
Abstract
The gut microbiota has been shown to play diverse roles in human health and disease although the underlying mechanisms have not yet been fully elucidated. Large cohort studies can provide further understanding into inter-individual differences, with more precise characterization of the pathways by which the gut microbiota influences human physiology and disease processes. Here, we aimed to profile the stool microbiome of children and adults from two population-based cohort studies, comprising 2,111 children in the age-range of 9 to 12 years (the Generation R Study) and 1,427 adult individuals in the range of 46 to 88 years of age (the Rotterdam Study). For the two cohorts, 16S rRNA gene profile datasets derived from the Dutch population were generated. The comparison of the two cohorts showed that children had significantly lower gut microbiome diversity. Furthermore, we observed higher relative abundances of genus Bacteroides in children and higher relative abundances of genus Blautia in adults. Predicted functional metagenome analysis showed an overrepresentation of the glycan degradation pathways, riboflavin (vitamin B2), pyridoxine (vitamin B6) and folate (vitamin B9) biosynthesis pathways in children. In contrast, the gut microbiome of adults showed higher abundances of carbohydrate metabolism pathways, beta-lactam resistance, thiamine (vitamin B1) and pantothenic (vitamin B5) biosynthesis pathways. A predominance of catabolic pathways in children (valine, leucine and isoleucine degradation) as compared to biosynthetic pathways in adults (valine, leucine and isoleucine biosynthesis) suggests a functional microbiome switch to the latter in adult individuals. Overall, we identified compositional and functional differences in gut microbiome between children and adults in a population-based setting. These microbiome profiles can serve as reference for future studies on specific human disease susceptibility in childhood, adulthood and specific diseased populations.
Similar content being viewed by others

Introduction
The human gut microbiome is dynamic, shaped by multiple factors and has been shown to play an important role in human health. Several studies have reported an association between alterations in the composition of the gut microbiome and various gastrointestinal (GI)1,2,3,4,5,6,7,8,9 and non-GI10,11,12,13,14,15 disease conditions in both children and adults. These changes in composition of the microbiome (known as dysbiosis) and their associations with health and disease have led to an increased interest in the complex microbial community of the gut16,17. However, our understanding of the relationship between the gut microbiome and health and disease still remains superficial at best. A fundamental issue hampering progress in this respect is that our lack of knowledge on the composition and variation of the gut microbiome across the human lifespan remains limited. So far, most microbiome studies have been relatively small in terms of sample size and have usually focused on specific diseases and phenotypes, with most publications having used a case-control study design, what makes the analyses more vulnerable to methodological challenges. A better understanding of the variation in microbiome composition (from stool or other sources) and its role in the etiology of chronic diseases can be achieved through the study of large and well-characterized population-based cohorts, which generate rich information on many different physiological parameters, disease status, medication use, dietary intake, and other layers of “omics” data from individual participants.
Up to now, microbiome studies have focused mainly on adult populations, whereas it has been shown that the intestinal microbiome undergoes dynamic changes in diversity and composition during the human lifespan and particularly during development; with the most substantial changes believed to occur throughout childhood18,19,20. Interestingly, emerging data suggest that early alterations in the gut microbiome are associated with an increased risk of developing diseases later in childhood and adulthood e.g., asthma21,22 and Crohn’s disease23. These studies were, however, limited by low sample sizes. If the field of microbiota dynamics is to move forward, it requires studies in well-characterized general population cohorts of different ages, including studies investigating pediatric cohorts. So far, a complete description of diversity, compositional and functional differences between children and adults in a large, homogeneous and population-based cohorts have not been reported.
The above-mentioned considerations prompted us to profile the stool microbiome composition of children and adults within two large, independent and extensively characterized population-based cohorts: the Generation R Study (GenR, visit at 9 years) and the Rotterdam Study (RS, sub-cohort RSIII-2). In this publication, we report our findings on the differences in gut microbiota between 2,111 children aged 9-12 years and 1,427 adults >40 years of age living in the same city with similar urban surroundings.
Methods
Study populations and sample collection
The Generation R Study (GenR) is a population-based prospective multi-ethnic pregnancy cohort study from fetal life until young adulthood conducted in the city of Rotterdam24. The study was designed to identify early environmental and genetic factors and causal pathways underlying normal and abnormal growth during development during childhood. GenR recruited 9,749 children undergoing several rounds of follow-up after birth. Stool sample collection started in 2012 at a mean age of 9.8 years (SD: 0.32). Ethics approval was obtained from the Medical Ethical Committee of Erasmus MC (MEC-2012-165) and written informed consent was obtained from all participants’ parents. All methods were performed in accordance with the Declaration of Helsinki.
The Rotterdam Study (RS) is a prospective population-based cohort study established in 1990 to study determinants of disease and disability in Dutch adult individuals. The original design and updates of this study have been described in detail25. RS consists of four sub-cohorts and comprises approximately 18,000 inhabitants of the Ommoord suburb in Rotterdam (which is predominantly populated by individuals of European ethnicity (about 96%)), aged ≥40 years. The collection of fecal samples started in 2012 among the RS-III sub-cohort comprising 3,932 participants. This study was approved by the Medical Ethical Committee of Erasmus MC (MEC-02-1015) and by the Ministry of Health, Welfare and Sport of the Netherlands. All subjects provided written consent prior to participation in the study. All methods were performed in accordance with the Declaration of Helsinki.
Stool samples were collected at home by the participants using a Commode Specimen Collection System (Covidien, Mansfield, MA). An aliquot of approximately 1 g was transferred to a 25 × 76 mm feces collection tube (Minigrip Nederland, Lelystad, The Netherlands) without preserving agent included and sent through regular mail to the Erasmus MC. A short questionnaire addressing date and time of defecation, current or recent antibiotics use (past year), recent probiotics use (past 3 days), and recent travel activities (past month), was filled out by the participants and included in the package. Upon arrival at Erasmus MC, samples were recorded and stored at −20 °C. The only modification in the collection protocol for the GenR cohort (as compared to RS) was that, in case of delay, the samples were stored by participants at 4 °C (home fridge) before mailing to Erasmus MC. This modification allowed to better preserve samples that were produced in the evening or during the weekend.
DNA isolation
Stool samples from the two cohorts were randomly taken from the −20 °C freezer and allowed to thaw for 10 minutes at room temperature prior to DNA isolation. Samples with inconsistent or lack of information on sample production and samples in which mold growth was observed were excluded (Supp. Fig. 1). An aliquot of approximately 300 mg was homogenized in stool stabilizing buffer according to the manufacturer’s protocol (Arrow Stool DNA; Isogen Life Science, De Meern, The Netherlands). Homogenized samples were bead beated in Lysing Matrix B tubes containing 0.1 mm silica beads (MP Biomedicals, LLC, Bio Connect Life Sciences BV, Huissen, The Netherlands) using the MagNA Lyser instrument (Roche Diagnostics, Almere, The Netherlands) at 7,000 rpm for 45 seconds. Samples were then centrifuged at 6,000 × g for 5 min and 0.5 ml of supernatant was subjected to automated DNA isolation (Arrow; DiaSorin S.P.A., Saluggia, Italy) according to the manufacturer’s protocol using setting ‘Stool DNA 2.0’ in batches of 12 samples per run. DNA concentration was measured using Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, Waltham, MA) and DNA was stored at −20 °C.
16S rRNA gene sequencing
The V3 and V4 variable regions of the 16 S rRNA gene were amplified using the 309F-806R primer pair and dual indexing (12 base pairs (bp) each on the forward and reverse primers) as previously described26. Amplicons were normalized using the SequalPrep Normalization Plate kit (Thermo Fischer Scientific) and pooled. The pools were purified prior to sequencing using Agencourt AMPure XP (Beckman Coulter Life Science, Indianapolis, IN) and the amplicon size and quantity of the pools were assessed on the LabChip GX (PerkinElmer Inc., Groningen, The Netherlands). PhiX Control v3 library (Illumina Inc., San Diego, CA) was spiked into (~10%) the pooled amplicon libraries and each pool was sequenced on an Illumina MiSeq sequencer (MiSeq Reagent Kit v3, 2 × 300 bp) at an average depth of 50,000 read-pairs per sample.
Data pre-processing, OTU picking and quality control
Phylogenetic multi-sample profiling was performed using an in-house developed analysis pipeline (microRapTor) based on QIIME (version 1.9.0)27 and UPARSE (version 8.1)28 software packages. Briefly, index sequences (12 bp) were removed from each read and concatenated to generate a unique index of 24 bp for each read-pair. Spacer and primer sequences were removed using TAGCleaner (version 0.16)29. Paired reads were merged using PEAR (version 0.9.6)30 with the following settings: minimum overlap of 10 bp (default) and an average read quality phred-score of 20 over a 30 bp sliding window. Merged reads shorter than 200 bp were discarded. Reads were de-multiplexed using QIIME including extra quality filtering steps: merged reads were truncated before three consecutive low-quality bases, ambiguous bases were not allowed. Chimeric reads were removed using UCHIME (version 8.1)28. Duplicate samples, samples with less than 10,000 reads, and samples from participants that have used antibiotics (self-reported) in one year prior to sample production were excluded (Supp. Fig. 1). The 16 S sequence reads of the remaining samples (2,214 for GenR and 1,544 for RS) were randomly subsampled at 10,000 reads per sample (after rarefaction analysis). Combined reads of all samples, in each cohort separately, were clustered into operational taxonomic units (OTUs) using UPARSE at a minimum cluster identity of 97%. The representative read from each OTU was then mapped to the SILVA rRNA database version 12831 using RDP Naïve Bayesian Classifier version 2.1232. OTUs containing less than 40 reads were removed as described by Benson et al.33. This threshold was established based on the correlation analysis of OTU tables of 5 pairs of technical replicates, of which DNA was amplified, sequenced and profiled twice (Supp. Fig. 2). The sequence data was then analyzed for α-diversity metrics (Shannon diversity Index, species richness and Inverse Simpson Index). Final OTU filtering was performed by removing OTUs with a total read count less than 0.005% of all reads and OTUs observed in less than 1% of the total number of samples of each cohort as described previously34. The final OTU table was divided into 5 sub-tables at different taxonomic levels (in QIIME environment): phylum, class, order, family, and genus.
Statistical analyses
All statistical analyses were performed in R35 using vegan36, phyloseq37 and MaAsLin38 packages. As MaAsLin performs paralleled multiple analyses, q < 0.05 (false discovery rate (FDR) multiple testing corrected) was used as significance threshold. All MaAsLin models were adjusted for technical covariates and other confounders as described in the sections below.
Technical covariates in the initial stool 16S datasets of the two cohorts
After generating the initial 16S datasets of both the RS and GenR cohorts, the effects of collection time (time in mail; TIM) and the yield of DNA isolation runs (Batch; Supp. Fig. 3) on the 16S profiles were assessed. To identify the impact of these technical covariates on the α-diversity and overall profiles, Shannon α-diversity and Bray-Curtis dissimilarity metrics were calculated in each cohort using ‘diversity’ and ‘vegdist’ functions in vegan, respectively. The effects of the covariates on α-diversity were assessed by linear regression ‘lm’ function in package stats. The basic model was adjusted for age and sex. Each of the two technical covariates were stepwise added in order to identify its contribution to the model. Then, likelihood ratio test was used to compare these nested models. Additionally, in GenR self-reported ethnicity was added to the model to inspect and compare the effect of this covariate. Subsequently, permutation analysis of variance (PERMANOVA) was performed to inspect the global effects of these technical covariates on the overall profiles using the ‘adonis’ function in vegan. Furthermore, for TIM, additive general linear regression analyses of single genus-level OTUs were performed in MaAsLin. For this, we used sub-sampled datasets that included only samples up to a certain TIM (up to 7 days). Regression analyses in MaAsLin were performed after arcsine square root transformation of the relative abundance data and were adjusted for age, sex and BMI to assess the effect of TIM exclusively.
Validation of the final stool 16S datasets of GenR and RS
Final datasets were constructed by excluding samples that had been in the mail for 3 (in RS) or 5 (for GenR) days (see Results section). Average relative abundances of the six major phyla were compared to those of other large cohorts. GenR profiles were compared to those reported by the Copenhagen Prospective Study on Asthma in Childhood (COPSAC) cohort (Copenhagen, Denmark; n = 156; age range = 4–6 years39), and RS profiles were compared to those reported by the Dutch LifeLines-DEEP (LLD) cohort (Groningen, the Netherlands; n = 1,135; age range = 20–90 years40,41) and the Belgian Flemish Gut Flora Project (FGFP) cohort (Leuven, Belgium; n = 1,106; age range = 20–80 years42). In addition, we performed association analyses of BMI with Shannon α-diversity and single OTUs at genus level to confirm this well-established association11,13,40 in our cohorts, and by comparing associated OTUs observed in our cohorts to those reported by the above-mentioned cohorts.
The LifeLines DEEP (LLD) study was approved by the ethics committee of the University Medical Centre Groningen, document number METC UMCG LLD: M12.113965 and all methods were performed in accordance with the Declaration of Helsinki. All participants signed an informed consent form prior to study enrolment.
Flemish Gut Flora Project (FGFP) procedures were approved by the medical ethics committee of the University of Brussels/Brussels University Hospital (approval 143201215505, 5/12/2012) and all methods were performed in accordance with the Declaration of Helsinki. A declaration concerning the FGFP privacy policy was submitted to the Belgian Commission for the Protection of Privacy. Participants were recruited through repeated announcements in print, audiovisual, and social media as well as through the FGFP website, where volunteers could enroll from January 2013 onwards (http://www.vib.be/nl/mens-en-gezondheid/darmflora-project/Pages/default.aspx).
The Copenhagen Prospective Study on Asthma in Childhood (COPSAC) was approved by all relevant authorities including the Danish Ethical Committee (H-B-2008-093), and the Danish Data Protection Agency (2015-41-3696) and all methods were performed in accordance with the Declaration of Helsinki. Both parents provided informed consent prior to participation.
Comparing the gut microbiome profiles and functions between children and adults
To compare gut microbiome profiles between children and adults, we selected samples from European participants. Ethnic background of GenR participants was assessed based on self-reported country of birth of four grandparents as described elsewhere43. Ethnic background of RS participants was assessed based on self-reported ethnic backgrounds of the four grandparents. We selected European participants by combining all European countries together with Americans, Oceanics and North Africans as evaluated elsewhere44. In short, the participant was deemed to be of non-Dutch origin if one parent was born abroad. If both parents were born abroad, the country of birth of the participant’s mother defined the ethnic background. Ethnicities of the parents were derived from the grandparents using the same protocol. The different ethnicities based on the parents country of birth or ethnic background were narrowed to three main ancestry groups: Europeans, including all European countries, together with Americans, Oceanics, and North Africans (with parents from Algeria, Egypt, Libya, Morocco, Sudan, Tunisia, and Western Sahara); Africans, including Sub‐Saharan Africans, Dutch Antilleans, and Surinamese Creoles; and Asians, including all Asian countries and Surinamese Hindustanis. Reads were subsampled at 10,000 reads per sample and pooled. Classification was performed (as described above) in one combined run. Shannon α-diversities were calculated and tested for significant differences between the two cohorts using Wilcoxon signed-rank test (1000 permutations). Between-sample Bray-Curtis dissimilarities were calculated and principal coordinate analysis (PCoA) was performed and tested for significant differences between the two cohorts using PERMANOVA. During the beta diversity analysis we adjusted for DNA isolation batch and time in mail since these were the technical covariates. Linear regression models intending to determine significantly different genera between both cohorts were performed in MaAsLin. During analysis we adjusted for BMI, sex, technical covariates (TIM and Batch), and multiple testing by FDR (q < 0.05).
To compare the functional metagenome of gut bacteria between children and adults, we used the PICRUSt (v.1.1.0) tool45 to obtain predicted bacterial functions. HUMAnN2 (v0.99) was used to identify the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways46. For the identification of the specific pathway biomarkers distinguishing the gut microbiome of the children from those of adults, we performed a linear discriminant effect size (LEfSe) analysis47 with the default settings: α (ANOVA) = 0.05 and logarithmic LDA (linear discriminant analysis) score = 2.0.
Results
Stool microbiota 16S rRNA data generation in the GenR and RS cohorts
Selection of subjects
For GenR 4,959, and for RS 2,440 participants were invited to provide a stool sample. In total, 2,921 (response rate = 59%) and 1,691 (response rate = 69%) were received at Erasmus MC for GenR and RS stool samples, respectively (Supp. Fig. 1). Excluding antibiotic users resulted in exclusion of 196 samples from GenR and 7 samples from RS. In GenR, individuals who used antibiotics in the last year had a significantly lower microbiome diversity and altered microbiome composition (Supp. Table 1). For probiotic use and recent travelling activity outside the Netherlands we could not detect significant effects on diversity or composition in the two cohorts (Supp. Table 1). After quality control (Supp. Fig. 1), 16S rRNA data of 2,214 subjects in the initial dataset of GenR and 1,544 subjects in the initial dataset of RS were included.
Assessing the influence of technical co-variates and sample exclusion
Since stool samples were collected at ambient temperature via regular mail, we assessed the effects of time in the mail (TIM) on the microbiome profiles. Furthermore, since DNA-yields varied across different DNA isolation batches (Supp. Fig. 3), we assessed its effect on the microbiome profiles as well.
Upon longer periods of TIM (except for day 7 in GenR), an increase in the relative abundance of phylum Proteobacteria was observed in the average profiles of both cohorts (Supp. Fig. 4). For RS only, an increase in phylum Bacteroidetes was observed between days 6 and 7. In contrast, we did not observe any substantial changes in phylum-level profiles with respect to the DNA-Batch variable (Supp. Fig. 4). Moreover, TIM had a small but significant negative effect on α-diversity in GenR and RS (beta = −0.02 alpha units/day, p = 9.3e-03 and beta = −0.03, p = 4.6e-03, respectively). Again, we did not observe an effect of Batch on α-diversity. Correlations of TIM and Batch with overall composition (β-diversity) in both cohorts were small but significant (R2 = 0.004, p = 0.001 and R2 = 0.002, p = 0.005 for TIM and R2 = 0.01, p = 0.001 and R2 = 0.005, p = 0.001 for Batch in GenR and RS, respectively). In taxonomy-based analyses (MaAsLin) at genus-level of both datasets for TIM, we observed increased abundances of genus Escherichia/Shigella upon prolonged times in the mail (Fig. 1). However, these differences were only significant after 3 days in the RS cohort and after 5 days in the GenR cohort (Fig. 1). Furthermore, we observed smaller decreases in the abundances of a number of genera, including Roseburia and Coprococcus, upon prolonged TIM.

Effect of ambient temperature on individual OTUs. Regression analysis of individual OTUs with time in mail (TIM) for samples in GenR (A) and RS (B). At each TIM. the initial OTU table was sub-sampled to contain only samples up to that TIM. Red bars indicate bacteria that decreased in abundance and green bars bacteria that increased upon increasing TIM. Q-values are indicated; only significantly abundant OTUs are presented.
In order to further evaluate the importance of TIM and Batch effects, we added them to linear models for the analysis of well-established association of α-diversity with BMI11,13,40 in both GenR and RS. Although, the estimates (betas) of the exposure (i.e., BMI) remained similar after various levels of adjustment, the model including sex, age and TIM (model 1) as co-variates provided a better fit to the data than the model including only sex and age (model 0; likelihood ratio test: p = 5.8e-03 and p = 5.5e-03 for GenR and RS, respectively). Including Batch, within model 1 (model 2), resulted in a better fit to the data in RS only (Table 1; p = 1.9e-04).
Based on the results of the effects of the technical covariates on the profiles in the RS and GenR cohorts, only stool samples that arrived at the research center within 3 days for RS and within 5 days for GenR, were included in the final 16 S datasets (Fig. 1). Furthermore, TIM and Batch were included as technical covariates in all further analyses.
Description and validation of final 16S rRNA datasets of the GenR and RS cohorts
After quality control and sample exclusion, the final 16 S datasets comprised 2,111 individuals in GenR and 1,427 subjects in RS. The average age of GenR children was 10 (range = 9 to 12) years and included participants from 13 self-reported ethnicities with Dutch being the most frequent one (62%; Table 2 and Supp. Fig. 5). The average age of RS participants in the final 16S dataset was 57 years (range = 46 to 88) and 82% were Dutch based on self-reported ethnicity (Table 2 and Supp. Fig. 5).
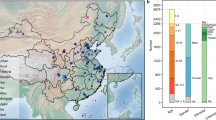
A total number of 661 OTUs were identified in the GenR cohort and 777 OTUs in the RS cohort (Fig. 2). Of these, 656 OTUs overlapped between the two cohorts, leaving 5 OTUs specific for GenR and 42 OTUs specific for RS. For both datasets, variations in overall microbiome profiles were driven by the relative abundances of the four major phyla Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria (Supp. Fig. 6). We observed higher Shannon α-diversities in RS (mean 4.02; SD = 0.50) than in the GenR cohort (mean 3.81; SD = 0.57; Fig. 2). Furthermore, we observed the absence of kingdom Archaea and a lower abundance of phylum Firmicutes in the GenR cohort as compared to RS (Fig. 2).

Characteristics of the final datasets of the two cohorts. GenR (A) and RS (B). Number of observed taxa at each taxonomy level Top: indicates the number of unique OTUs identified in each taxonomic clade, top A: RS cohort, top B: GenR cohort. Bottom: Donut plots indicate the average relative abundances of the top major phyla in each cohort. Donut plots of the COPSAC cohort (children aged 6 years) and doughnut plots of FGFP and LLD cohorts (adults) are plotted for comparison with the abundance in GenR and RS.
To validate our datasets, we compared the averaged phylum level profiles of our cohorts with those of other cohorts of similar characteristics (i.e., age range, ethnic background). In addition, we tested the well-established association between BMI and alpha diversity11,13,40, and we determined the association between single OTUs and BMI and compared these results with other cohorts.
To compare GenR and RS 16S datasets with other populations, we used the 16S datasets of the COPSAC39, LLD40,41 and FGFP42 cohorts. The average phylum-level profiles of GenR were similar to those of COPSAC, and the average phylum-level profiles of RS were similar to those of LLD and FGFP (Fig. 2). The lower abundance of phylum Firmicutes in the GenR cohort was also observed in the COPSAC cohort39.
Further, in order to validate our microbiome datasets, we investigated the association of gut microbiome with BMI. We excluded subjects of non-Northern European origin from both GenR and RS, which resulted in a dataset of 1,712 GenR samples and a dataset of 1,371 RS samples.
α-diversity was negatively associated with BMI after adjusting for age, sex and technical covariates in both cohorts (beta = −0.019, SE = 2.3e-03, p = 1.1e-03 for GenR and beta = −0.015, SE = 2.8e-03, p = 1.8e-06 for RS). We compared this association with observations reported in similar cohorts of Northern European origin (LLD and FGFP). The negative correlation of α-diversity with BMI was in line with findings reported previously40. At genus-level, we identified 27 BMI-associated genera in GenR and 33 BMI-associated genera in RS (Table 3, Supp. Table 2), of which 14 genera overlapped across both cohorts. In the RS cohort, we confirmed 6 genera found to be associated with BMI in the LLD and FGFP cohorts (Table 3). In GenR we only confirmed two genera (Alistipes and Barnesiella) to be associated with BMI in RS, LLD and FGFP cohorts. As previously reported40,48 we also observed the association between increased abundance of genus Akkermansia and lower BMI.
Comparison of RS and GenR stool microbiome diversities, compositions and functions
To analyze differences in gut microbiome compositions between children and adults, both datasets were combined after exclusion of the non-Northern European samples. The combined dataset included 3,082 samples: 1,371 from RS and 1,712 from GenR and the final OTU table contained 173 genera. Although Shannon α-diversity was not associated with age in each cohort separately (linear model; GenR: beta = −0.003, p = 0.91; RS: beta = 0.001, p = 0.63), Shannon α-diversity was significantly higher in the RS cohort than in the GenR cohort in the combined dataset (p < 2.2e-16, mean = 5.9, sd = 0.74 for RS and mean = 5.58, sd = 0.82 for GenR). Also, the overall compositions (Bray Curtis distances) differed significantly between both groups (Fig. 3B; PERMANOVA; R2 = 0.06; p = 0.001). We observed a 2 to 3 times lower abundance of phylum Firmicutes in the GenR cohort as compared with RS (p < 2.2e-16). Furthermore, we observed higher abundances of the gram-negative classes (Bacteroidia, Negativicutes and some classes from phylum Proteobacteria) in children. Regression analysis, in MaAsLin, on the relative abundances of the individual genera showed higher relative abundances of 59 genera in RS compared to GenR and higher relative abundances of 20 genera in GenR compared to the RS cohort (q < 0.05; Supp. Table 3). The largest differences were observed for genera from family Lachnospiraceae including Blautia, Lachnospiraceae, Anaerostipes, Dorea, Fusicatenibacter, Coprococcus, Roseburia, Ruminoclosteridium, Butyricicoccus and Lachnoclostridium that were more abundant in RS, and genera from families Ruminococcaceae and Bacteroidaceae including Bateroides, Faecalibacterium, Alistipes, Barnesiella, Parabacteroides, Bifidobacterium and Odoribacter that were more abundant in GenR (Fig. 3C). The most abundant genera were Bacteroides in GenR (children) cohort and Blautia in the RS (adults) cohort (Fig. 3D).

Comparison of the gut microbiome diversity and composition between adults (RS) and children (GenR). (A) boxplots of the Shannon diversity Index. (B) ordination plot of the gut microbiome composition in the two cohorts based on Bray-Curtis dissimilarities. The centroid and dispersion of each cohort is represented by the cohort name and ellipses, respectively. Clustering of RS and GenR was tested for significance using PERMANOVA. (C) Circular representation of the taxonomic tree of the microbiome compositions of the two cohorts. Each node represents one taxon at different taxonomic level. Orange nodes are the taxa that were observed with higher abundance in the GenR cohort and green nodes represent the taxa that were higher abundant in the RS cohort. (D) The genera represented the most in each cohort. On the x-axis the arcsine squared root transformed coefficients of the most significantly abundant genera in each cohort are shown. Orange bars represent GenR and green bars represent RS. Minus signs in the x-axis are used only for visualization.
To assess functional differences in the gut microbiome between children and adults, we predicted the functional content based on the 16S rRNA data using PICRUSt. Analyses of the differences in the predicted functional metagenomics data between children and adults showed significant overrepresentation of 25 pathways in children and 25 pathways in adults (Fig. 4, Supp. Table 4). A remarkable difference between GenR and RS was the predominance of catabolic pathways in GenR (Val, Leucine, iso-Leucine degradation; ko00280) and its opposite in RS (Val, Leucine, iso-Leucine biosynthesis; ko00290). Putative colonization-related pathways like biofilm formation (ko05111), flagellar assembly (ko02040) and LPS biosynthesis (ko00540) were only enriched in GenR cohort.

Predicted functional composition of metagenomes based on 16S rRNA gene sequencing data from GenR and RS cohorts. LEfSe based on the PICRUSt dataset revealed differentially enriched metabolic pathways associated with GenR (orange) or RS (green).
Discussion
In this publication we report on the 16S stool microbiota profiles of 3,538 subjects from two large, deeply phenotyped and well-characterized population-based cohorts: the Generation R (GenR) Study and the Rotterdam Study (RS). The 16S microbiome datasets originated from both children (GenR: n = 2,111) and adults (RS: n = 1,427) populations.
In general, the stool microbiota within RS had similar profiles as profiles in the previously published LLD and FGFP cohorts40,41,42. Similarly, our GenR stool microbiota possessed similar profiles as those observed in the COPSAC cohort39.
Further, both of our cohorts replicated the previously reported negative association between α-diversity and BMI40. Additionally, BMI was also associated with community composition (Bray-Curtis) in both RS and GenR cohorts, as reported for the LLD and FGFP cohorts. Also, Alistipes and Barnesiella were reported as negatively associated with BMI in all four of these cohorts. Interestingly, the lower Firmicutes abundance observed in the GenR and COPSAC children cohorts compared to RS, LLD and FGFP adult cohorts suggests an age-specific phenomenon is responsible for this difference. However, understanding these differences merit further investigation. Thus, as both our RS and GenR datasets contain many characteristics that are similar to previously published large cohort studies, we conclude that our datasets allow valid investigations into the composition and variation of the gut microbiota across child and adult subjects.
Comparing the gut microbiome composition between children and adults in a combined dataset of 3,083 children and adults showed significant clustering of the cohorts based on overall composition (PCoA beta diversity) and significant different α-diversities. The lower diversity in GenR suggested a lack of ‘maturation’ of the gut microbiome in children. However, we did not observe a significant age-related change in alpha diversity within each cohort separately. This might be due to the narrow age-range in both cohorts (9.8 ± 0.32 in GenR, 56.8 ± 5.9 in RS, Table 2). At genus level, the relative abundances of genus Bacteroides were higher in GenR than in RS. Members of the genus Bacteroides are specialized at utilizing both plant and host-derived polysaccharides49,50,51,52. As compared to other genera, Bacteroides have a large number of genes specialized to metabolize various glycans. They also have environmental sensors that control their expression upon exposure to glycans. The predicted metagenomics data showed indeed higher abundances of glycan degradation pathways in children’s gut microbiome than in adults. In contrast, the relative abundances of the genus Blautia were higher in RS than in GenR. Blautia digest complex carbohydrates like whole-grains53 and its abundance has been shown to be reduced in patients with colorectal cancer54 and in children with type 1 diabetes55. Its abundance has also been reported to be inversely related to bone mineral density in human56. In fact, the relative abundance of Blautia and Bacteroides, has been implicated as a determinant of the so-called ‘healthy microbiome’ due to differences in the metabolic functions of these genera54,55,57,58.
With respect to metabolic functions, we identified many predicted carbohydrate pathways that were significantly enriched in adult versus child metagenome datasets, with many of these carbohydrate pathways involving the metabolism of simple sugars such as fructose and mannose. However, whether these differences in microbiota profiles can be explained by possible differences in energy demand and metabolic programming between adults and children remains to be proven.
Other interesting differences in the predicted functional pathways between children and adults, were observed in antibiotics synthesis pathways. The vancomycin biosynthesis pathways were significantly higher in the gut microbiome of children as compared to adults. Vancomycin is a glycopeptide with activity against gram-positive bacteria, while gram-negative bacteria are resistant to this drug because of their distinct cell-wall59,60. On the other hand, biosynthesis of ansamycin was higher in adults than in children. Ansamycin is another antibiotic with activity against gram-positive, as well as gram-negative bacteria61. These findings indicate potential differential anti-bacterial activities between the microbiota of children as compared to adults, which appears to be in line with our observation of higher relative abundances of gram-negative bacteria in the gut microbiota of children. However, the exact role of antibiotic production in modulating the microbiota profiles of children and adults remains to be shown.
Another difference identified by predictive functional pathway analysis was the differential biosynthesis of vitamin B classes by the gut microbiota of children and adults. Children’s gut microbiomes are associated with pathways related to increased biosynthesis of vitamins B2, B6, and B9 (folate), while in adults vitamin B1 and B5 biosynthesis pathways appear to be increased. This predictive finding could be linked to age-dependent differences in, for example, the requirement for folate, which is particularly important for cell division and growth during pregnancy and childhood.
In children, the enrichment of putative colonization-related pathways involving biofilm formation, flagella assembly and LPS biosynthesis, may provide insights into the development of intra-microbiome and host-microbiome interactions, and also be linked to our observation of higher relative abundances of gram-negative bacteria in children - LPS is a major constituent of the cell wall of gram-negative bacteria.
Finally, observing photosynthesis pathway (ko00195) in the functional data could be explained by the presence of Cyanobacteria in our datasets, as Cyanobacteria are photosynthetic bacteria. Likely, the photosynthesis pathways detected in the PICRUSt analysis were derived from these Cyanobacteria. The presence of Cyanobacteria in the gut may have a dietary origin62.
As with all studies, the current study was not free of limitations. One of the main discussion points involves the fact that stool samples were not directly frozen and immediately stored at −80 °C. For logistical reasons relating to the large scale cohorts recruited, we depended on postal delivery of home-collected stool samples to the research laboratory. Several studies have addressed the effects on microbiota composition when sample collection is performed at room temperature63,64,65,66,67,68, and observed that longer periods of storage at ambient temperature may affect microbiota profile composition and decrease diversity in the samples. We, therefore, analyzed the effects of stool sample collection at ambient temperature in both cohorts. Within our own initial datasets, we addressed this issue by using several approaches and observed increased abundances of Escherichia/Shigella when stool samples were mailed to the research laboratory and received after 3 days (RS) or after 5 days (GenR) in the post. Therefore, to avoid the influence of any delayed processing time, we excluded samples that had been in the mail for longer than 3 days in RS and 5 days in GenR cohorts.
Next to the increase in abundance of Escherichia/Shigella upon prolonged times in the mail, we observed several taxa that decreased over time (Coprococcus and Roseburia). These decreases were, however, relatively low and likely a consequence of the compositionality of the data: if one OTU increases, other OTUs decrease. We decided to adjust for these technical artifacts in further analyses by including time in mail as a technical cofactor.
The actual explanation for the difference in microbiota composition stability over time between RS and GenR cohorts is likely due to the fact that GenR participants were asked to keep their samples in their home fridge at 4 °C (for posting on Monday) if they were produced at the weekend, allowing better preservation of samples compared to RS participants who mailed their samples over the weekend. As well as the exclusion of 3 day and 5 day samples, we also included TIM as a technical covariate in all analyses.
Next to excluding samples that had been in the mail for too long, we also excluded recent antibiotic users from our datasets (196 samples from GenR and 7 samples from RS). Antibiotic use had a significant effect on alpha diversity and overall composition in GenR (Supplementary Table 1), whereas in RS the number of users was too small to have a detectable effect. Probiotic use and travel abroad were also recorded, but we could not detect significant effects on alpha diversity and composition in both cohorts. Given the small effect size of probiotic use, this study might be underpowered to conclude a lack of effect.
Recent studies indicate that the method of DNA isolation is the main source of technical variance in microbiome studies69,70,71. We, therefore, analyzed the effect of DNA isolation throughout the 391 runs (134 runs for RS and 257 runs for GenR) that were performed in this study and observed a batch effect causing a reduction in average DNA yield per sample in a proportion of the hundreds of runs performed. As we could not trace any clear cause for this technical artifact we introduced a “Batch” variable that allowed us to discriminate between low yield and the high yield runs. This “Batch” variable was significantly associated with overall profiles in the GenR and RS cohorts and was included as technical covariate in all analyses. Another, more general limitation when comparing different cohort datasets, is the accuracy of replication of 16S profiles, since sample collection, sample storage, DNA isolation, PCR amplification, amplified 16S rRNA variable region, the sequencing technique used and downstream bioinformatics may differ between different cohorts. For example, out of 33 associated OTUs with BMI, we could only replicate 6 in LLD and FGFP. In addition, geographical differences and lack of repetition may limit the accuracy of replicating microbiota profiles from different countries. In addition, although (besides using harmonized sample collection and dataset generation) we adjusted for the potential confounders in the analyses comparing children and adults, we should be aware that we cannot totally discard the presence of residual stratification as a consequence of the fact that both populations were derived from different cohorts.
To conclude, in this study we performed microbiota profiling on the stools of 2,111 children in the age-range of 9 to 12 years and 1,427 adult individuals in the range of 46 to 88 years of age. We observed a clear distinction between the gut microbiomes of children as compared to adults, including differences in microbiota diversity and Firmicutes and Bacteroidetes abundances. These changes were associated with predicted shifts in functional properties, including in energy metabolism, antibiotic production and the production of essential B-vitamins. These observations are likely due to the development of human-gut microbiome interactions with age. As both GenR and the RS cohort have been deeply characterized24,25 we here presented two valuable datasets for studying the possible association of the human stool microbiome and, life style, environmental factors, and health and disease outcomes. In addition, as the participants of both cohorts have been genotypes, also association between host genetics and host microbiome could be investigated.
URLs. The Rotterdam Study: http://www.ergo-onderzoek.nl/wp/; Generation R study: https://www.generationr.nl/researchers/; QIIME: http://qiime.org/; USEARCH: https://www.drive5.com/usearch/; PEAR: https://sco.h-its.org/exelixis/web/software/pear/; TAGcleaner: http://tagcleaner.sourceforge.net/; Vegan R package: https://cran.r-project.org/web/packages/vegan/index.html; phyloseq: https://joey711.github.io/phyloseq/; PICRUSt: http://picrust.github.io/picrust/; HUMAnN2: http://huttenhower.sph.harvard.edu/humann2; LEfSe: https://bitbucket.org/biobakery/biobakery/wiki/lefse.
Data availability
All relevant summary data supporting this study are available within the article and its supplementary information files, or additional unpublished data can be provided by the corresponding author upon reasonable request. Due to ethical and legal restrictions, individual-level data of the Generation R Study and Rotterdam Study cannot be made publicly available. These data are available upon request to the data manager of the Rotterdam Study Frank van Rooij (f.vanrooij@erasmusmc.nl) or of the Generation R Study Claudia Kruithof (c.kruithof@erasmusmc.nl) and subject to local rules and regulations. This includes submitting a proposal to the management team of RS, where upon approval, analysis needs to be done on a local server with protected access, complying with GDPR regulations.
References
Carroll, I. M., Chang, Y.-H., Park, J., Sartor, R. B. & Ringel, Y. Luminal and mucosal-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Gut Pathog. 2, 19 (2010).
Carroll, I. M. et al. Molecular analysis of the luminal- and mucosal-associated intestinal microbiota in diarrhea-predominant irritable bowel syndrome. Am. J. Physiol. Gastrointest. Liver. Physiol. 301, G799–G807 (2011).
Frank, D. N. et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm. Bowel Dis. 17, 179–184 (2011).
Frank, D. N. et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 104, 13780–13785 (2007).
Kassinen, A. et al. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology 133, 24–33 (2007).
Mai, V. et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PloS One 6, e20647–e20647 (2011).
Malinen, E. et al. Analysis of the fecal microbiota of irritable bowel syndrome patients and healthy controls with real-time PCR. Am. J. Gastroenterol. 100, 373 (2005).
Ringel, Y. & Carroll, I. M. Alterations in the intestinal microbiota and functional bowel symptoms. Gastrointest. Endosc. Clin. N. Am. 19, 141–150 (2009).
Swidsinski, A., Loening-Baucke, V., Verstraelen, H., Osowska, S. & Doerffel, Y. Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology 135, 568–579 (2008).
Kalliomäki, M., Carmen Collado, M., Salminen, S. & Isolauri, E. Early differences in fecal microbiota composition in children may predict overweight. Am. J. Clin. Nutr. 87, 534–538 (2008).
Ley, R. E., Turnbaugh, P. J., Klein, S. & Gordon, J. I. Human gut microbes associated with obesity. Nature 444, 1022–1023 (2006).
Penders, J. et al. Gut microbiota composition and development of atopic manifestations in infancy: the KOALA Birth Cohort Study. Gut 56, 661–667 (2007).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Vael, C. & Desager, K. The importance of the development of the intestinal microbiota in infancy. Curr. Opin. Pediatr. 21, 794–800 (2009).
Wang, M. et al. Reduced diversity in the early fecal microbiota of infants with atopic eczema. J. Allergy Clin. Immunol. 121, 129–134 (2008).
Floch, M. H. Advances in Intestinal Microecology. Nutr. Clin. Pract. 27, 193–194 (2012).
Ringel-Kulka, T. Targeting the intestinal microbiota in the pediatric population. Nutr. Clin. Pract. 27, 226–234 (2012).
Palmer, C., Bik, E. M., DiGiulio, D. B., Relman, D. A. & Brown, P. O. Development of the human infant intestinal microbiota. PLoS Biol. 5, e177–e177 (2007).
Tiihonen, K., Ouwehand, A. C. & Rautonen, N. Human intestinal microbiota and healthy ageing. Ageing Res. Rev. 9, 107–116 (2010).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012).
Kozyrskyj, A. L., Ernst, P. & Becker, A. B. Increased risk of childhood asthma from antibiotic use in early life. Chest 131, 1753–1759 (2007).
Risnes, K. R., Belanger, K., Murk, W. & Bracken, M. B. Antibiotic exposure by 6 months and asthma and allergy at 6 years: findings in a cohort of 1,401 US children. Am. J. Epidemiol. 173, 310–318 (2011).
Hviid, A., Svanström, H. & Frisch, M. Antibiotic use and inflammatory bowel diseases in childhood. Gut 60, 49–54 (2011).
Kooijman, M. N. et al. The Generation R Study: design and cohort update 2017. Eur. J. Epidemiol. 31, 1243–1264 (2016).
Ikram, M. A. et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur. J. Epidemiol. 32, 807–850 (2017).
Fadrosh, D. W. et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2, 6 (2014).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Schmieder, R., Lim, Y. W., Rohwer, F. & Edwards, R. TagCleaner: Identification and removal of tag sequences from genomic and metagenomic datasets. BMC bioinformatics 11, 341–341 (2010).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620 (2014).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Benson, A. K. et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 107, 18933–18938 (2010).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59 (2013).
R Foundation for Statistical Computing, Vienna, Austria. A language and environment for statistical computing, https://www.R-project.org (2010).
Oksanen, J. et al. Vegan: community ecology package, http://CRAN.R-project.org/package=vegan (2013).
McMurdie, P. J. & Holmes, S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217–e61217 (2013).
Morgan, X. C. et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13, R79–R79 (2012).
Stokholm, J. et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat. Commun. 9, 141 (2018).
Fu, J. et al. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ. Res. 117, 817–824 (2015).
Zhernakova, A. et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016).
Falony, G. et al. Population-level analysis of gut microbiome variation. Science 352, 560–564 (2016).
Medina-Gomez, C. et al. Bone mass and strength in school-age children exhibit sexual dimorphism related to differences in lean mass: The Generation R Study. J. Bone Miner. Res. 31, 1099–1106 (2016).
Medina-Gómez, C. et al. BMD loci contribute to ethnic and developmental differences in skeletal fragility across populations: assessment of evolutionary selection pressures. Mol. Biol. Evol. 32, 2961–2972 (2015).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013).
Abubucker, S. et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 8, e1002358–e1002358 (2012).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
Everard, A. et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 110, 9066–9071 (2013).
Anderson, K. L. & Salyers, A. A. Biochemical evidence that starch breakdown by Bacteroides thetaiotaomicron involves outer membrane starch-binding sites and periplasmic starch-degrading enzymes. J. Bacteriol. 171, 3192–3198 (1989).
Chung, W. S. F. et al. Modulation of the human gut microbiota by dietary fibres occurs at the species level. BMC Biol. 14, 3 (2016).
Koropatkin, N. M., Cameron, E. A. & Martens, E. C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 10, 323–335 (2012).
Xu, J. et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 5, e156–e156 (2007).
Martínez, I. et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. 7, 269–280 (2013).
Chen, W., Liu, F., Ling, Z., Tong, X. & Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PloS One 7, e39743–e39743 (2012).
Murri, M. et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 11, 46 (2013).
Hou, A. Y., Kaczmarek, J. L., Khan, N. A. & Holscher, H. D. Dietary fiber and the human gastrointestinal microbiota as predictors of bone health. FASEB J. 31, lb322 (2017).
Bajaj, J. S. et al. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G675–G685 (2012).
Hong, P.-Y., Croix, J. A., Greenberg, E., Gaskins, H. R. & Mackie, R. I. Pyrosequencing-based analysis of the mucosal microbiota in healthy individuals reveals ubiquitous bacterial groups and micro-heterogeneity. PloS One 6, e25042–e25042 (2011).
Nikaido, H. Outer membrane barrier as a mechanism of antimicrobial resistance. Antimicrob. Agents Chemother. 33, 1831–1836 (1989).
Shivaramaiah, H. S., Relhan, N., Pathengay, A., Mohan, N. & Flynn, H. W. Endophthalmitis caused by gram-positive bacteria resistant to vancomycin: clinical settings, causative organisms, antimicrobial susceptibilities, and treatment outcomes. Am. J. Ophthalmol. Case Rep. 10, 211–214 (2018).
Wrona, I. E., Agouridas, V. & Panek, J. S. Design and synthesis of ansamycin antibiotics. C. R. Chim. 11, 1483–1522 (2008).
Huttenhower, C. et al. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Carroll, I. M., Ringel-Kulka, T., Siddle, J. P., Klaenhammer, T. R. & Ringel, Y. Characterization of the fecal microbiota using high-throughput sequencing reveals a stable microbial community during storage. PloS One 7, e46953–e46953 (2012).
Dominianni, C., Wu, J., Hayes, R. B. & Ahn, J. Comparison of methods for fecal microbiome biospecimen collection. BMC Microbiol. 14, 103 (2014).
Gorzelak, M. A. et al. Methods for improving human gut microbiome data by reducing variability through sample processing and storage of stool. PloS One 10, e0134802–e0134802 (2015).
Lauber, C. L., Zhou, N., Gordon, J. I., Knight, R. & Fierer, N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol. Lett. 307, 80–86 (2010).
Sinha, R. et al. Collecting fecal samples for microbiome analyses in epidemiology studies. Cancer Epidemiol. Biomarkers Prev. 25, 407–416 (2016).
Tedjo, D. I. et al. The effect of sampling and storage on the fecal microbiota composition in healthy and diseased subjects. PloS One 10, e0126685–e0126685 (2015).
Costea, P. I. et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 35, 1069–1076 (2017).
Santiago, A. et al. Processing faecal samples: a step forward for standards in microbial community analysis. BMC Microbiol. 14, 112 (2014).
Sinha, R. et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat. Biotechnol. 35, 1077–1086 (2017).
Acknowledgements
The generation and management of stool microbiome data for the Generation R Study (Focus @9) and the Rotterdam Study (RSIII-2) was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Rotterdam, The Netherlands. We thank Nahid El Faquir and Jolande Verkroost-Van Heemst for their help in sample collection and registration, and Kamal Arabe, Hedayat Razawy and Karan Singh Asra for their help in DNA isolation and sequencing. Furthermore, we thank drs. Jeroen Raes and Jun Wang (KU Leuven, Belgium) for their guidance in 16S rRNA profiling and dataset generation. Djawad Radjabzadeh was funded by an Erasmus MC mRACE grant “Profiling of the human gut microbiome”. The authors are very grateful to the study participants of the Rotterdam Study, the staff from the Rotterdam Study (particularly L. Buist and J.H. van den Boogert) and the participating general practitioners and pharmacists. The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMW), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. The GWAS datasets are supported by the Netherlands Organization of Scientific Research NWO Investments (nr. 175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project nr. 050-060-810. FR and CMG received funding from ZonMW-VIDI, project nr. 016.136.367. The Generation R Study is conducted by the Erasmus Medical Center in close collaboration with the School of Law and Faculty of Social Sciences of the Erasmus University Rotterdam, the Municipal Health Service Rotterdam area, Rotterdam, the Rotterdam Homecare Foundation, Rotterdam and the Stichting Trombosedienst & Artsenlaboratorium Rijnmond (STAR-MDC), Rotterdam. We acknowledge the contribution of children and parents, general practitioners, hospitals, midwives and pharmacies in Rotterdam. The general design of Generation R Study was made possible by financial support from the Erasmus Medical Center, Rotterdam, the Erasmus University Rotterdam, the Netherlands Organization for Health Research and Development (ZonMW), the Netherlands Organization for Scientific Research (NWO), the Ministry of Health, Welfare and Sport and the Ministry of Youth and Families. This project also received funding from the European Union’s Horizon 2020 research and innovation programme under the following grant agreements: [No. 633595 (DynaHEALTH) and No. 733206 (LIFECYCLE)]. Furthermore, Generation R received additional funding from the European Research Council (ERC Consolidator Grant, ERC-2014-CoG-648916).
Author information
Authors and Affiliations
Contributions
R.K., A.G.U., H.A.M. conceptualized the study. R.K., D.R., P.V.D.W., S.A.B., J.C.K.D.J., M.A.E.J., M.P.P., H.A.M., V.W.V.J. and M.A.I. were responsible for project administration and sample collection. P.V.D.W. and D.R. were responsible for wet laboratory work and documentation of the samples. R.K., A.G.U., M.P.P. and H.A.M. were responsible for funding acquisition. D.R., R.K., A.G.U., S.R.K., J.P.H. and P.V.D.W. designed the methodology. D.R. was responsible for software, data generation, carried out the formal analyses, wrote the original draft of the manuscript and visualized the results. R.K., A.G.U., H.A.M., and S.R.K. supervised the project. C.G.B. and C.M.G. contributed to the data generation and analyses. R.K., C.G.B., C.M.G., S.A.B., J.C.K.D.J., S.R.K., M.P.P., J.P.H., F.R., J.B.J.V.M., A.G.U. reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Radjabzadeh, D., Boer, C.G., Beth, S.A. et al. Diversity, compositional and functional differences between gut microbiota of children and adults. Sci Rep 10, 1040 (2020). https://doi.org/10.1038/s41598-020-57734-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57734-z
This article is cited by
-
Navigating the microbial maze: unraveling the connection between gut microbiome and pediatric kidney and urinary tract disease
Pediatric Nephrology (2024)
-
Gut microbiome studies in CKD: opportunities, pitfalls and therapeutic potential
Nature Reviews Nephrology (2023)
-
Migration Spurs Changes in the Human Microbiome: a Review
Journal of Racial and Ethnic Health Disparities (2023)
-
Linking gastrointestinal microbiota and metabolome dynamics to clinical outcomes in paediatric haematopoietic stem cell transplantation
Microbiome (2022)
-
ABO genotype alters the gut microbiota by regulating GalNAc levels in pigs
Nature (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
