
Abstract
Somatic mutation profiling in gastric cancer (GC) enables main driver mutations to be identified and their clinical and prognostic value to be evaluated. We investigated 77 tumour samples of GC by next-generation sequencing (NGS) with the Ion AmpliSeq Hotspot Panel v2 and a custom panel covering six hereditary gastric cancer predisposition genes (BMPR1A, SMAD4, CDH1, TP53, STK11 and PTEN). Overall, 47 somatic mutations in 14 genes were detected; 22 of these mutations were novel. Mutations were detected most frequently in the CDH1 (13/47) and TP53 (12/47) genes. As expected, somatic CDH1 mutations were positively correlated with distant metastases (p = 0.019) and tumours with signet ring cells (p = 0.043). These findings confirm the association of the CDH1 mutations with diffuse GC type. TP53 mutations were found to be significantly associated with a decrease in overall survival in patients with Lauren diffuse-type tumours (p = 0.0085), T3-T4 tumours (p = 0.037), and stage III-IV tumours (p = 0.013). Our results confirm that the detection of mutations in the main driver genes may have a significant prognostic value for GC patients and provide an independent GC-related set of clinical and molecular genetic data.
Similar content being viewed by others

Introduction
Gastric cancer (GC) is the third leading cause of cancer-related deaths worldwide after lung cancer and breast cancer. The incidence of GC is particularly high in East Asia, including China, Japan and Korea, and in South America1. Based on the Lauren classification, GC is divided into two main types, namely, intestinal and diffuse, which have different epidemiological, morphological and clinical features. Intestinal GC commonly appears in elderly patients with multifocal atrophic gastritis, which is accompanied by intestinal metaplasia or dysplasia. Diffuse GC is more common in younger patients, and its association with atrophic gastritis or intestinal metaplasia is not obvious. Clinical differences between these two types reflect different mechanisms of the development and molecular pathogenesis of tumours2. However, Lauren’s classification is not closely associated with treatment and prognosis, necessitating the development of a classification combining clinical, morphological, and molecular features of GC in response to certain therapeutic modalities.
Comprehensive studies, including analyses of the genome, epigenome, proteome and transcriptome, offered an entirely different view on the tumour, moving it out of a single plane and into a multidimensional spatial image. The ability to determine the tumour-specific spectrum of genetic and epigenetic changes enables us to expand our understanding of the molecular pathogenesis of the tumour and to obtain information about the potential of targeted therapies. Mutational profiling is one way to classify tumours depending on the mutation spectrum into specific molecular subtypes that differ from the standard morphological classification. The results of recent studies, such as TCGA (The Cancer Genome Atlas) and ACRG (Asian Cancer Research Group), indicate that GC could be divided into four molecular subtypes with different mechanisms of pathogenesis. These subtypes are not completely consistent with the standard morphological classification and the Lauren classification3,4.
As whole-genome research methods are difficult to introduce into clinical practice, it is necessary to provide a reduced set of the most informative diagnostic and prognostic clinical markers. Furthermore, validation of the molecular subtypes of GC in large patient groups with different ethnic and racial backgrounds is clearly required. At present, it is already clear that there can be no universal classification for GC, and national genetic and epigenetic features should be considered.
A number of genes have been identified as driver genes in gastric cancer. However, the association between somatic mutations and clinical features has not been thoroughly elucidated to date. It is therefore important to profile the somatic mutation patterns of driver genes and potential driver genes in gastric cancer. Research investigating the somatic mutation profiles of cancer-related genes reveals the main driver mutations that determine the clinical behaviour of a tumour, its aggressiveness, invasion and metastasis, and the direction of targeted antitumour therapy. It was determined that GC is not enriched with known driver mutations. Therefore, the targeted drugs that are useful in the treatment of other tumours are not effective in GC therapy, and despite the development of novel drugs for GC, trastuzumab and ramucirumab (targeting HER2 and VEGFR2, respectively) are the only targeted therapies approved to date5.
Germline mutations in some driver genes determine predisposition to the development of hereditary gastric cancer. Mutations in CDH1 are responsible for the development of early hereditary diffuse GC, as are mutations in TP53 (Li-Fraumeni syndrome), STK11 (Peutz–Jeghers syndrome), SMAD4 or BMPR1A (gastrointestinal polyposis) and PTEN (Cowden syndrome)6. Thus, it is advisable to combine the BMPR1A, SMAD4, CDH1, TP53, STK11 and PTEN genes into a targeted sequencing panel that will provide significant information on the mutational profile of gastric tumours in both hereditary and sporadic cancer, which can be associated with the clinical and pathomorphological features of the disease. Screening for mutations in these genes might be important to determine germline mutations in patients with both early manifestation and/or family history, as well as for somatic profiling of sporadic GC.
Research examining the main driver mutations in the tumour is critical for accurate personalized medicine. A specific profile of somatic mutations and their combinations may indicate more aggressive behaviour, invasion and metastasis and may represent a diagnostic or prognostic marker.
To investigate the GC mutation profile and determine its prognostic value, we conducted a study of 77 GC tumour samples using next-generation sequencing (NGS) on both the Ion AmpliSeq Cancer Hotspot Panel v2, covering mutation hotspots in 50 cancer-related genes, and a custom panel covering six hereditary gastric cancer predisposition genes (BMPR1A, SMAD4, CDH1, TP53, STK11 and PTEN).
Results
Spectrum of identified somatic mutations
NGS analysis of tumour samples from 77 gastric cancer patients revealed 47 somatic mutations in 14 of the 51 genes initially selected for this study, either because the genes harboured the oncogenic mutational “hot spots” or because they were associated with the development of hereditary GC. In this paper, we report as mutations only the genetic variants that are either notably rare or absent in populations (assessed with gnomAD) or have previously been classified as pathogenic/likely pathogenic in other studies. Genetic variants with MAF > 0.0001 were excluded from the analysis. For genetic variants not previously reported in human mutation databases, we performed in silico pathogenicity estimation (see below).
DNA sequencing with our hereditary GC panel revealed a total of 36 mutations (Table 1), with the most frequently mutated genes being CDH1 (13/36) and TP53 (12/36), and the other genes being distributed as follows: SMAD4 (6/36), STK11 (3/36), PTEN (1/36), and BMPR1A (1/36). One of the TP53 gene mutations, NM_000546.5:c.743 G > A:p. R248Q, was detected in two cases, which is in line with TCGA data, where this mutation is also recurrent.
CDH1 mutations were found in 11 patients. Although none of the CDH1 mutations was recurrent in our study, some were found in the same codons and had similar in silico pathogenicity predictions. These mutations include c.641 T > C and c.641 T > A, which cause leucine 214 substitution to proline and glutamine, respectively, and are both predicted to alter protein function, or c.1199 A > T and c.1198 G > A, which change aspartic acid 400 to valine and asparagine, respectively, and apparently lead to loss of protein function and alterations in posttranslational modifications.
Upon sequencing with the CHPv2 panel, 11 mutations in 8 genes were detected: PIK3CA (2/11), RB1 (3/11), and one mutation each in CDKN2A, SMO, KRAS, EGFR, KIT, and KDR. The results are presented in Table 2.
Clinical relevance of gastric cancer somatic mutational status
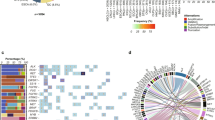
In the tumour samples of 32/77 (42%) patients, we identified at least one somatic mutation, whereas no mutations that met the selection criteria were found in the remaining 45/77 (58%) patients (Fig. 1). We found no associations of overall tumour somatic mutational status (absence of mutations in the genes under study vs presence of at least one mutation) with patients’ age, gender, 5-year overall survival, lymph node metastases and distant metastases or such tumour characteristics as size, stage, Lauren type and presence of signet ring cells (Table 3).

Distribution of mutations in the 77 gastric tumours under study. Each column denotes an individual tumour, and each row represents a gene. Only genes with at least one mutation found in our cohort are depicted.
We further investigated the clinical significance of somatic mutations in the CDH1 and TP53 genes in patients with GC. The results are presented in Table 3. We found no significant differences in the groups with CDH1 and TP53 mutations regarding gender, age, tumour localization, lymph node metastasis, distant metastasis, stage, and Lauren type. As expected, somatic CDH1 mutations were positively correlated with distant metastases (p = 0.019). CDH1 mutations were also observed significantly more frequently in tumours with signet ring cells (p = 0.043).
To investigate the prognostic value of the detected mutations, we conducted a study of the overall survival (OS) of GC patients within the 5-year interval after surgery. OS associations with tumour mutational status were studied in groups with or without lymph node metastasis, distant metastasis, different tumour stages (I-II vs III-IV), Lauren classification as diffuse or intestinal, gender and age.
Regarding the overall tumour somatic mutational status, we detected no difference in OS of patients carrying at least one mutation in the genes under study vs those with no mutations in the same genes (Fig. 2). OS assessed on the whole CG patient cohort under study was also independent of somatic mutation to either the CDH1 gene or the TP53 gene, which is in line with TCGA Provisional data estimated by cBioPortal.

Absence of association between tumour mutational status and overall survival in GC patients. The blue graph describes a subcohort of GC patients with at least one somatic mutation, and the red graph is for the patients with no somatic mutations identified in the genes under study.
We further analysed OS in different clinical groups with respect to the presence of TP53 and CDH1 mutations in tumours. OS appeared to be significantly decreased in the groups of patients with tumours harbouring TP53 mutations with diffuse Lauren type (p < 0.0085; Fig. 3a), with T3-T4 tumours (p = 0.037; Fig. 4b), and with stage III-IV tumours (p = 0.013; Fig. 5b).

Somatic TP53 mutational status and overall survival in GC patients with diffuse and intestinal Lauren tumour types. (a) Diffuse type. (b) Intestinal type.

Somatic TP53 mutational status and overall survival in GC patients with T1-T2 tumours (a) and with T3-T4 tumours (b).

Somatic TP53 mutational status and overall survival in GC patients with different tumour stages. (a) Stages I-II. (b) Stages III-IV.
Evaluation of pathogenicity for genetic variants not annotated in human mutation databases
The pathogenicity of 25 missense genetic variants that were either identified in our study for the first time, had been previously reported in populations but lacked annotations in the human mutation databases, or were ambiguously annotated in terms of clinical significance (conflicting interpretations were presented regarding pathogenicity or uncertain significance), was assessed using the prediction programs PolyPhen2, PROVEAN, SNPs&GO and MutPred2, I-Mutant 3.0 and MutPred-LOF. The results are presented in Supplementary Tables 1 and 2.
According to the PolyPhen2 HumDiv, 6 substitutions are benign; among these substitutions, only one is in CDH1, and one is in TP53, while others may be damaging. PolyPhen2-HumVar classified 10 substitutions as benign, but it should be borne in mind that this program is better in predicting pathogenicity for Mendelian disorders. PROVEAN indicates that 16 substitutions are deleterious, and 9 of them are neutral. SNPs&GO indicates that 22/25 substitutions are pathogenic with a different reliability index.
MutPred2 and MutPred-LOF software predict how single-nucleotide substitutions can affect the molecular mechanisms in a cell. In our set of somatic genetic variants obtained from gastric tumours, MutPred2 and MutPred-LOF predicted significant disruption of various molecular mechanisms for 13/25 variants with p-values < 0.05 (Supplementary Table 2). The I-Mutant 3.0 tool predicts a decrease in protein stability for 23/25 mutant variants, whereas for two mutations (CDH1:c.A1199T:p.4. D400V and TP53:c.517 G > T:p. V173L), the stability of a mutant protein molecule was predicted to be increased.
The results of this study indicate that the assessed substitutions may be considered to be pathogenic based on the estimates provided by the bioinformatic tools generally used to predict the effects of genetic variants.
Discussion
In our cohort of gastric tumours, somatic mutations were most frequently observed in the CDH1 and TP53 genes. Mutations in these genes were previously described as the most frequent in other studies, although the reported percentages vary between cohorts7,8. Discrepancies in the reported frequencies of the mutations detected in these genes may be explained by different approaches to attribution of a genetic variant to the list of deleterious mutations, as well as by ethnic characteristics of the patients. It was demonstrated that the frequencies of somatic mutations of certain genes (e.g., APC, ARID1A, KMT2A, PIK3CA and PTEN) differ between Caucasian and Asian GC patients9. According to TCGA data, CDH1 mutations were identified in 11% of all GCs, and ТР53 mutations were determined in 50% of non-hypermutated GCs and in 71% of chromosomally unstable (CIN) samples10.
In our study, we used two NGS panels to screen for somatic mutations in GS samples. One panel is the Ion AmpliSeq Cancer Hotspot Panel v2, which has been previously used for GC genotyping in a clinical cancer research11. Using this panel, we identified 11 mutations in 8 of the 50 genes studied in our 77 GC samples. Additionally, 36 mutations were identified using a Hereditary Gastric Cancer Panel (HGC), designed by our group, with full coverage of the coding regions of the TP53, CDH1, PTEN, BMPR1A, SMAD4 and STK11 genes associated with the development of hereditary GC. All identified mutations were located in the driver genes for GC. The application of our oligogene NGS panel for mutational profiling of GC may enable identification of hereditary cases in clinical practice. Furthermore, detection of CDH1 and TP53 mutations can serve as a surrogate marker to distinguish the chromosomally unstable GC, which is enriched for TP53 mutations, from the genomically stable subtype, which is enriched for CDH1 mutations3. In addition, this test may assist in distinguishing MSS-TP53+ and MSS-TP53−, as well as MSS-ETM, enriched for CDH1 mutations, when used for ACRG (Asian Cancer Research Group) GC type detection4.
The pivotal role played by CDH1 mutations in the development of GC is undoubted12. In our study, somatic CDH1 mutations were positively correlated with distant metastases (p = 0.019). CDH1 mutations were also significantly more frequent in tumours with signet ring cells (p = 0.043). However, we have identified no independent prognostic value of somatic CDH1 mutations, which is in line with TCGA Provisional data estimated by cBioPortal.
Inactivation of CDH1 during GC development occurs in the tumour because of genetic and epigenetic changes. Loss of CDH1 expression appears after inactivation of both copies, where one allele can be mutant, and the second allele is inactivated by promotor methylation in approximately 50% of cases. We have previously demonstrated that CDH1 promoter methylation is associated with the diffuse Lauren type and locally advanced GC13. Other studies have shown the prognostic relevance of CDH1 mutations in diffuse GC, where the presence of somatic mutations in this gene was associated with a decrease in patient survival, regardless of the stage of the disease14.
In our cohort, a group of 35 patients had early manifestation of GC, age of disease onset ranging from 26 to 49 years, and no family cancer history. We assumed that this group of patients might harbour a certain mutation spectrum or enrichment with germline mutations in the genes associated with the development of hereditary GC. However, we found no significant differences in the somatic mutation profile and no enrichment with germline mutations for this group. Cho et al. found a significant increase in the frequency of somatic mutations of CDH1 and TGFBR1 in patients from Korea with early manifestations of GC before 45 years (P < 0.001 for CDH1 and P = 0.014 for TGFBR1)15. We assessed data for patients with manifestations of GC before 45 years in our cohort and found no significant increase in the frequency of CDH1 somatic mutations in this group.
Currently, patients with early manifestations of GC (before 45–50 years) are classified into an independent cancer group of early-onset GC (EOGC) with specific clinical and molecular characteristics. This group features prevalence among women, multifocal growth and diffuse phenotype of the tumour without intestinal metaplasia. A molecular profile of the tumour is characterized by the absence or low level of microsatellite instability (MSI), rare loss of heterozygosity (LOH), retained expression of RUNX3, amplifications of 17q, 19q and 20q, and high expression of low-molecular-weight cyclin E isoforms16. The reported prognosis for this group varies from better to poorer survival depending on the study17,18. Our study did not reveal a statistically significant difference in OS for patients with early GC onset, but we detected a decrease in the survival rate of patients in this group in the presence of somatic TP53 mutations.
Although TP53 mutations in GC have long been studied, the clinical relevance of these mutations for the prognosis of the disease and treatment of patients has not been fully determined. A large number of studies have presented diametrically opposite results, which may be explained by the specific characteristics of patient cohorts, including the ethnicity of the studied groups. At present, the frequency of mutant alleles of this gene is used to stratify patients into molecular subtypes, to predict the course of the disease and to control the response to chemotherapy. It was demonstrated that the mutant allele frequency (AF) decreases during chemotherapy in GC patients19.
Discrepancies in the estimations of the clinical significance of the somatic mutations presented by different research teams may not only be caused by ethnic factors but also by differences in the criteria used to attribute a genetic variant to a class of pathogenic or possibly pathogenic mutations. Standards and guidelines for the interpretation of sequence variants widely used in medical genetics, such as those provided by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology20, cannot be directly extrapolated to cancer somatic variants. In silico predictors of pathogenicity for missense variants are 65–80% accurate when examining known disease variants, and some are intended for analysis of Mendelian disorders20. In our study, we adopted the extremely low general population frequency of a genetic variant as a key criterion of its inclusion in the downstream analysis of the clinical significance of somatic mutations in a cohort of GC patients. More specifically, genetic variants with MAF > 0.0001 were excluded from analysis, which means that the remaining alternative alleles were either never found in gnomAD or were observed extremely rarely. At present, this criterion may be considered rather stable; with at least 60,000 human exomes annotated21, we are not expecting dramatic fluctuations in allele frequencies in the human population in the foreseeable future.
Materials and Methods
Patients and tumour samples
The study included 77 patients with locally advanced GC who were treated in N.N. Burdenko Facultative Surgery Clinic, I.M. Sechenov First Moscow State Medical University from 2007 to 2015. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Ethics Committee of I.M. Sechenov First Moscow State Medical University. Written informed consent was obtained from each participant in this study. All patients underwent surgical treatment, and resected tumour samples were used in the study. GC was confirmed in all patients by morphological examination of the surgical material. According to the Lauren classification, intestinal GC was confirmed in 29/77 cases, and diffuse GC was confirmed in 38/77 cases. The distribution of patients in clinical groups is presented in Table 4.
Mutation screening by NGS
Five to seven 10-μm paraffin sections were manually dissected to ensure that each tumour sample contained at least 70% of neoplastic cells. Genomic DNA was isolated from archived samples using a QIAamp DNA FFPE Tissue kit, as recommended by QIAGEN (Germany).
Deep sequencing was performed using the Ion Torrent platform (Life Technologies) following established protocol22. The protocol includes the preparation of libraries of genomic DNA fragments, clonal emulsion PCR, sequencing and bioinformatic analysis of results. DNA fragment libraries were prepared using Ion Ampliseq ultra-multiplex PCR technology.
A commercially available oligonucleotide panel Ion AmpliSeq Cancer Hotspot Panel v2 (Life Technologies) was used as a set of multiplex primers. The panel consists of 207 primer pairs for simultaneous amplification of mutation hotspots in 50 cancer-related genes (in alphabetical order): ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, JAK2, JAK3, IDH1, IDH2, KDR/VEGFR2, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, and VHL23.
Additionally, we utilized our original, customized panel, comprised of six hereditary GC-related genes (HGC panel). An HGC panel with 218 primer pairs was designed to amplify all coding regions, noncoding regions of the terminal exons, and putative splice site gene regions for six human genes: BMPR1A, SMAD4, CDH1, TP53, STK11, and PTEN. The panel was designed using the Ion Ampliseq Designer v.3.6, which minimizes the number of oligonucleotide pair pools that are necessary to completely cover the target genomic sequences. The total length of human genome sequences covered by the HGC panel is 42,320 bp.
Multiplex PCR and subsequent stages of the fragment library preparation were undertaken using an Ion AmpliSeq Library Kit 2.0 (Life Technologies) according to the manufacturer’s protocol. Aliquots from the prepared libraries were subjected to clonal amplification on microspheres in the emulsion on the Ion OneTouch instruments using the Ion OneTouch 200 Template Kit v2 DL (Life Technologies). Effective products of the emulsion PCR, the microspheres coated with the target amplicons, were purified from empty microspheres on the Ion OneTouch Enrichment System. Sequencing was performed on the Ion Torrent PGM genomic sequencer using an Ion PGM 200 Sequencing Kit (Life Technologies). The results were analysed with Torrent Suite software consisting of Base Caller (the primary analysis of the sequencing results); Torrent Mapping Alignment Program - TMAP (alignment of the sequences to the reference genome GRCh37/hg19); and Torrent Variant Caller (analysis of variations in nucleotide sequences). Genetic variants were annotated with ANNOVAR software24. Visual data analysis, manual filtering of sequencing artefacts and sequence alignment were performed using the Integrative Genomics Viewer (IGV)25.
To investigate the hereditary cancer syndrome genes involved in Li-Fraumeni syndrome, Peutz–Jeghers syndrome, Cowden syndrome, hereditary GC and hereditary gastrointestinal polyposis, we designed a panel consisting of 218 primer pairs for PCR amplification of all coding sequences of the human genes BMPR1A, SMAD4, CDH1, TP53, STK11 and PTEN, as well as exon flanks and terminal untranslated regions (UTRs). A primer panel was designed with Ion Ampliseq Designer v.3.6 software. The total length of human genome sequences covered by this panel is 42320 bp.
Validation of mutations detected by next-generation sequencing
Validation of the mutations detected by NGS screening was performed to exclude sequencing artefacts in tumour samples. The direct sequencing of individual PCR products from primers that flank areas of specific mutations was performed on the automatic genetic analyser ABI PRISM 3500 (Life Technologies, Carlsbad, United States) according to the manufacturer’s protocols.
Statistical analysis
Samples were compared using Fisher’s exact test. OS was calculated by the Kaplan–Meier product limit method from the date of surgery until death by any cause. Statistica 7.0 software was used for data processing. The results obtained from our tumour tissue collection were compared to the most comprehensively characterized collection of stomach adenocarcinoma samples (TCGA, Provisional) with the assistance of The cBioPortal (www.cbioportal.org), an open access resource for interactive exploration of multidimensional cancer genomics datasets26.
Pathogenicity prediction for novel mutations
To predict the pathogenicity of identified novel missense variants, PolyPhen2, PROVEAN, SNPs&GO and MutPred2 tools were used. I-Mutant 3.0 software was used to calculate the stability of the mutant protein. Loss of protein function effects were assessed with MutPred-LOF software. The effect of nonsynonymous substitutions on the structure was illustrated using the Project HOPE3D portal.
Data availability
Raw sequence reads data are available at the NCBI BioProject, Accession: PRJNA560649, ID: 560649 (https://www.ncbi.nlm.nih.gov/bioproject/560649).
References
Bray, F. et al. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 68, 394–424, https://doi.org/10.3322/caac.21492 (2018).
Jang, B. G. & Kim, W. H. Molecular pathology of gastric carcinoma. Pathobiology. 78, 302–310, https://doi.org/10.1159/000321703 (2011).
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 513, 202–229, https://doi.org/10.1038/nature13480 (2014).
Cristescu, R. et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 21, 449–456, https://doi.org/10.1038/nm.3850 (2015).
Apicella, M., Corso, S. & Giordano, S. Targeted therapies for gastric cancer: failures and hopes from clinical trials. Oncotarget. 8, 57654, https://doi.org/10.18632/oncotarget.14825 (2017).
Colvin, H., Yamamoto, K., Wada, N. & Mori, M. Hereditary Gastric Cancer Syndromes. Surg. Oncol. Clin. N. Am. 24, 765–777, https://doi.org/10.1016/j.soc.2015.06.002 (2015).
Kuboki, Y. et al. Comprehensive analyses using next-generation sequencing and immunohistochemistry enable precise treatment in advanced gastric cancer. Ann. Oncol. 27, 127–133, https://doi.org/10.1093/annonc/mdv508 (2016).
Pan, X. et al. Landscape of somatic mutations in gastric cancer assessed using next-generation sequencing analysis. Oncol. Lett. 16, 4863–4870, https://doi.org/10.3892/ol.2018.9314 (2018).
Jia, F. et al. Discordance of somatic mutations between Asian and Caucasian patient populations with gastric cancer. Mol. Diagn. Ther. 21, 179–185, https://doi.org/10.1007/s40291-016-0250-z (2017).
Katona, B. W. & Rustgi, A. K. Gastric Cancer Genomics: Advances and Future Directions. Cell Mol. Gastro-enterol Hepatol. 3, 211–217, https://doi.org/10.1016/j.jcmgh.2017.01.003 (2017).
Bria, E. et al. Comprehensive molecular portrait using next generation sequencing of resected intestinal-type gastric cancer patients dichotomized according to prognosis. Sci. Rep. 6, 22982, https://doi.org/10.1038/srep22982 (2016).
Luo, W., Fedda, F., Lynch, P. & Tan, D. CDH1 gene and hereditary diffuse gastric cancer syndrome: molecular and histological alterations and implications for diagnosis and treatment. Front. Pharmacol. 9, 1421, https://doi.org/10.3389/fphar.2018.01421 (2018).
Nemtsova, M. V. et al. Implication of Gastric Cancer Molecular Genetic Markers in Surgical Practice. Curr. Genomics. 18, 408–415, https://doi.org/10.2174/1389202918666170329110021 (2017).
Li, X. et al. Distinct Subtypes of Gastric Cancer Defined by Molecular Characterization Include Novel Mutational Signatures with Prognostic Capability. Cancer Res. 76, 1724–1732, https://doi.org/10.1158/0008-5472.CAN-15-2443. (2016).
Cho, S. Y. et al. Sporadic Early-Onset Diffuse Gastric Cancers Have High Frequency of Somatic CDH1 Alterations, but Low Frequency of Somatic RHOA Mutations Compared with Late-Onset Cancers. Gastroenterology. 153, 536–549, https://doi.org/10.1053/j.gastro.2017.05.012 (2017).
Skierucha, M. et al. 1. Molecular alterations in gastric cancer with special reference to the early-onset subtype. World J. Gastroenterol. 22, 2460–2474, https://doi.org/10.3748/wjg.v22.i8.2460 (2016).
Pisanu, A., Podda, M., Cois, A. & Uccheddu, A. Gastric cancer in the young: is it a different clinical entity? A retrospective cohort study. Gastroenterol. Res. Pract. 2014, 125038, https://doi.org/10.1155/2014/125038 (2014).
Bautista, M. C., Jiang, S. F., Armstrong, M. A., Postlethwaite, D. & Li, D. Impact of age on clinicopathological features and survival of patients with noncardia gastric adenocarcinoma. J. Gastric Cancer. 14, 238–245, https://doi.org/10.5230/jgc.2014.14.4.238 (2014).
Zhang, S. The association of mutant allele frequency of TP53 with response of chemotherapy in advanced gastric cancer patients. J. Clin. Oncol. 37(no. 4_suppl), 42–42, https://doi.org/10.1200/JCO.2019.37.4_suppl.42 (2019).
Richards, S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424, 1038/gim.2015.30 (2015).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536, 285–291, https://doi.org/10.1038/nature19057 (2016).
Scarpa, A. et al. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PLoS One. 8, e80478, https://doi.org/10.1371/journal.pone.0080478 (2013).
Butler, K. S., Young, M. Y., Li, Z., Elespuru, R. K. & Wood, S. C. Performance characteristics of the AmpliSeq Cancer Hotspot panel v2 in combination with the Ion Torrent Next Generation Sequencing Personal Genome Machine. Regul. Toxicol. Pharmacol. 74, 178–186, https://doi.org/10.1016/j.yrtph.2015.09.011 (2016).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variations from next-generation sequencing data. Nucl. Acids Res. 38, e164, https://doi.org/10.1093/nar/gkq603 (2010).
Robinson, J. T. et al. Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26, https://doi.org/10.1038/nbt.1754 (2011).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404, https://doi.org/10.1158/2159-8290.CD-12-0095 (2012).
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (projects 18-015-00333 and 18-29-09020) and the Ministry of Science and Higher Education of the Russian Federation.
Author information
Authors and Affiliations
Contributions
M.V.N., A.S.T. and V.V.S. conceived the experiments, E.B.K., E.A.A. and D.S.M. conducted the experiments, I.I.B. and T.V.K. performed clinical investigations, A.I.K., I.V.B. and D.V.Z. analysed the results, and S.I.K. administered the project. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nemtsova, M.V., Kalinkin, A.I., Kuznetsova, E.B. et al. Clinical relevance of somatic mutations in main driver genes detected in gastric cancer patients by next-generation DNA sequencing. Sci Rep 10, 504 (2020). https://doi.org/10.1038/s41598-020-57544-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57544-3
This article is cited by
-
Prospective Identification of Prognostic Hot-Spot Mutant Gene Signatures for Leukemia: A Computational Study Based on Integrative Analysis of TCGA and cBioPortal Data
Molecular Biotechnology (2023)
-
Genetic mosaicism in the human brain: from lineage tracing to neuropsychiatric disorders
Nature Reviews Neuroscience (2022)
-
Mucoepidermoid carcinoma (MEC) and adenosquamous carcinoma (ASC), the same or different entities?
Modern Pathology (2022)
-
Circulating tumor DNA: a noninvasive biomarker for tracking ovarian cancer
Reproductive Biology and Endocrinology (2021)
-
Comprehensive tumor molecular profile analysis in clinical practice
BMC Medical Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
