic state and ER stress.
Electrical stimulation can lead to oxidative stress in myotubule cells via regulating the phosphorylation of AMPK24. Rapid, transient electrical stimulation increased the level of ROS in cardiac myocytes. In addition, previous studies showed higher ROS levels in SCNT embryos, which could inhibit embryo development25. Antioxidants are often added to culture medium to increase antioxidant capacity in embryo culture26,27. Melatonin is an antioxidant and free-radical scavenger that may be beneficial for the production of developmentally competent embryos when electrofusion is employed5. Melatonin effectively reduced oxidative stress and improves the development and hatchability rates of preimplantation fertilized embryos and SCNT embryos in cows, pigs, goats, mice and other species5,18,28,29. The results in the present study showed that greater electrical stimulation induced higher ROS level in SCNT embryos, which were reduced by incubation with melatonin.
Electromagnetic stimulation can lead to specific activation of the histone acetyltransferase, Brd2, resulting in histone H3K27 acetylation17. Electrical stimulation causes rat muscle dysfunction by altering miRNA-related signaling pathways30. Also, electrical stimulation modulates the gap junction-mediated cardiac cell-cell communication by regulating histone acetylase and deacetylase activities16. Chromatin structures were changed in BlCa cells exposed to ROS, and high levels of ROS increased H3K9me3 expression in human BlCa tissues31. H3K9me3 is highly conserved in most animals and is associated with the transcriptional inhibition of euchromatin, repression of chromatin, and formation of heterochromatin and plays a pivotal role in mammalian development1,32. The elevated level of H3K9me3a is a major epigenetic obstacle to the process of somatic cell nuclear reprogramming1. Our previous study reported that the inhibition of SUV39H1 can downregulate H3K9me3 and improve the efficiency of SCNT33. Importantly, embryonic genome activation (EGA) is the most important event during reprogramming, and EGA occurs around the 8-cell stage in rabbit embryos34,35,36. In this study, we found that electrical stimulation increased the level of H3K9me3 in SCNT embryos at the 2-, 4- and 8-cell stages, while stronger electrical stimulation caused H3K9me3 to be expressed at even higher levels. Su et al. demonstrated that melatonin has a positive role on modifying the epigenetic state of embryo via scavenging ROS5. And in this study, we found that electrofusion changed the epigenetics of SCNT embryos, and these epigenetic changes can be protected by melatonin.
Electrical stimulation such as in the treatment of tumors can induce endoplasmic reticulum (ER) stress and cause cell apoptosis37. When cells were subjected to stimulation, the unfolded protein response (UPR) was activated via the PERK, ATF6, and IRE1 pathways38. Pro-apoptotic factors, such as the CHOP, ASK1, JNK and Bcl2 protein families, were activated and overexpressed, inducing apoptosis and cell death38,39. Electrical stimulation can disrupt intracellular membranes, including those of the ER, mitochondria, and nucleus, thereby inducing apoptotic pathways39,40. In this study, we found that there were no significant differences in the expression levels of IRE-1 and CHOP in 1-, 2-, and 4-cell stage embryos among the 3.2 kV/cm SCNT group, 1.6 kV/cm SCNT group and the control group. It was conjectured that before the EGA stage, the embryonic genes were not activated, and the expression of ER stress genes was mainly derived from maternal storage. With the development of the EGA stage, embryonic genes were activated, and gene expression was affected by the epigenetic state of the embryos. We also demonstrated that H3K9me3 expression levels were influenced by electrical stimulation. We found that the expressions of IRE-1 and CHOP were markedly increased in the 3.2 kV/cm SCNT group and the 1.6 kV/cm SCNT group at the 8-cell, 16-cell, morula, and blastocyst stages. The results indicated that the ER stress at the post-EGA stage was regulated by embryonic genes whose control depended on the epigenetic state of the SCNT embryos. Previous studies reported that melatonin improved the meiotic maturation of porcine oocytes by reducing ER stress, and melatonin can also alleviate lipopolysaccharide-induced placental cellular stress response in mice41,42. These positive functions were thanks to the inhibition of oxidative stress function of melatonin41,42. In the present study, we also found that treatment with melatonin significantly ameliorated SCNT embryo ER stress induced by electrical stimulation.
The fragmentation rate is an important index for assessing the developmental competence of embryo43. Abnormal cleavage can cause defects in chromosome segregation, the appearance of haploid cells, and impaired embryonic development44. The total cell numbers in blastocysts and the apoptotic index are also important indicators of embryonic developmental competence45,46,47,48,49. In this study, we found that incubation with melatonin significantly reduced the fragmentation rate, increased the total cell number in blastocysts and decreased apoptosis, indicating that melatonin protects against electrical stimulation-induced damage in rabbit SCNT embryos. The positive effects of melatonin may be a result of the reduction of ROS, a decrease in ER stress and the preservation of normal epigenetic modifications.
To conclude, we discovered that electrofusion induced oxidative stress, increased H3K9me3, and led to ER stress in rabbit SCNT embryos, while melatonin protected against those changes. Our findings are of signal importance for improving the efficiency of SCNT and for optimizing the application of electrical stimulation in other biomedical areas.
Material and Methods
Reagents and media
Unless otherwise stated, culture media and reagents used in this study were purchased from Sigma (St. Louis, MO, USA).
Animal preparation
The experimental protocol was in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and was approved by the Laboratory Animal Care Committee of Xi’an Jiaotong University. New Zealand white rabbits, approximately 3 kg in weight and 12 months old were used. Rabbits were raised in the Laboratory Animal Center of Xi’an Jiaotong University Health Science Center at a temperature of 24 ± 1 °C, relative humidity of 55 ± 5%, and a 12 h light:12 h dark photoperiod. The animals were given access to water and food ad libitum.
Collection of oocytes
Female rabbits were super-ovulated by subcutaneous injection of 80 IU of pregnant mare serum gonadotropin (PMSG; Sansheng, Ningbo, China) and after 96 h, 100 IU of chorionic gonadotropin (CG; Sansheng, Ningbo, China) was administered by intravenous injection. After 12 h, the rabbits were euthanized and the oviducts and uteri were removed and flushed with 5 mL of Dulbecco’s phosphate-buffered saline (DPBS) to recover the oocytes. The oocytes were observed under a microscope and transferred into DPBS containing 5% fetal bovine serum (FBS).
Preparation of donor cells
A small piece of ear skin (1 × 1 cm) from a three year-old New Zealand white rabbit was removed, washed three times with DPBS containing penicillin and streptomycin, cut into small pieces, and cultured for approximately 10 days in DMEM containing 10% FBS, penicillin and streptomycin to obtain fibroblasts. The fibroblasts were cultured in saturation humidity with 5% CO2 at 38.5 °C. When the cells were approximately 80% confluent, the fibroblasts were subcultured and transferred into a 3.5-cm dish with fresh culture medium. Fibroblasts were purified through two passages. Fibroblasts from 3–4 passages were cultured to confluence in 96-well plates (Corning, NY, USA) and then incubated in DMEM supplemented with 0.5% FBS for 3 days. The cells were detached with DPBS containing 0.25% (w/v) trypsin and 1 mM ethylenediaminetetraacetic acid (EDTA) for 1 min, trypsinization was terminated by addition of DMEM containing 10% FBS, and cells were pelleted by centrifugation at 350 g for 10 min. The pellets were resuspended in M199 containing 7.5 m g/mL cytochalasin B and 10% FBS for SCNT.
Somatic cell nuclear transfer (SCNT)
SCNT was conducted as previously described8. Oocytes containing the first polar body were selected and stained with 7.5 μg/mL Hoechst 33342 in Earle’s balanced salt solution-complete medium (Thermo Fisher Scientific; Waltham, MA, USA) for 20 min. The oocytes were then placed in M199 containing 7.5 μg/mL cytochalasin B for 10 min. The nuclei of oocytes were localized by UV illumination for 1 to 2 s and removed using an 18 μm outer diameter micropipette. For electrofusion, a donor cell was drawn up and injected into the perivitelline space of an enucleated oocyte. The microelectrode consisted of a 25 cm long copper wire with a diameter of 100 microns passed through a 20 cm long plastic tube with an outer diameter 3.5 mm and inner diameter 2.5 mm. The copper wire was fixed to the plastic tube with glue so that electrostatic interference from the plastic tubing and the micromanipulation system and operators could be avoided. The copper wire was soldered with an electric soldering iron to a 5 cm long platinum wire curved at about 135 degrees. A second electrode was made in the same way, and each was attached to a micromanipulator controlled by the micromanipulation system (Eppendorf, Saxony, Germany). The embryos were incubated three times with fusion medium (BTX; Holliston, MA, USA) for 3 min, and then transferred to a fusion medium droplet. The complex was gently clamped with microelectrodes to maintain contact with the cell membrane. The electrofusion conditions were three 20 μs DC pulses at 1.6 kV/cm or 3.2 kV/cm with an ElectroSquare Porator (BTX; ECM830). After fusion, the somatic embryos were washed three times with DPBS and transferred into embryo culture medium for one hour at 38.5 °C, 5% CO2, and 100% humidity. For the non-electrofusion group, a donor cell was drawn up and injected into the enucleated oocyte, washed three times with DPBS, transferred into embryo culture medium and incubated for one hour at 38.5 °C, 5% CO2, and 100% humidity.
Embryos activation and culture
The fused embryos or injected embryos were treated with ionomycin for 5 min and then washed three times with DPBS. The embryos were treated for 1 h with 2 mM 6-dimethylaminopurine (6-DMAP) and 5 μg/mL cycloheximide (CHX; Abcam) in Earle’s balanced salt solution (EBSS)-complete medium. The embryos were then washed three times with DPBS. The cloned embryos were subsequently cultured in SOF medium (Caisson, Wuhan, China) containing 6% BSA at 38.5 °C in 5% CO2 at 100% humidity. Melatonin was added to the culture medium at concentrations of 0, 10−3, 10−6, and 10−9 M and was present throughout the development period. After 4 h of culture, the reconstructed embryos were examined microscopically and the number of cells counted. We examined the embryos again after 24 hours of culture and counted the number of embryos undergoing cleavage and the number of fragmented embryo. After four days in culture, the numbers of blastocysts were counted, and samples of blastocysts from the pool were collected for the measurement of ROS, immunofluorescence staining of H3K9me3, mRNA expression levels using real-time PCR, apoptosis detection, and counting of total cell numbers per blastocyst. The reconstruction rate was determined by the normalization of the number of reconstructed embryos to that of donor oocytes. The cleavage rate was determined by the normalization of the number of cleavage embryos to that of reconstructed embryos. The fragmentation rate was defined as the ratio of the number of fragmented embryos to that of cleavage embryos. The blastocyst rate was defined as the ratio of the number of blastocysts to the number of cleavage embryos.
Measurement of reactive oxygen species in embryos
A reactive oxygen species assay kit (Beyotime, Shanghai, China) was used to quantitate the ROS levels. Embryos were incubated in serum-free culture medium containing 10 mM dichlorodihydrofluorescein diacetate (DCHF-DA) at 37 °C for 20 min. The embryos were then washed three times in serum-free culture medium and fluorescence examined using the Nikon Eclipse Ti-S microscope (Nikon, Tokyo, Japan). Images were captured using a digital camera, analyzed with Image-Pro Plus software (Media Cybernetics, Bethesda, MD), and background fluorescence was subtracted from the experimental readings. Experiments were performed in triplicate and fifteen embryos were analyzed for each group at each stage.
Quantitation of H3K9me3 by immunofluorescence
Embryos (15 per group at each stage) were incubated in pronase and acidic Tyrode’s solution to remove the zona pellucida and mucin coat. Embryos were then fixed in 4% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100 for 20 min, and blocked in 1% BSA at 4 °C overnight. Embryos were stained with anti-H3K9me3 antibody (Abcam, Cambridge, MA, USA) at 4 °C overnight, washed thoroughly, and incubated with secondary antibody, Alexa Fluor 488-labeled goat anti-mouse IgG (Beyotime), for 1 h at room temperature. The embryos were counterstained with DAPI (Beyotime), mounted, and fixed with a drop of Antifade Mounting Medium (Beyotime). The immunofluorescent signals of the samples were detected using a laser scanning confocal microscope (Zeiss, Jena, Germany). The mean intensity of fluorescence was measured using Image-Pro Plus 6.0 software (Media Cybernetics). Each nucleus was manually outlined and the mean fluorescence intensities for H3K9me3- and DAPI-stained samples were recorded, and divided by the acquisition times of the corresponding signal. The total fluorescent intensities for H3K9me3 and DAPI were then obtained according to the amount of the embryos.
Real-time PCR
Total RNA was extracted from embryos using the Cells-to-Signal Kit (Invitrogen, Carlsbad, CA, USA) and the PrimeScriptTM RT Reagent Kit (TaKaRa, Tokyo, Japan) was used to synthesize cDNA. Real-time PCR was performed using the CFX96 RT-PCR detection system (Bio-Rad, Hercules, CA, USA) with SYBR Premix Ex TaqTM II (TaKaRa, Tokyo, Japan). The thermocycling conditions were as follows: 1 min at 95 °C, followed by 40 cycles of 5 s at 95 °C and 30 s at 60 °C, and a melting curve ranging from 65 °C to 95 °C, 5 s per 0.5 °C increment. The specificity of the real-time PCR reaction was confirmed by single peaks in the melt curves. Data were collected during each extension. Amplification efficiencies of genes were obtained using the slopes of the standard curves. The geometric mean of the Ct values of H2AFZ and HPTR1 was adopted to normalize the gene expression as previously described. The primers were designed using Primer3 and sequences are shown in Table 3. Twenty embryos per group at each stage were processed in each replication and measurements of gene expression were performed in triplicate.
Apoptosis measurement and counting of total cells per blastocyst
The DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) was used according to the manufacturer’s instructions to quantitate apoptosis. Blastocysts (15 per group) were fixed in 4% paraformaldehyde at room temperature for 2 h, permeabilized in 0.5% Triton X-100 at room temperature for 5 min, and incubated with FITC-conjugated dUTP and terminal deoxynucleotidyl transferase in the dark at 37 °C for 1 h. The tailing reaction was terminated by addition of 2 × SSC in the dark for 15 min at room temperature. The embryos were then incubated with PBS containing 25 µg/mL RNase A in the dark at room temperature for 30 min. After DAPI staining and washing with PBS in the dark, the blastocysts were placed on a slide, mounted with a coverslip, and examined using the Nikon Eclipse Ti-S fluorescence microscope. Images were captured with a digital camera and analyzed using Image-Pro Plus software. The total number of cells per blastocyst was indicated by DAPI staining.
Statistical analysis
Statistical analysis was conducted using the SPSS software package (SPSS Inc., Chicago, IL, USA). The reconstructed rate, the cleavage rate, the fragmentation rate, and the blastocyst rate were analyzed using χ2 tests. The relative intense of H3K9me3, apoptotic index, and total cell numbers were analyzed using one-way ANOVA. The equal variance was analyzed by the Levene median test and ANOVA, and multiple pairwise comparisons were performed by Tukey’s test. A P value less than 0.05 (P < 0.05) was considered statistically significant. Error bars on the means represent ± SEM.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Matoba, S. & Zhang, Y. Somatic Cell Nuclear Transfer Reprogramming: Mechanisms and Applications. Cell stem Cell 23, 471–485, https://doi.org/10.1016/j.stem.2018.06.018 (2018).
Narbonne, P., Miyamoto, K. & Gurdon, J. B. Reprogramming and development in nuclear transfer embryos and in interspecific systems. Curr. Opin. Genet. Dev. 22, 450–458, https://doi.org/10.1016/j.gde.2012.09.002 (2012).
Ogura, A., Inoue, K. & Wakayama, T. Recent advancements in cloning by somatic cell nuclear transfer. Philos. Trans. R. Soc. London. Ser. B, Biol. Sci. 368, 20110329, https://doi.org/10.1098/rstb.2011.0329 (2013).
Qu, P. et al. Effects of embryo-derived exosomes on the development of bovine cloned embryos. PLoS one 12, e0174535, https://doi.org/10.1371/journal.pone.0174535 (2017).
Su, J. et al. Melatonin significantly improves the developmental competence of bovine somatic cell nuclear transfer embryos. J. pineal Res. 59, 455–468, https://doi.org/10.1111/jpi.12275 (2015).
D’Souza, A. A. & Shegokar, R. Polyethylene glycol (PEG): a versatile polymer for pharmaceutical applications. Expert. Opin. drug. delivery 13, 1257–1275, https://doi.org/10.1080/17425247.2016.1182485 (2016).
Hoffman, A. N., Bamba, R., Pollins, A. C. & Thayer, W. P. Analysis of polyethylene glycol (PEG) fusion in cultured neuroblastoma cells via flow cytometry: Techniques & optimization. J. Clin. neuroscience: Off. J. Neurosurgical Soc. Australas. 36, 125–128, https://doi.org/10.1016/j.jocn.2016.10.032 (2017).
Jia, L. et al. Use of oocytes selected by brilliant cresyl blue staining enhances rabbit cloned embryo development in vitro. Zygote 27, 166–172, https://doi.org/10.1017/S0967199419000200 (2019).
Qu, P. et al. Sperm-borne small RNA regulate alpha-tubulin acetylation and epigenetic modification of early bovine somatic cell nuclear transfer embryos. Molecular human reproduction, https://doi.org/10.1093/molehr/gaz023 (2019).
Liu, J. et al. Effects of interval between fusion and activation, cytochalasin B treatment, and number of transferred embryos, on cloning efficiency in goats. Theriogenology 76, 1076–1083, https://doi.org/10.1016/j.theriogenology.2011.05.013 (2011).
Kong, Q. & Liu, Z. Electrofusion of 2-Cell Embryos for Porcine Tetraploid Embryo Production. Methods Mol. Biol. 1874, 361–371, https://doi.org/10.1007/978-1-4939-8831-0_21 (2019).
Popova, E., Bader, M. & Krivokharchenko, A. Effects of electric field on early preimplantation development in vitro in mice and rats. Hum. Reprod. 26, 662–670, https://doi.org/10.1093/humrep/deq.379 (2011).
Koo, O. J. et al. Electrical activation induces reactive oxygen species in porcine embryos. Theriogenology 70, 1111–1118, https://doi.org/10.1016/j.theriogenology.2008.06.031 (2008).
Hajian, M., Hosseini, S. M., Ostadhosseini, S. & Nasr-Esfahani, M. H. Comparative Stepwise Pattern of Reactive Oxygen Species Production during In Vitro Development of Fertilized and Nuclear Transferred Goat Embryos. Int. J. Fertil. Steril. 11, 93–98, https://doi.org/10.22074/ijfs.2017.5049 (2017).
Hwang, I. S., Bae, H. K., Cheong, H. T. & Mitochondrial, D. N. A. damage in bovine somatic cell nuclear transfer embryos. J. veterinary Sci. 14, 235–240, https://doi.org/10.4142/jvs.2013.14.3.235 (2013).
Meraviglia, V. et al. Acetylation mediates Cx43 reduction caused by electrical stimulation. J. Mol. Cell. cardiology 87, 54–64, https://doi.org/10.1016/j.yjmcc.2015.08.001 (2015).
Yoo, J. et al. Electromagnetized gold nanoparticles mediate direct lineage reprogramming into induced dopamine neurons in vivo for Parkinson’s disease therapy. Nat. Nanotechnol. 12, 1006–1014, https://doi.org/10.1038/nnano.2017.133 (2017).
An, Q. et al. Melatonin supplementation during in vitro maturation of oocyte enhances subsequent development of bovine cloned embryos. Journal of cellular physiology, https://doi.org/10.1002/jcp.28357 (2019).
Kim, E. Y. et al. Establishment of bovine embryonic stem cell lines using a minimized feeder cell drop. Cell. reprogramming 14, 520–529, https://doi.org/10.1089/cell.2012.0038 (2012).
Wakayama, S., Kishigami, S. & Wakayama, T. Improvement of Mouse Cloning from Any Type of Cell by Nuclear Injection. Methods Mol. Biol. 1874, 211–228, https://doi.org/10.1007/978-1-4939-8831-0_12 (2019).
Dong, W. et al. Developmental toxicity from exposure to various forms of mercury compounds in medaka fish (Oryzias latipes) embryos. PeerJ 4, e2282, https://doi.org/10.7717/peerj.2282 (2016).
Butts, C. D. et al. Urine cortisol concentration as a biomarker of stress is unrelated to IVF outcomes in women and men. J. Assist. Reprod. Genet. 31, 1647–1653, https://doi.org/10.1007/s10815-014-0359-0 (2014).
Haritou, M., Yova, D. & Loukas, S. Agents facilitating the electric field-induced fusion of intact rabbit erythrocytes. Bioelectrochemistry 52, 229–238 (2000).
Evers-van Gogh, I. J. et al. Electric Pulse Stimulation of Myotubes as an In Vitro Exercise Model: Cell-Mediated and Non-Cell-Mediated Effects. Sci. Rep. 5, 10944, https://doi.org/10.1038/srep10944 (2015).
Ge, L., Li, C., Wang, Z., Zhang, Y. & Chen, L. Suppression of Oxidative Stress and Apoptosis in Electrically Stimulated Neonatal Rat Cardiomyocytes by Resveratrol and Underlying Mechanisms. J. cardiovascular pharmacology 70, 396–404, https://doi.org/10.1097/FJC.0000000000000534 (2017).
Uhm, S. J., Gupta, M. K., Das, Z. C., Kim, N. H. & Lee, H. T. 3-Hydroxyflavone improves the in vitro development of cloned porcine embryos by inhibiting ROS production. Cell. reprogramming 13, 441–449, https://doi.org/10.1089/cell.2011.0017 (2011).
Liang, S., Jin, Y. X., Yuan, B., Zhang, J. B. & Kim, N. H. Melatonin enhances the developmental competence of porcine somatic cell nuclear transfer embryos by preventing DNA damage induced by oxidative stress. Sci. Rep. 7, 11114, https://doi.org/10.1038/s41598-017-11161-9 (2017).
Pang, Y. W. et al. Melatonin protects against paraquat-induced damage during in vitro maturation of bovine oocytes. J. pineal Res. 66, e12532, https://doi.org/10.1111/jpi.12532 (2019).
Soto-Heras, S. et al. Beneficial effects of melatonin on in vitro embryo production from juvenile goat oocytes. Reproduction, fertility, Dev. 30, 253–261, https://doi.org/10.1071/RD17170 (2018).
Pan, L. L. et al. Electrical Stimulation Improves Rat Muscle Dysfunction Caused by Chronic Intermittent Hypoxia-Hypercapnia via Regulation of miRNA-Related Signaling Pathways. PLoS one 11, e0152525, https://doi.org/10.1371/journal.pone.0152525 (2016).
Whongsiri, P. et al. Oxidative stress and LINE-1 reactivation in bladder cancer are epigenetically linked through active chromatin formation. Free. Radic. Biol. Med. 134, 419–428, https://doi.org/10.1016/j.freeradbiomed.2019.01.031 (2019).
Chawalit, S. et al. Trichostatin A and ascorbic acid assist in the development of porcine handmade cloned embryos via different physiologic pathways. Reprod. Sci. 19, 976–986, https://doi.org/10.1177/1933719112440049 (2012).
Zhang, J. et al. MicroRNA-125b is a key epigenetic regulatory factor that promotes nuclear transfer reprogramming. J. Biol. Chem. 292, 15916–15926, https://doi.org/10.1074/jbc.M117.796771 (2017).
Reis E. Silva, A. R. et al. Alteration of DNA demethylation dynamics by in vitro culture conditions in rabbit pre-implantation embryos. Epigenetics 7, 440–446, https://doi.org/10.4161/epi.19563 (2012).
Reis Silva, A. R. et al. Dynamics of DNA methylation levels in maternal and paternal rabbit genomes after fertilization. Epigenetics 6, 987–993, https://doi.org/10.4161/epi.6.8.16073 (2011).
Henderson, G. R. et al. Candidate gene expression patterns in rabbit preimplantation embryos developed in vivo and in vitro. J. Assist. Reprod. Genet. 31, 899–911, https://doi.org/10.1007/s10815-014-0233-0 (2014).
Jia, J., Xiong, Z. A., Qin, Q., Yao, C. G. & Zhao, X. Z. Picosecond pulsed electric fields induce apoptosis in a cervical cancer xenograft. Mol. Med. Rep. 11, 1623–1628, https://doi.org/10.3892/mmr.2014.2953 (2015).
Song, S., Tan, J., Miao, Y. & Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 233, 3867–3874, https://doi.org/10.1002/jcp.26137 (2018).
Li, W. et al. Selenoprotein W as a molecular target of d-amino acid oxidase is regulated by d-amino acid in chicken neurons. Metallomics: Integr. biometal Sci. 10, 751–758, https://doi.org/10.1039/c8mt00042e (2018).
Love, M. R., Palee, S., Chattipakorn, S. C. & Chattipakorn, N. Effects of electrical stimulation on cell proliferation and apoptosis. J. Cell. Physiol. 233, 1860–1876, https://doi.org/10.1002/jcp.25975 (2018).
Park, H. J. et al. Melatonin improves the meiotic maturation of porcine oocytes by reducing endoplasmic reticulum stress during in vitro maturation. Journal of pineal research 64, https://doi.org/10.1111/jpi.12458 (2018).
Wang, H. et al. Melatonin alleviates lipopolysaccharide-induced placental cellular stress response in mice. J. pineal Res. 50, 418–426, https://doi.org/10.1111/j.1600-079X.2011.00860.x (2011).
Halvaei, I. et al. Ultrastructure of cytoplasmic fragments in human cleavage stage embryos. J. Assist. Reprod. Genet. 33, 1677–1684, https://doi.org/10.1007/s10815-016-0806-1 (2016).
Minasi, M. G. et al. Correlation between aneuploidy, standard morphology evaluation and morphokinetic development in 1730 biopsied blastocysts: a consecutive case series study. Hum. Reprod. 31, 2245–2254, https://doi.org/10.1093/humrep/dew183 (2016).
Zullo, G. et al. Crocetin improves the quality of in vitro-produced bovine embryos: Implications for blastocyst development, cryotolerance, and apoptosis. Theriogenology 86, 1879–1885, https://doi.org/10.1016/j.theriogenology.2016.06.011 (2016).
Han, J. et al. Deoxynivalenol exposure induces autophagy/apoptosis and epigenetic modification changes during porcine oocyte maturation. Toxicol. Appl. pharmacology 300, 70–76, https://doi.org/10.1016/j.taap.2016.03.006 (2016).
Zhang, L., Li, L., Xu, J., Pan, M. H. & Sun, S. C. HT-2 toxin exposure induces mitochondria dysfunction and DNA damage during mouse early embryo development. Reprod. Toxicol. 85, 104–109, https://doi.org/10.1016/j.reprotox.2019.02.011 (2019).
Bosco, L., Chiarelli, R., Roccheri, M. C., Matranga, D. & Ruvolo, G. Relationship between apoptosis and survival molecules in human cumulus cells as markers of oocyte competence. Zygote 25, 583–591, https://doi.org/10.1017/S0967199417000429 (2017).
Song, B. S. et al. Inactivated Sendai-virus-mediated fusion improves early development of cloned bovine embryos by avoiding endoplasmic-reticulum-stress-associated apoptosis. Reproduction, fertility, Dev. 23, 826–836, https://doi.org/10.1071/RD10194 (2011).
Acknowledgements
The authors wish to thank the research group at the Laboratory Animal Centre, Xi’an Jiaotong University Health Science Centre for their work in this study. Funding was provided by the Natural Science Foundation of Shaanxi Province under grant 2014FWPT07; Wuzhong Innovation and Entrepreneurship Talent Project of Suzhou under grant WC201526; and the National Natural Science Foundation of China under grant No. 81270348.
Author information
Authors and Affiliations
Contributions
E.L. and P.Q. designed the study. P.X., C.S., H.Q., S.L., S.F., Y.D., and F.H. performed the experiment. S.G., Y.X., Y.D., and P.Q. analyzed the data. P.Q. and E.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qu, P., Shen, C., Du, Y. et al. Melatonin Protects Rabbit Somatic Cell Nuclear Transfer (SCNT) Embryos from Electrofusion Damage. Sci Rep 10, 2186 (2020). https://doi.org/10.1038/s41598-020-59161-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59161-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
