
Abstract
Present work aims to utilize systems biology and molecular modelling approach to understand the inhibition kinetics of Leishmania major GLO I and identifying potential hit followed by their validation through in vitro and animal studies. Simulation of GLO I inhibition has shown to affect reaction fluxes of almost all reactions in the model that led to increased production of various AGEs and free radicals. Further, in vitro testing of C1 and C2, selected through molecular modelling revealed remarkable morphological alterations like size reduction, membrane blebbing and loss in motility of the parasite, however, only C1 showed better antileishmanial activity. Additionally, C1 showed apoptosis mediated leishmanicidal activity (apoptosis-like cell death) along with cell-cycle arrest at sub-G0/G1 phase and exhibited potent anti-leishmanial effect against intracellular amastigotes. Furthermore, decrease in parasite load was also observed in C1 treated BALB/c female mice. Our results indicate that C1 has healing effect in infected mice and effectively reduced the parasitic burden. Hence, we suggest C1 as a lead molecule which on further modification, may be used to develop novel therapeutics against Leishmaniasis.
Similar content being viewed by others

Introduction
Pathogenic parasites trypanosomatids belong to the class Kinetoplastida and have been extensively studied for a long time due to their association with many neglected tropical diseases1,2 in humans as well as in animals and plants. They are predominantly possessing a monoxenous life-cycle (restricted to only invertebrates), yet, several of them have dixenous life-cycle in which their transmission to vertebrates is facilitated by a sandfly vector. Among these parasites, many species of Leishmania causes different clinical forms of leishmaniasis known as cutaneous leishmaniasis (CL), mucocutaneous leishmaniasis (MCL), and the most life threatening one, visceral leishmania (VL). CL is the most common clinical forms of leishmaniasis and is caused by various species of Leishmania including Leishmania major. This disease has always been neglected as a major public health problem due to its non-fatality, however, major consequences of the disease consists of skin lesions leading to scars. Current chemotherapies are inadequate and suffer by various problems like toxicity, side effects and emerging drug resistance. These issues have raised concerns among researchers for developing more effective and affordable drugs as well as potential therapeutic molecular drug targets.
Recent studies show that instead of focusing on single enzyme and/or protein, researchers nowadays are putting their efforts in analysing metabolic networks to identify potential drug targets in many organisms3,4,5,6. Kinetic modeling, genome scale metabolic modeling, and constraint-based methods etc. are such techniques that have been extensively used in many studies for finding the importance of enzymes in various pathways including redox metabolism7,8,9,10,11,12,13. Among these techniques, kinetic modelling requires experimental kinetic parameters and knowledge of rate laws. There are several studies in which kinetic modelling has been used to study the kinetics of gene transfer and antibiotic resistance in several bacteria14,15.
In trypanosomatids, a unique redox metabolism exists that help the parasites to deal with the oxidative stresses that can be host originated (inside parasite infected macrophages) or parasite originated. In both the cases, the stress is posed mainly by free radicals generated via many pathways. Additionally, several reactive metabolites, viz., glyoxals, apart from their nature to modify macromolecules, can also play important role in free radicals formation. Methylglyoxal (MGO), a dicarbonyl glyoxal, is one of the highly reactive metabolites having affinity towards amino groups of proteins and nucleic acids. It is a spontaneous product of glycolysis pathway and formed from dihydroxyacetone (DHAP) and/or glyceraldehyde-3-phosphate (GA3P)16,17. MGO is found to irreversibly modify ~0.1–2% arginine residues of proteins and one in ten bases of DNA thereby causing their inactivation and leading to the formation of advanced glycation end products (AGEs)18,19,20,21,22,23,24,25. Therefore, MGO needs to be neutralized before producing any harm to the cells. The most important pathway for its detoxification is glyoxalase pathway that consists of two enzymes, glyoxalase I (GLO I) and glyoxalase II (GLO II)26,27. GLO I, first, isomerizes hemithioacetal (HTA), a spontaneous product formed after MGO-T[SH]2 conjugation, to S-D-lactoyaltrypanothione followed by GLO II mediated conversion to D-lactate. In this pathway, GLO I mediated step is described as rate limiting and the enzyme is found to have essential role in the survival of Leishmania28. In our previous work, we also identify GLO I as an important drug target through systems biology approach29. Hence, in current work, we seek to understand the kinetics of GLO I through in silico kinetic modeling followed by the identification of potential hit compounds by molecular modeling and dynamics simulation approaches. The identified potential hits were further tested experimentally to check their in vitro and in vivo potency against Leishmania major.
Materials and Method
Kinetic modelling and sensitivity analysis
Construction of all kinetic models was done using CellDesigner v4.430. After drawing, these models were saved as Systems Biology Markup Language (SMBL) file and imported in COPASI (COmplex Pathway Simulator) v4.19.14031 followed by assignment of appropriate kinetic laws. All reactions were considered as irreversible for maintaining uniformity in the kinetic model (Table 1). The concentrations, time units and reaction fluxes in the model are in molar (M), seconds and M/s, respectively. All the kinetic parameters were obtained/calculated from published literature (Supplementary File 1, Table 2, S1). To observe the effect of different parameters on the models, parameter scanning and sensitivity analyses were performed using deterministic LSODA ODE (ordinary differential equations) solver32.
Molecular modelling and pharmacophore model development
Benzimidazoles and thiosemicarbazones derivatives were retrieved from various online databases (PubChem, ZINC, DrugBank) and prepared by applying OPLS2005 force field followed by their ADME/Tox screening in QikProp module (Schrodinger, LLC). Receptor preparation and docking was done in protein Preparation Wizard module and Glide, respectively, with default parameters.
MD simulation using Desmond (v 4.4) was performed on selected complexes solvated in an orthorhombic box filled with SPC water molecules and neutralized by adding Na+ ions at random places. The entire system was minimized using steepest decent algorithm until a gradient threshold of 25 Kcal/mol/Å was reached. The Martyna-Tobias-Klein scheme was used for pressure coupling. Electrostatic forces were calculated using the PME algorithm33. All runs were performed at 300 K at constant volume and temperature (NPT ensemble) under certain periodic boundary conditions (explicit solvent simulations with periodic boundary conditions using orthorhombic box distance (Å) 10.0, 10.0, 10.0; Na+ ions: 15) for a period of 10 ns –100 ns with the time steps of 2 fs.
Pharmacophore hypotheses describing the important inherent chemical features responsible for the biological activity were generated using Discovery Studio software.
In vitro culture of parasites and culture condition
Wild type Leishmania major promastigotes (MHOM/IL/67/JERICHOII) were grown in RPMI-1640 media supplemented with 20% heat inactivated fetal bovine serum (FBS) (GIBCOBRL, Grand Island, NY) and antibiotics at 27 °C. For all experiments, parasite cultures in logarithmic (exponential) growth phase were considered.
Analysis of drug sensitivity by colorimetric MTT assay
Anti-leishmanial activity assay on L. major promastigotes by colorimetric MTT assay
The compounds were dissolved in DMSO to prepare a stock of 100 mM and further diluted in R20 (RPMI with 20% (v/v) heat inactivated FBS) to make fresh working solution of 1 mM concentration. The final concentration of DMSO was kept below 0.4% (v/v). Exponential growth phase promastigotes were resuspended in fresh medium to achieve 1 × 106 cells/ml and seeded in 96-well culture plates in triplicates. The concentration of compounds was used ranging from 1 to 1000 μM. The plates were incubated at 27 °C for 48 h followed by PBS wash. The viability of the parasites was evaluated by using quantitative colorimetric MTT (3-[4,5-methylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide) assay to assess the metabolic activity of the cells. Briefly, 20 μl of MTT labelling reagent (5 mg/ml) and 180 μl PBS was added to each well including the controls (non-treated) and incubated in dark for a time period of 4 h at 27 °C. As an indicator of cell viability, the formazan crystals, were dissolved in 100 μl of DMSO per well to obtain a blue solution. The absorbance was read spectrophotometrically at 570 nm. A decrease in the amount of MTT converted indicates toxicity to the cell. Tetrazolium dye reduction is generally assumed to be dependent of NAD(P)H dependent cellular oxidoreductase enzymes present largely in cytosolic compartment of viable cells. Therefore, the reduction of MTT depends on the cellular metabolic activity due to NAD(P)H flux. These enzymes reduce the tetrazolium dye (MTT) to its insoluble formazan purple coloured crystals inside the cells. Cells with low metabolism (or dead cells) reduce very little or negligible MTT in comparison to rapidly dividing cells34. Hence, in our work, cells treated with C1 showed less MTT reduction thereby less formazan formation and low spectrophotometric absorbance demonstrating cytotoxicity of C1 to the parasite. The IC50 (drug concentrations at which 50% of parasites were inhibited) was calculated by regression analysis. The results are expressed as the means and standard deviations of three independent experiments.
Macrophage infections and drug susceptibility assay against intracellular amastigote forms
Raw cell line macrophages were seeded (1 × 105 cells/ml) onto coverslips and incubated at 37 °C for 12 h. After washing with PBS (pH 7), cells were infected with stationary phase L. major promastigotes (1 × 106 cells/ml) at a macrophage to parasite ratio of 1:10 and incubated at 37 °C for 24 h for proper internalization of parasite. Further, the cells were washed with PBS and given appropriate treatments with C1 in all groups except control and incubated for at 37 °C. After 48 h incubation, cells were given PBS wash followed by DAPI staining. Amastigote IC50 was calculated by computing parasite infectivity index in untreated and treated groups by counting the number of amastigotes inside the infected macrophage (amastigotes/macrophage)35.
Mammalian cell cytotoxicity
The cytotoxicity assay for compounds was performed on macrophage cell line (Raw 264.7 cell line). The macrophage cells were seeded in 96 well plates (1 × 105 cells/ml) in DMEM media (GIBCO BRL, Grand Island, NY) supplemented with 10% heat inactivated FBS and 100 μg/ml penicillin-streptomycin. Cells were incubated at 37 °C, 5% CO2 for a period of 24 h. This was followed by the media replacement with fresh medium containing the compound (80–200 µM), and the cells again were incubated for 48 h at same culture condition. The assay was performed as mentioned above and %Cell viability in relation to untreated controls was calculated
Detection of phosphatidylserine exposure
For detecting phosphatidylserine exposure, double staining with FITC Annexin V apoptosis detection kit with PI (Propidium Iodide), Biolegend, was used. Briefly, the untreated and treated promastigotes (1 × 106 cells/ml) were incubated at 27 °C for 48 h followed by PBS washing and centrifuged at 14,00 g for 10 min. The pellets were then resuspended in the standard provided with the kit followed by double staining with annexin V-FITC and PI as per the manufacturer’s instructions. The parasites were incubated in dark for 15–20 min at 26 °C before recording the intensity of labelled parasites by capturing 10,000 events on FACS Calibur flow cytometer (Becton Dickinson). For analysis, CellQuest software was used and the percentage of positive cells was plotted and assessed in the form of respective dot plots and histograms.
DNA content analysis for cell cycle
The untreated and treated promastigotes (1 × 106 cells/ml) were incubated at 27 °C for 48 h and washed with PBS and centrifuged at 14,00 g for 10 min. Pelleted cells were dissolved and fixed in 70% cold ethanol and incubated overnight at −20 °C. The fixed cells were again washed and resuspended in PBS and stained with PI (1 mg/ml). The cells were incubated for 20 min in dark at room temperature and the intensity of PI was analysed with FACS Calibur (Becton Dickinson) flow cytometer and CellQuest software by acquiring 10,000 cells.
Morphological analysis
Briefly, 1 × 106 L. major promastigotes were cultured with or without respective drugs at their IC50 concentrations and incubated for 48 h at 27 °C. Further, parasites were washed with PBS and smeared onto the poly L-lysine coated coverslips. Air dried cells were fixed in 2.5% glutaraldehyde solution by keeping the cells for a minimum time period of 3 h. The fixed cells were then washed thrice in PBS and dehydrated in acetone gradient (30%, 50%, 70%, 90%, and 100%; each for 5 min). Finally, cells were air dried, coated with platinum using Q150T Turbo-Pumped Sputter Coater and observed under Nova NanoSEM field emission scanning electron microscope operating at 15 kV.
Cloning, expression and purification of L. major GLO I
The construct containing the L. major GLO I gene (437 bp) was procured from Invitrogen, Thermofisher Scientific. The synthesised gene was inserted into pET28a(+)_AF090 vector. The construct was reconstituted and confirmation of GLO I insertion was done using restriction enzymes NotI and NdeI. Competent Escherichia coli BL21 (DE3) cells were prepared and the construct was transformed at 42 °C for 30 sec by heat shock treatment. These cells were grown on kanamycin (50 µg/ml) containing Luria Berttini (LB) agar medium plates followed by overnight incubation at 37 °C. Kanamycin resistant single colony of BL21 (GLO I + ve) was inoculated in LB broth with kanamycin and incubated at 37 °C for 2–3 h or until an absorbance of 0.6 at 600 nm was reached. The GLO I expression was induced by 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) and again incubated at 27 °C for 4 h. The expression of GLO I was checked on 12% SDS-PAGE36.
Cell were centrifuged at 4,696 g for 30 min and lysed by resuspending the pellet in Ni-NTA lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole pH 8.0) with lysozyme and incubated for 30 min on ice followed by sonication (four 30 sec on/off cycle). Further, the homogenate was centrifuged and the supernatant was poured into Ni-NTA column. The column was washed twice with Ni-NTA wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0). GLO I was eluted with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mm imidazole, pH 8.0) in fractions. The purity of GLO I was checked on 12% SDS-PAGE using standard protocol.
In vivo screening
Animal study was conducted according to the protocols approved by the Institutional Animal Ethical Committee of National Centre for Cell Science (NCCS, Registration number: 7/GO/ReBi/S/99/CPCSEA; Date of registration: 09/03/1999) under the Project number: EAF/2017/B-312. BALB/c female mice, 8 week old with 20–25 mg body weight were used. For infection, stationary phase L.major promastigotes (2 × 107 cells/ml) were injected subcutaneously in the left hind limb footpad of the mice. Compound C1 was initially prepared in DMSO followed by its dilution in Milli-Q water. Mice were divided in groups (5 mice per group) and administered orally for 10 days as follows: control group with Milli-Q water; C1 groups with four different doses of C1 (5, 10, 20 and 40 mg/kg/body weight/per day). Mice were euthanized 12 days post treatment and treatment potency was determined by estimating parasite load in draining lymph nodes by limiting dilution assay as per the Taswell’s method37.
The statistical significance of differences in parasite load amongst groups was evaluated using one-way ANOVA with Tukey’s correction.
Glyoxalase I assay
The activity of LmGLO I was assayed spectrophotometrically at room temperature. For this, the initial rate of formation of S-D-lactoyl trypanothione was measured at 240 nm as described previously38 with slight modifications. Briefly, DTT (3 mM) was used to reduce trypanothione disulfide (1 mM) (BOC Sciences) at 60 °C for 20 min. For preparing the assay mixture, 200 mM methylglyoxal (Sigma), 300 mM reduced trypanothione and 20 mM NiCl2 were added to 100 mM MOPS buffer to reach the final volume of 500 µl39. The assay mixture was then incubated for 10 min followed by the addition of LmGLO I (100 nM) and C1 inhibitor in various concentrations ranging from 0–320 µM for calculating the IC50. All the assays were performed in triplicate.
Ethics statement
All mice were maintained in compliance with the Institutional Animal Ethics guidelines. All animal experiments were approved by the Institutional Animal Ethics Committee (IAEC) (7/GO/c/99/CPCSEA) of the National Centre for Cell Science (IAEC Project Number-EAF /2017/B-312).
Results and Discussion
Models reconstruction and kinetic simulation
Initially, two basic models for glyoxalase pathway were constructed. In wild type model (noninhibited), M1a, the reactions for synthesis and degradation of MGO were included. Hence, for constructing the perturbed model (inhibited), M1b, MGO mediated substrate level inhibition reaction for GLO I24,25 was introduced in M1a and assigned with appropriate rate law (Tables 1, 2, Table S1, Fig. 1a,b). For both models previously determined rate constants (except hypothetical Ki for GLO I) were used. The kinetics of degradation of MGO in both models was observed through time course simulation in COPASI.

Two basic models constructed to study the inhibition kinetics of GLO I. (a) M1a (NI: noninhibited), and (b) M1b (I: inhibited) models.
Time course simulation of two models showed maximum activity of GLO I enzyme in terms of the degradation of MGO and synthesis of SDLTSH in M1a model when compared to M1b model (Figs. 2, S1). The effect of higher concentration of MGO in the system was observed by increasing its initial concentration upto 10 folds. As assumed, increased MGO in M1b model has led to the similar level of accumulation of MGO due to the perturbation introduced in the model. This has, consequently, decreased the GLO I activity, thereby, affected the rate of synthesis of SDLTSH and D-lactate (DL) (Fig. 2). It was reported earlier that MGO damages proteins by modifying Arg and Lys residues, forms (AGEs and promotes the formation of free radicals40,41,42. Hence, to understand the effect of MGO accumulation on other metabolites in the system, we extended our models to larger scale and added several reactions (Table 1) of AGEs, in terms of modified Arg and Lys (MGO-Arg and MGO-Lys), and free radical formation (M2a and M2b) (Fig. S2). Interestingly, when comparison was made between inhibited and non-inhibited models, the perturbation in extended model demonstrated many folds increase in the formation of many metabolites including MGO-Lys and free radicals (MGO•-, •OH, O2•-, NO2•) (Fig. 3, Table 3).

Prediction of the effect of increased conc of MGO in M1a and M1b.

Effect of increased conc of MGO on other metabolites in extended model in M2a and M2b.
The robustness analysis, to check the performance and reliability of the kinetic model, was confirmed by varying the values of GLO I reaction (Vmax and Km) (Table S2). Our constructed kinetic model has shown negligible variation in the predicted concentration of SDLTSH. Further, to evaluate the relevance of the GLO I as a potential therapeutic target, sensitivity analysis of the models was performed by using GLOIVmax. It was observed that reaction fluxes of all reactions were found to be affected in perturbed M2b model (Fig. 4). In M2a model, most of the reaction were observed independent of GLO I reaction rate as most of the reactions have not shown any sensitivity towards GLOIVmax, however, the perturbation via GLO I reaction demonstrated the opposite suggesting its global impact on reaction fluxes of almost all reactions, thereby, causing the concentration fold increase of the metabolites in M2b. These observations suggest that free radical formation and other reactions are somehow connected to MGO level and decrease in GLO I activity should affect their formation. Our observations clearly show that the glyoxalase enzyme GLO I is an important therapeutic target. Therefore, it is suggested that GLO I plays an important role in the regulation of redox metabolism by detoxifying MGO. This view is also supported by an earlier study28.

Illustration of Sensitivity analysis of M2a (NI: non-inhibited) and M2b (I: inhibited) models. For sensitivity analyses in these models, Vmax of GLO I was used to observe its influence on the flux of other reactions. It was observed that reaction fluxes of all reactions were affected in perturbed (inhibited) model.
Molecular modelling and pharmacophore features selection
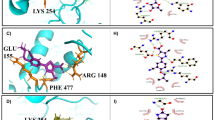
Benzimidazoles are bicyclic compounds, consisting of fusion of benzene and imidazole, and are known to have potent inhibitory activity43,44,45,46, including anti-viral, anti-bacterial, anti-leishmanial47,48,49,50. Similarly, thiosemicarbazones have also been broadly studied due to their role as antibacterial, antiprotozoal, and antiviral activity51,52,53,54,55. After ADME/Tox screening and docking, top 8 GLOI-ligand complexes were selected (Table S3) and subjected to MD simulation. Simulation results showed stable interaction between ligands and GLO I active site. RMSD of ligand bound GLO I were plotted to understand their relative stability (Fig. S3). Although, hydrogen bonds plots between ligand-GLO I have demonstrated that all ligands were interacting with the active site by forming at least one H-bond, rmsd in terms of interaction diagram of ligand-GLO I has suggested that only two complexes, C1 and C2 (Benzimidazole derivatives), were stable throughout the simulation cycle without much fluctuation. Furthermore, the ligand distance from Ni2+ was observed to be within the range (Fig. S3). These two compound-GLO I complexes were further subjected to extended MD simulation for up to 100 ns to inspect their structural and interaction stability. Our results revealed that only C1-GLO I complex exhibited stable behaviour in terms of rmsd and that C2 tends to deviate from GLO I and fluctuated even after coming into the contact of GLO I (Fig. 5). Same was noted in case of C2-GLO I complex when the two structures from 10 ns and 100 ns were superposed (Fig. 6). Hence, our molecular modelling and simulation analysis suggested that C1 can, further, be used as an active hit against Leishmania.

Evaluation of time course simulation of C1 (a) and C2 (b) with GLO I complex for 100 ns.

Superposed structures of (a) C1- and (b) C2-GLOI. ligand and metal ion shown in orange and green color represents structure after 10 ns and 100 ns, respectively.
Next, based on C1 binding modes in GLO I active site, initially, a 4-point pharmacophore model was developed using Discovery studio software (Fig. 7). Due to the importance in catalysis56,57, the active site metal ion present in the binding pocket of GLO I (Fig. 7a) was manually chosen as ‘metal binding’ feature (Fig. 7b). Finally, then this pharmacophore models was used for database screening against ZINC drug-like database (https://zinc.docking.org/substances/subsets/). The screened compounds were then docked against GLO I. Table S4 shows top 10 compounds selected from pharmacophore based virtual screening.

(a) Depiction of binding interaction between C1 and GLO I. (b) Pharmacophore features selection on the basis of C1-GLO I binding modes. A 5 point pharmacophore model was designed automatically using Discovery studio.
Expression and purification of LmGLO I
After reconstitution and transformation of construct (Fig. S4) into BL21 strain, the cells were grown at 37 °C for 24 hrs. The plasmid DNA was isolated and digested by restriction enzyme followed by its gel electrophoresis (Fig. S5a). Further, gloI gene was expressed by giving 1 mM IPTG induction for different time point upto 5 h as described in material and methods section (Fig. S5b). The purified GLO I protein through Ni-NTA resin column was found to be ~24 kDa in size39,58.
Anti-promastigote activity of compounds
Compounds C1 and C2 were procured and tested against L. major promastigotes and raw cell line of macrophages post 48 h of treatment and their IC50 values were calculated. Between the two compounds, C2 showed little inhibition and its IC50 against Leishmania could not be calculated (IC50 > 1000 μM). However, C1 exhibited promising antileishmanial activity (IC50 105 μM) (Fig. 8a) and ~80% cell viability on macrophage (Fig. 8b). Furthermore, in SEM analysis, promastigotes treated with both the compounds have shown remarkable morphological changes. Both the compounds were able to cause size reduction, membrane blebbing and loss in motility in the parasite (Fig. 9).

Testing the potency and cytotoxicity of compounds. (a) IC50 calculation of C1 against L. major parasite. (b) Cytotoxicity of the C1 over Raw macrophage cell line. (c) IC50 calculation of intracellular L. major amastigotes.

Morphological analysis of untreated and treated Leishmanial promastigotes through SEM. (a) untreated, (b,c) C1 treated, and (d,e) C2 treated. Both compounds (C1 and C2) caused reduction in size, membrane blebbing and flagella loss causing less motility in the parasite.
C1 efficiently eliminates L. major amastigotes from macrophage
C1 has also shown effective parasite clearance property in case of intracellular amastigotes (Fig. 8c). It was observed that C1 was more effective and exhibited slightly higher potency against the intracellular amastigotes (IC50 57.64 µM) compared to L. major promastigotes (IC50 105 µM).
C1 induces sub-G0/G1 phase cell-division arrest and phosphatidylserine externalization
Our results revealed that C1 treated promastigotes showed 17.12% increase in sub-G0/G1 phase when compared with untreated parasites (Fig. 10). This increase was accompanied by decrease in the number of cells in S and G2/M phase (4.78% and 0.11%, respectively) in comparison to untreated cells (18.57% and 4.95%, respectively). While in case of MIL treated parasite lower values were obtained (sub-G0/G1: 10.19%, S: 1.06%, and G2/M: 0.18%). These findings suggest that C1 arrests parasites in sub-G0/G1 phase and induces cell death resembling apoptosis59,60,61,62,63. We could also observe that approximately 8.69% treated parasite population was annexinV-positive and PI-negative (Fig. 11). Therefore, the anti-leishmanial activity of C1 was observed as apoptosis driven due to the lower percentage of PI-positive population (4.23%). Similarly, benzimidazoles have reported to cause cell cycle arrest and trigger apoptosis-like phenomenon in Leishmania64,65. Taken together, increased proportion of cells arrested in sub-G0/G1 phase and higher early apoptotic percentage confirmed that C1 induces apoptosis in Leishmanial promastigotes.

Cell cycle analysis of L. major. (a) Untreated; and treated (b) MIL; (c) C1.

Apoptosis analysis of untreated and treated (MIL and C1) L. major.
Efficacy of C1 in L. major infected mice
As mentioned above, the drug C1 was administered orally in different groups (5, 10, 20 and 40 mg/kg/body weight/day). The swelling in the foot pad was monitored post treatment and it was observed that in the groups who received C1 did not show any difference in the lesion size unlike the control group administered with water showing gradual increase in lesion size (Fig. 12a). To confirm the efficacy of C1, treated and untreated mice were euthanized and their lymph nodes were collected to find parasitic burden. It was noted that in all treated groups the parasitic load found to be decreased as compared top untreated mice (Fig. 12b). Moreover, it was noted that at higher dose (40 mg) the compound showed little toxic effect that caused weakening of mice. Our results indicate that C1 has healing effect in infected mice and reduced the parasitic burden in mice. Henceforth, we suggest C1 as potent anti-leishmanial hit and by further modification it could be possible to develop novel and potent compounds.

(a) Foot pad size difference in C1 treated and untreated groups. (b) Parasite load assay of the C1 treated and untreated groups. Every dot indicates one mice. P-value was calculated by comparing treated groups with control and significance is indicated in the figure as ***p < 0.0001.
Glyoxalase I assay
An IC50 of 18.34 µM for C1 against LmGLOI was obtained after the data fitting to sigmoidal dose-response curves with nonlinear regression in GraphPad Prism 5 (Fig. 13). To see the potency of C1, this IC50 was further converted to Ki value using the Cheng-Prusoff equation using online IC50 to Ki conversion tool66. For this published Km value of LmGLOI was used. Our Ki value for C1 (2.52 µM) was found to be lower than that of the previously investigated inhibitors against LmGLOI67 suggesting the potency of C1 against LmGLOI.

Illustration of inhibitory potency of C1 against LmGLOI.
Conclusion
Taken together, our study demonstrates the amalgamation of systems biology and structural modelling to understand the effect of GLO I inhibition kinetics and to use this knowledge into exploring potent lead compounds. Our model reveals that the perturbation introduced in the form of GLO I inhibition had noticeable effect on MGO detoxification that ultimately led to increased formation of free radicals (Fig. 14). Moreover, via molecular modelling we were able to screen C1 as hit compound that also exhibited good anti-leishmanial activity in vitro. This compound caused morphological alteration in the parasite and was also found to induce cell cycle arrest and apoptosis in L. major. Furthermore, in vivo studies on mice model have revealed the parasite clearance potency of the compound and proved the overall efficacy of C1.

Depiction of generation and effect of free radicals. Inside mitochondria, source of generation of ROS (reactive oxygen species) are Fumarase reductase (FR) and Ubiquinone 9 (UQ) carrying single electron reaction in ETC in mitochondrial membrane contributing to the generation of superoxide anion free radicals (O2•−) which are usually detoxified by other membrane bound complexes (eg., cytc, etc., not shown in the figure) by transporting the electron to oxygen. Superoxide anion free radicals contribute to form other ROS and damage various proteins, DNA and lipids. Similarly, leishmanial Nitric oxide synthase (NOS), in unfavorable circumstances, synthesizes NO• ions that further contribute to the formation of RNI (reactive nitrogen species). ROS and RNI also inactivate Fe-S cluster proteins (Fumarase reductase (FR) and Aconitase) by leaching out their iron centres. ROS and RNI also decrease GSH (glutathione) level thereby directly affecting the synthesis of trypanothione (T[SH]2) and activation of various T[SH]2 dependent proteins and enzymes. These reactive species also inactivates glyoxalase I (GLOI), an important rate limiting enzyme in a two-step methylglyoxal (MGO), a highly reactive metabolite and a by-product of glycolysis, detoxification pathway. Further, inactivation of GLOI enzyme can also be carried out by drugs or inhibitors leading to the increased level of MGO. MGO, when in excess, irreversibly modifies proteins (including GLOI) by reacting with their Arg and Lys residues and forms advanced glycation end products (AGEs), (leading to the degradation of proteins), and MGO•- free radicals.
References
Narain, J. P. et al. Elimination of neglected tropical diseases in the South-East Asia Region of the World Health Organization. Bull World Health Organ. 88, 206–210 (2010).
WHO. Accelerating Work to Overcome the Global Impact of Neglected Tropical Dieases: A Roadmap for implementation, http://www.who.int/neglected_diseases/NTD_RoadMap_2012_Fullversion.pdf. (2011).
Hornberg, J. J., Bruggeman, F. J., Bakker, B. M. & Westerhoff, H. V. Metabolic control analysis to identify optimal drug targets. Prog. Drug Res. 64(171), 173–189 (2007).
Moreno-Sanchez, R. et al. Metabolic control analysis indicates a change of strategy in the treatment of cancer. Mitochondrion. 10, 626–639 (2010).
Klipp, E., Wade, R. C. & Kummer, U. Biochemical network-based drug-target prediction. Curr. Opin. Biotechnol. 21, 511–516 (2010).
Kolodkin, A. et al. Emergence of the silicon human and network targeting drugs. Eur. J. Pharm. Sci. 46, 190–197 (2012).
Wang, R. S., Oldham, W. M., Maron, B. A. & Loscalzo, J. Systems Biology Approaches to Redox Metabolism in Stress and Disease States. Antioxid. Redox Signal. 29, 953–972 (2018).
Pillay, C. S., Hofmeyr, J. H., Mashamaite, L. N. & Rohwer, J. M. From top-down to bottom-up: computational modeling approaches for cellular redoxin networks. Antioxid. Redox Signal. 18, 2075–2086, https://doi.org/10.1089/ars.2012.4771 (2013).
Beiting, D. P. & Roos, D. S. A systems biological view of intracellular pathogens. Immunol. Rev. 240, 117–128 (2007).
Toledano, M. B., Kumar, C., Le Moan, N., Spector, D. & Tacnet, F. The system biology of thiol redox system in Escherichia coli and yeast: differential functions in oxidative stress, iron metabolism and DNA synthesis. FEBS Lett. 581, 3598–3607 (2007).
Ho, T. C. Kinetic Modeling of Large‐Scale Reaction Systems. Sci. Eng. 50, 287–378 (2008).
Chavali, A. K., Whittemore, J. D., Eddy, J. A., Williams, K. T. & Papin, J. A. Systems analysis of metabolism in the pathogenic trypanosomatid Leishmania major. Mol. Syst. Biol. 4, 177 (2008).
Fatumo, S. et al. Estimating novel potential drug targets of Plasmodium falciparum by analysing the metabolic network of knock-out strains in silico. Infect. Genet. Evol. 9, 351–358 (2009).
Knopoff, D. A. & Sanchez Sanso, J. M. A kinetic model for horizontal transfer and bacterial antibiotic resistance. Int. J. Biomath. 10, 1750051 (2017).
Lu, N. X. et al. A kinetic model of gene transfer via natural transformation of Azotobacter vinelandii. Environ. Sci.: Water Res. Technol. 1, 363–374 (2015).
Thornalley, P. J. The glyoxalase system in health and disease. Mol. Aspects Med. 14, 287–371 (1993).
Phillips, S. A. & Thornalley, P. J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 212, 101–105 (1993).
Thornalley, P. J. et al. Quantitative screening of advanced glycation end products in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 375, 581–592 (2003).
Vander Jagt, D. L. The glyoxalase system. In: Dolphin D, Poulson R, Avramovic O, editors. Glutathione: chemical biochemical and medical aspects, Part A. New York: John Wiley & Sons. p. 597–641 (1989).
Ayoub, F. M., Allen, R. E. & Thornalley, P. J. Inhibition of proliferation of human leukaemia 60 cells by methylglyoxal in vitro. Leuk. Res. 17, 397–401 (1993).
Egyud, L. G. & Szent-Gyorgyi, A. Cancerostatic action of methylglyoxal. Science. 160, 1140 (1968).
Westwood, M. E., Argirov, O. K., Abordo, E. A. & Thornalley, P. J. Methylglyoxal-modified arginine residues-a signal for receptor-mediated endocytosis and degradation of proteins by monocytic THP-1 cells. Biochim. Biophys. Acta. 1356, 84–94 (1997).
Chakraborty, S., Karmakar, K. & Chakravortty, D. Cells producing their own nemesis: understanding methylglyoxal metabolism. IUBMB Life. 66, 667–78 (2014).
Lo, T. W., Westwood, M. E., McLellan, A. C., Selwood, T. & Thornalley, J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. Chem. 269, 32299–32305 (1994).
Gangadhariah, M. H., Mailankot, M., Reneker, L. & Nagaraj, R. H. Inhibition of methylglyoxal-mediated protein modification in glyoxalase I overexpressing mouse lenses. J. Ophthalmol. 2010, 274317 (2010).
Neuberg, C. The destruction of lactic aldehyde and methylglyoxal by animal organs. Biochem. Z. 49, 502–506 (1913).
Sousa Silva, M. et al. The glyoxalase pathway in protozoan parasites. Int. J. Med. Microbiol. 302, 225–229 (2012).
Chauhan, S. C. & Madhubala, R. Glyoxalase I Gene Deletion Mutants of Leishmania donovani Exhibit Reduced Methylglyoxal Detoxification. PLoS ONE. 4, e6805 (2009).
Chauhan, N. & Singh, S. Integrative Computational Framework for Understanding Metabolic Modulation in Leishmania. Front. Bioeng. Biotechnol. 7, 336, https://doi.org/10.3389/fbioe.2019.00336 (2019).
Funahashi, A., Tanimura, N., Morohashi, M. & Kitano, H. CellDesigner: a process diagram editor for gene-regulatory and biochemical networks. BIOSILICO. 1, 159–162 (2003).
Hoops, S. et al. COmplex Pathway SImulator. Bioinformatics. 22, 3067–3074 (2006).
Hindmarsh, A. C. ODEPACK, a systematized collection of ODE solvers. Scientific computing. 55–64 (1983).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–92 (1993).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 65, 55–63 (1983).
Vermeersch, M. et al. In vitro susceptibilities of Leishmania donovani promastigote and amastigote stages to antileishmanial reference drugs: practical relevance of stage-specific differences. Antimicrob. Agents Chemother. 53, 855–9 (2009).
Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. UK Laemmli. Nature. 227, 5259 (1970).
Taswell, C. Limiting dilution assays for the determination of immunocompetent cell frequencies. I. Data analysis. The Journal of Immunology. 126, 1614–9 (1981).
Racker, E. The mechanism of action of glyoxalase. J. Biol. Chem. 190, 685–696 (1951).
Vickers, T. J., Greig, N. & Fairlamb, A. H. A trypanothione-dependent glyoxalase I with a prokaryotic ancestry in Leishmania major. PNAS. 101, 13186–13191 (2004).
Kalapos, M. P. The tandem of free radicals and methylglyoxal. Chem. Biol. Interact. 171, 251–271 (2008).
Desai, K. M. & Wu, L. Free radical generation by methylglyoxal in tissues. Drug Metabol. Drug Interact. 23, 151–173 (2008).
Fraval, H. N. & McBrien, D. C. The effect of methyl glyoxal on cell division and the synthesis of protein and DNA in synchronous and asynchronous cultures of Escherichia coli B/r. J. Gen. Microbiol. 117, 127–134 (1980).
Singh, N. et al. Benzimidazole: A short review of their antimicrobial activities. Int. J. Curr. Pharm. Res. 1, 110–118 (2012).
Ansari, K. F. & Lal, C. Synthesis, physicochemical properties and anti-microbial activity of some new benzi-midazole derivatives. Eur. J. Med. Chem. 44, 4028–4033 (2009).
Kazimierczuk, Z. et al. Synthesis and antiproto-zoal activity of some 2-(trifluoromethyl)-1H-benzimidazole bioisosteres. Acta. Biochim. Pol. 49, 185–195 (2002).
Grocer, H., Kus, C., Boykin, D. W., Yildiz, S. & Altanlar, N. Synthesis and Anti-fungal Properties of Some Benzimidazole Derivatives. Bioorg. Med. Chem. 10, 2589–2596 (2002).
Tuncbilek, M., Kiper, T. & Altanlar, N. Synthesis and in vitro anti-microbial activity of ome novel substituted benzimidazole derivatives having potent activity against MRSA. Eur. J. Med. Chem. 44, 1024–1033 (2009).
Oh, S. et al. Synthesis and biological evaluation of 2,3-dihydroimidazo[1,2-a]benzimidazole derivatives against Leishmania donovani and Trypanosoma cruzi. Eur. J. Med. Chem. 84, 395–403 (2014).
Tonelli, M. et al. Benzimidazole derivatives endowed with potent antileishmanial activity. J. Enzyme Inhib. Med. Chem. 33(1), 210–226, https://doi.org/10.1080/14756366.2017.1410480 (2018).
De Luca, L. et al. Discovery of benzimidazole-based Leishmania mexicana cysteine protease CPB2.8ΔCTE inhibitors as potential therapeutics for leishmaniasis. Chem. Biol. Drug Des. 92(3), 1585–1596, https://doi.org/10.1111/cbdd.13326 (2018).
Kizilcikli, I. et al. Antimicrobial activity of a series of thiosemicarbazones and their Zn(II) and Pd(II) complexes. Folia microbiologica. 52, 15–25 (2007).
Mendes, I. et al. Organotin(IV) complexes of 2-pyridineformamide-derived thiosemicarbazones: antimicrobial and cytotoxic effects. Eur. J. Med. Chem. 43, 1454–1461 (2008).
Bharti, N., Athar, F., Maurya, M. & Azam, A. Synthesis, characterization and in vitro anti-amoebic activity of new palladium(II) complexes with 5-nitrothiophene-2-carboxaldehyde N(4)-substituted thiosemicarbazones. Bioorg. Med. Chem. 12, 4679–4684 (2004).
Aguirre, G. et al. In vitro activity and mechanism of action against the protozoan parasite Trypanosoma cruzi of 5-nitrofuryl containing thiosemicarbazones. Bioorg. Med. Chem. 12, 4885–4893 (2004).
Finkielsztein, L. et al. New 1-indanone thiosemicarbazone derivatives active against BVDV. Eur. J. Med. Chem. 43, 1767–1773 (2008).
Creighton, D. J. & Hamilton, D. S. Brief history of glyoxalase I and what we have learned about metal ion-dependent, enzyme-catalyzed isomerizations. Arch. Biochem. Biophys. 387, 1–10 (2001).
Clugston, S. L., Yajima, R. & Honek, J. F. Investigation of metal binding and activation of Escherichia coli glyoxalase I: kinetic, thermodynamic and mutagenesis studies. Biochem. J. 377, 309–16 (2004).
Padmanabhan, P. K. et al. Glyoxalase I from Leishmania donovani: A potential target for anti-parasite drug. Biochem. Biophys. Res. Commun. 337, 1237–1248 (2005).
Krishan, A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell. Biol. 66, 188–193 (1975).
Marquis, J. F., Drolet, M. & Olivier, M. Consequence of Hoechst 33342-mediated Leishmania DNA topoisomerase-I inhibition on parasite replication. Parasitology. 126, 21–30 (2003).
Marquis, J. F., Makhey, D., LaVoie, E. J. & Olivier, M. Effects of topoisomerases inhibitors protoberberine on Leishmania donovani growth, macrophage function, and infection. J. Parasitol. 89, 1048–1052 (2003).
Liang, Y., Zhou, Y., Deng, S. & Chen, T. Microwave-Assisted Syntheses of Benzimidazole-Containing Selenadiazole Derivatives That Induce Cell-Cycle Arrest and Apoptosis in Human Breast Cancer Cells by Activation of the ROS/AKT Pathway. Chem. Med. Chem. 11, 2339–2346 (2016).
He, L. J. et al. Facile construction of fused benzimidazole-isoquinolinones that induce cell-cycle arrest and apoptosis in colorectal cancer cells. Bioorg. Med. Chem. 26, 3899–3908 (2018).
Marquis, J. F., Hardy, I. & Olivier, M. Resistance mechanism development to the topoisomerase-I inhibitor Hoechst 33342 by Leishmania donovani. Parasitology. 131, 197–206 (2005).
Takasawa, R. et al. TLSC702, a Novel Inhibitor of Human Glyoxalase I, Induces Apoptosis in Tumor Cells. Biol. Pharm. Bull. 39(5), 869–73, https://doi.org/10.1248/bpb.b15-00710 (2016).
Cer, R. Z., Mudunuri, U., Stephens, R. & Lebeda, F. J. IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 37(Web Server issue), W441–W445, https://doi.org/10.1093/nar/gkp253 (2009).
Ariza, A. et al. Specificity of the trypanothione-dependent Leishmania major glyoxalase I: structure and biochemical comparison with the human enzyme. Mol. Microbiol. 59, 1239–48 (2006).
Acknowledgements
NC acknowledges her fellowship from DST-SERB NPDF (PDF/2016/001346) from Department of Science and Technology, Ministry of Science and Technology, Government of India. The authors would also like to thank the Director, National Centre for Cell Science (NCCS) for supporting the Bioinformatics and High Performance Computing Facility (BHPCF) at NCCS, Pune, India. FACS and Animal house Facilities at NCCS are also acknowledged by the authors.
Author information
Authors and Affiliations
Contributions
Nutan Chauhan (N.C.) has performed kinetic modeling and also performed experimental validation for the same. Shailza Singh (S.S.) designed, conceived, conceptualized the whole idea and subsequently helped with the design of experiments to validate the computational approach.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chauhan, N., Singh, S. Multiscale Process Modelling in Translational Systems Biology of Leishmania major: A Holistic view. Sci Rep 10, 785 (2020). https://doi.org/10.1038/s41598-020-57640-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57640-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
