
Abstract
Recently, genome-editing tools have come into common use in the field of rat research, and consequently, many genetically modified rat strains have been preserved and archived as frozen embryos. Although there have been many reports published on the topic of rat sperm cryopreservation, no report has yet provided satisfactory and acceptable protocols for the cryopreservation of rat sperm. In this study, we developed methods for both the cryopreservation of transgenic rat sperm and in vitro fertilisation using frozen sperm, which yielded high fertilisation rates.
Similar content being viewed by others


Introduction
Over the past ten years, genome-editing techniques have been used to produce many genetically modified rat strains1. Consequently, the cryobanking of gametes and embryos from these strains is rapidly becoming an important issue. Frozen embryos and sperm from many rat strains have been preserved in rat resource centres worldwide, including the National BioResource Project - Rat, Kyoto University, Japan; the Rat Resource & Research Center, University of Missouri, USA; and the Gene Editing Rat Resource Center, Medical College of Wisconsin, USA.
In general, embryo freezing is a reliable method for preserving rat strains. Using the conventional embryo freezing method, 400–500 embryos per strain are required from the oviducts of 30–50 mated females. Moreover, if only a few genetically modified males are available to be used for mating, it would take from 2 to 4 months to obtain sufficient embryos for cryopreservation because the number of females used for mating at any one time is limited and the males have to mate with females multiple times. On the other hand, a large number of spermatozoa can be cryopreserved immediately after being collected from the epididymides of males. Thus, sperm freezing is a much simpler, more efficient and more economical alternative to embryo freezing for preserving genetically modified rat strains.
Since the publication in 2001 of the first successful report concerning the cryopreservation of rat sperm2, many papers on the topic have been published3,4,5,6,7,8,9,10,11,12,13. However, only one of those papers has reported the successful production of pups derived from embryos that were obtained via in vitro fertilisation using frozen rat sperm5. Meanwhile, satisfactory and acceptable protocols for rat sperm cryopreservation and subsequent IVF have not yet been established.
Recently, we established a rat sperm cryopreservation method that results in high fertilisation rates via in vitro fertilisation using frozen/thawed sperm. In this paper, we restrict ourselves to a description of the detailed procedure routinely used for freezing rat sperm.
Results
In vitro fertilisation of either fresh or frozen rat sperm
High fertilisation rates were achieved with both fresh and frozen rat sperm (Table 1). However, the fertilisation rates achieved using cryopreserved sperm were lower than the rates achieved with fresh sperm (P < 0.05). Fresh sperm also had higher percentages of motile and progressive motile sperm than frozen sperm (Fig. 1).

Motile parameters of fresh and cryopreserved rat sperm Parameters of sperm motility were analysed by a computer-assisted sperm analyser. (A) Motility is the ratio of sperm moving 5 μm/s to total sperm. (B) Progressive motility is the ratio of sperm moving at 50 μm/s and with straightness greater than 50% to total sperm. (C) Path velocity (VAP) is the average velocity of the smoothed sperm path. (D) Progressive velocity (VSL) is the average velocity measured in a straight line from the beginning to the end of the sperm track. (E) Lateral amplitude (ALH) is the mean width of the head oscillation as the sperm swims. (F) Beat frequency (BCF) is the frequency of sperm heads crossing the average sperm path in either direction. (G) Straightness (STR) is a measure of the departure of the average sperm path from a straight line (ratio of VSL/VAP). Values are given as the mean ± SD (n = 4). *p < 0.05 compared with fresh sperm.
Developmental ability of fertilised rat oocytes in vitro and in vivo
Almost all fertilised oocytes developed to 2-cell embryos 28 hours after insemination. More than 60% of 2-cell embryos derived from cryopreserved and fresh sperm developed to blastocysts, half of which were blastocysts with green fluorescent protein (GFP) signals (Table 2 and Fig. 2). A total of 46 normal pups (26 pups with GFP signals) and 48 normal pups (24 pups with GFP signals) were obtained from cryopreserved and fresh sperm, respectively (Table 3 and Fig. 3). There were no differences between the development of the fertilised oocytes derived from either cryopreserved or fresh sperm either in vitro or in vivo.

Blastocysts derived from in vitro fertilisation using cryopreserved rat sperm. Wild-type blastocysts and GFP-positive blastocysts (green) are shown. GFP-positive blastocysts are labelled with asterisks.

Live pups derived from in vitro fertilisation with cryopreserved rat sperm. Wild-type pups and GFP-positive pups (green) are shown.
Discussion
The current study is the first report to show that transgenic rat spermatozoa can be successfully cryopreserved and that the oocytes fertilised by cryopreserved spermatozoa can develop into normal offspring after in vitro fertilisation and transfer of the fertilised oocyte.
Although the success of in vitro fertilisation using fresh sperm was demonstrated by Toyoda et al. in 197414, there were no successful cases of in vitro fertilisation using frozen sperm for many years after that. It was 2009 before the first account of successful in vitro fertilisation using frozen Wistar rat sperm, which was reported by Seita et al.5. Their data showed that when isobutylmethylxanthine (IBMX)-treated frozen/thawed sperm were used for in vitro fertilisation, the rates of pronuclear formation and blastocyst formation were significantly higher (pronuclear formation: 50%, blastocyst formation: 20%, birth rate: 49%) than what they achieved when frozen/thawed sperm were used without IBMX treatment. However, since then, there have been no papers showing that frozen sperm can retain sufficient sperm motility to produce pups via in vitro fertilisation and embryo transfer.
In the current study, we addressed the problem of poor reproducibility of rat sperm cryopreservation and in vitro fertilisation. We have combined the best of our knowledge and experience in rat reproductive technology. We demonstrated fertilisation rates of more than 80% using cryopreserved transgenic rat sperm (Table 1), and 60% of 2-cell embryos developed to the blastocyst stage (Table 2). Furthermore, 58% of transferred fertilised oocytes developed into pups (Table 3).
Rat sperm is known to be extremely sensitive to environmental changes, such as shifts in viscosity and osmotic stress15,16. As such, if a cryopreservation agent (CPA) containing sperm is diluted immediately after thawing, the cryopreserved sperm may be damaged. In our previous study, we found that the fertilising ability of mouse sperm that were diluted using mHTF immediately after thawing was much lower than the rate of sperm that were diluted slowly17. The rat sperm CPA in the current study has high viscosity and an osmolarity of 370–380 mOsm. On the other hand, mHTF used for dilution has low viscosity and an osmolarity of 300–310 mOsm. Thus, we think that it is very important to preincubate the straw containing the sperm suspension in a 37 °C water bath for fifteen minutes (Fig. 4I) and to slowly dilute the cells using the swim-up method (Fig. 4J) after thawing; both strategies are described in this paper because they prevent sharp changes in viscosity and osmolarity between the CPA and the diluent. In addition, suppressing the movement of sperm at 0 °C before freezing may be another important factor (Fig. 4C–F). Further investigation of the above factors is required to comprehend the improvement of quality of cryopreserved rat sperm described here.

Sperm cryopreservation and in vitro fertilisation in rat.
Over the past ten years, many transgenic and targeted mutant rats have been produced worldwide1. Moreover, even more genetically modified rats will be produced using genome-editing techniques in the near future18. However, rats are approximately ten times larger than mice, and the number of rats that can be kept in an animal room is limited. Thus, we would need a large capital investment and substantial space to keep hundreds of rats at our centre, and preservation of transgenic rat strains will become a major issue in the near future. Moreover, opportunities to exchange these rat strains will increase rapidly because of collaborations between institutes. As we have mentioned previously, a large number of spermatozoa can be collected from just one male. Ergo, if the sperm collected from 2–3 males is cryopreserved, we can produce more than 300 pups at a later date using that sperm via in vitro fertilisation and embryo transfer. This method may facilitate a reduction in the number of rats required to maintain rat strains. Therefore, we strongly believe that sperm freezing represents an extremely powerful tool for storing a large number of rat strains with induced mutations, and we further believe that we will see application of the technique in the exchange of mutant strains between labs worldwide.
Materials and Methods
Animals
Male transgenic rats (SD-Tg(CAG-EGFP)4Osb) were purchased from Slc Japan (Shizuoka, Japan) (http://www.anim.med.kyoto-u.ac.jp/NBR/strains/Strains_d.aspx?StrainID=559&s_geneAffected=GFP&s_References=GFP&s_livingAnimals=1) and were used as sperm donors at 11–12 weeks of age; Slc:SD female rats (purchased from Slc) were used as oocyte donors at 4–5 weeks of age. Meanwhile, Crl:CD (SD) female rats purchased from Charles River Japan (Kanagawa, Japan) were used as recipients of fertilised oocytes at 10–15 weeks of age. All animals were housed under a twelve-hour dark/light cycle (light from 07:00 to 19:00) at 22 °C ± 1 °C, with ad libitum access to food and water. The Animal Care and Use Committee of Kumamoto University School of Medicine approved the protocols for animal experiments and all methods were performed in accordance with the relevant guidelines and regulations.
Sperm cryoprotective agent
Preparation of a cryopreservation agent (CPA) for sperm was carried out essentially following the method described by Nakatsukasa et al.2,3. A solution of 8% (w/v) lactose (Sigma, St Louis, MO) and 23% (v/v) chicken egg yolk, with 1,000 units/mL of penicillin G and 1 mg/mL of streptomycin sulfate, was mixed in distilled water. After adjusting the pH of the solution to 7.4 with 10% tris aminomethane solution, the solution was centrifuged at 1,600 g for 15 minutes, and the upper layer of the solution was collected. Then, 0.7% Equex Stm (ES; Nova Chemical Sales, Inc., Scituate, Mass.) and 0.1% ATP (Adenosine 5′-triphosphate disodium salt hydrate Grade II; Sigma) were added to the solution7,8, and the solutions was mixed on a stirring plate. Finally, the CPA was divided into aliquots and stored in a freezer at −20 °C before use.
Sperm freezing
In each experiment, one male transgenic rat was used for sperm freezing.
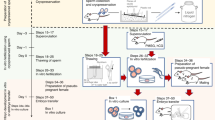
Figure 4 summarises the sperm freezing process.
- 1.
A male rat was euthanised via cervical dislocation, and then its two cauda epididymides were removed aseptically and transferred into a 300 μL drop of CPA in a 35 mm culture dish at room temperature (Fig. 4A).
- 2.
Ten to twelve deep cuts were made in the epididymides using scissors under a microscope (Fig. 4B).
- 3.
The dish was placed on a tin plate, which was set on top of crushed ice, and the tin was incubated there for ten minutes. (Fig. 4C).
- 4.
During equilibration of the sperm in the CPA, a connector with a 1 mL syringe was attached to the end of a straw (Fig. 4D) and placed on the aforementioned tin plate. Fifteen straws can be prepared from the two epididymides of a single male.
- 5.
A total of 30 μL mHTF (KYUDO Co. Ltd, Saga, Japan) at 0 °C was aspirated carefully into the straw along with 10 mm air. Then, 150 μL sperm suspension was aspirated, and the plunger of the syringe was drawn up until the 30 μL mHTF reached the cotton plug in the straw. The tip of the straw was then sealed using an impulse sealer (Fig. 4E).
- 6.
Fifteen sealed straws were placed on a tin plate, which was set on top of crushed ice and incubated for thirty minutes (Fig. 4F).
- 7.
The straws were transferred to a float (Styrofoam frame). The Styrofoam frame holding the straws was floated promptly on liquid nitrogen inside a Styrofoam box and was kept there for ten minutes (Fig. 4G).
- 8.
After ten minutes, the straws were immersed in liquid nitrogen. The straws were then transferred to a triangular cassette, which was stored in the liquid nitrogen tank for 4–8 weeks (Fig. 4H).
Sperm thawing
In each experiment, 3–4 of the fifteen cryopreserved straws were used for sperm thawing.
- 1.
One millilitre of mHTF was placed in a 1.5 mL tube, and the medium was equilibrated in an incubator (37 °C, 5% CO2) for 30 minutes before use.
- 2.
A frozen straw was removed from the liquid nitrogen tank and placed in the floating container, after which it was preincubated in a 37 °C water bath for 15 minutes to thaw the cell (Fig. 4I).
- 3.
The straw was removed from the water bath and dried using a paper towel.
- 4.
The straw was cut in the area between the sperm suspension and the seal and then placed in a tube containing mHTF. Then, after cutting off the cotton plug at its end, the straw was inserted into the straw connector, and only the sperm suspension was transferred into the bottom of the tube by pushing down the plunger of the 1 mL syringe. The tube was then laid on its side and kept in a humidified incubator (37 °C, 5% CO2) for 30 minutes (Fig. 4J).
- 5.
After 30 minutes, the tube was inverted slowly 2–3 times and then centrifuged at 300 g for 60 seconds (Fig. 4K). Then, 50 μL sediment containing sperm pellets was sucked from the bottom of the tube using a 200 μL pipette with a large opening (Cat. No. 4290–00 S, Funakoshi Co., Ltd. Tokyo, Japan). The sediment was transferred to a 200 μL drop of mHTF covered with paraffin liquid in a culture dish (Fig. 4L).
- 6.
After 30 minutes, 125 μL medium containing dead sperm was removed from the mHTF drop (Fig. 4M), and the sperm suspension was preincubated for two hours prior to insemination.
In vitro fertilisation and development of fertilised oocytes
In each experiment, in vitro fertilisation was carried out using fresh sperm and cryopreserved sperm taken from four male rats. Before the in vitro fertilisation, sperm motility was analysed by using a computer-assisted sperm analyser (IVOS Sperm Analyzer, Hamilton-Thorne Research Co. Ltd., USA). Sperm were incubated in mHTF for 120 minutes at 37 °C under 5% CO2 in air and analysed using the IVOS system. We analysed the following sperm motility parameters: percentage of motile sperm (motile sperm that moved more than 5 µm/s), percentage of motile sperm with progressive motility (motile sperm with progressive motility were denoted by a path velocity >50 µm/s and a straightness ratio >50%) and a marker of hyperactivation [lateral amplitude of head (ALH): this is the average value of the maximum swing width of the sperm head]. In addition, path velocity (VAP), progressive velocity (VSL), beat frequency (BCF) and straightness (STR) were measured. Approximately 500 sperm were analysed in each experiment. In the case of cryopreserved sperm, sperm suspensions with greater than 30% total motility and 10% progressive motility measured via computer-assisted sperm analysis (Integrated Visual Optical System, Hamilton Thorn Inc., Beverly, MA, United States) prior to insemination were used for in vitro fertilisation (Supplementary Movie 1).
In vitro fertilisation was performed following procedures described previously19,20. Immature females were induced to ovulate via injections of 30 IU equine chorionic gonadotropin (ASKA Pharmaceutical Co. Ltd, Japan) and 30 IU human chorionic gonadotropin (hCG, ASKA Pharmaceutical Co. Ltd, Japan), which were administered 54–56 hours apart. Between 15–16 hours after the hCG injection, females were sacrificed via cervical dislocation, their oviducts were promptly collected, and all intact cumulus oocyte complexes were released from the collected oviducts into the preincubated sperm suspension (sperm concentration: 500–1200 cells/μL) for insemination. As a control, superovulated oocytes obtained using the same method were introduced to a fresh sperm suspension and were incubated for 2 hours (sperm concentration: 500 cells/μL).
Twenty hours after insemination, the oocytes were observed under an inverted microscope, and fertilisation rates were calculated as the total number of fertilised oocytes (oocytes with either two pronuclei and a sperm tail or with a sperm tail in the cytoplasm were judged as fertilised oocytes) were divided by the total number of inseminated oocytes and multiplied by 100.
In each experiment, 20 fertilised oocytes were transferred into the oviducts of ICRL:CD (SD) females (ten embryos/oviduct) on the day on which a vaginal plug was identified (day one of pseudopregnancy); embryos were transferred through the wall of the oviduct21. After 22–23 days, the number of pups was recorded. The remainder of the fertilised oocytes were cultured for an additional eight hours, and 100 of the fertilised oocytes that had developed to the 2-cell stage were cultured to the blastocyst stage in mR1ECM22. GFP signals in developed blastocysts were observed under a fluorescence microscope (BioRevo BZ-9000, Keyence Co. Ltd., Japan).
Statistical analysis
Statistical analysis was performed using Prism version 5.0 (GraphPad). The results are expressed as the mean ± standard deviation. Group results were compared using Student’s t-test after arcsine transformation of the percentages; P < 0.05 was considered statistically significant.
Change history
25 February 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Meek, S., Mashimo, T. & Burdon, T. From engineering to editing the rat genome. Mamm. Genome 28, 302–314 (2017).
Nakatsukasa, E., Inomata, T., Ikeda, T., Shino, M. & Kashiwazaki, N. Generation of live rat offspring by intrauterine insemination with epididymal spermatozoa cryopreserved at −196 °C. Reproduction 122, 463–467 (2001).
Nakatsukasa, E. et al. Cryopreservation of spermatozoa from closed colonies, and inbred, spontaneous mutant, and transgenic strains of rats. Comp. Med. 53, 639–641 (2003).
Yamashiro, H. et al. Freezability of rat epididymal sperm induced by raffinose in modified Krebs-Ringer bicarbonate (mKRB) based extender solution. Cryobiology 55, 285–294 (2007).
Seita, Y., Sugio, S., Ito, J. & Kashiwazaki, N. Generation of live rats produced by in vitro fertilization using cryopreserved spermatozoa. Biol. Reprod. 80, 503–510 (2009).
Hagiwara, M. et al. Cellular biophysics during freezing of rat and mouse sperm predicts post- thaw motility. Biol. Reprod. 81, 700–706 (2009).
Yamashiro, H. et al. Lactate and adenosine triphosphate in the extender enhance the cryosurvival of rat epididymal sperm. J. Am. Assoc. Lab. Anim. Sci. 49, 160–166 (2010a).
Yamashiro, H. et al. Extracellular ATP and dibutyryl cAMP enhance the freezability of rat epididymal sperm. J. Am. Assoc. Lab. Anim. Sci. 49, 167–172 (2010b).
Kim, S., Agca, C. & Agca, Y. Changes in rat spermatozoa function after cooling, cryopreservation and centrifugation processes. Cryobiology. 65, 215–223 (2012).
Nakata, M. et al. Successful production of offspring using cryopreserved sperm via nonsurgical artificial insemination in rats. J. Reprod. Dev. 58, 501–504 (2012).
Seita, Y. et al. Full-term development of rats from oocytes fertilized in vitro using cryopreserved ejaculated sperm. Cryobiology 63, 7–11 (2011).
Kim, S., Hooper, S., Agca, C. & Agca, Y. Post-thaw ATP supplementation enhances cryoprotective effect of iodixanol in rat spermatozoa. Reprod. Biol. Endocrinol. 14, 5 (2016).
Fujiwara, K., Kamoshita, M., Kato, T., Ito, J. & Kashiwazaki, N. Generation of rats from vitrified oocytes with surrounding cumulus cells via in vitro fertilization with cryopreserved sperm. Anim. Sci. J. 88, 180–184 (2017).
Toyoda, Y. & Chang, M. C. Fertilization of rat eggs in vitro by epididymal spermatozoa and the development of eggs following transfer. J. Reprod. Fertil. 36, 9–22 (1974).
Chulavatnatol, M. Motility initiation of quiescent spermatozoa from rat caudal epididymis: effects of pH, viscosity, osmolality and inhibitors. Int. J. Androl. 5, 425–436 (1982).
Si, W., Benson, J. D., Men, H. & Critser, J. K. Osmotic tolerance limits and effects of cryoprotectants on the motility, plasma membrane integrity and acrosomal integrity of rat sperm. Cryobiology 53, 336–348 (2006).
Nakagata, N. & Takeshima, T. High fertilizing ability of mouse spermatozoa diluted slowly after cryopreservation. Theriogenology 37, 1283–1291 (1992).
Honda, A. et al. Efficient derivation of knock-out and knock-in rats using embryos obtained by in vitro fertilization. Sci. Rep. 9, 11571 (2019).
Nakagata, N. Cryopreservation of unfertilized rat oocytes by ultrarapid freezing. Jikken Dobutsu. 41, 443–447 (1992).
Anzai, M. et al. Cryopreservation of in vitro fertilized embryos from transgenic rat by ultrarapid freezing. Jikken Dobutsu. 43, 247–50 (1994).
Nakagata, N. Embryo transfer through the wall of the fallopian tube in mice. Jikken Dobutsu 41, 387–388 (1992).
Miyoshi, K., Abeydeera, L. R., Okuda, K. & Niwa, K. Effects of osmolarity and amino acids in a chemically defined medium on development of rat one-cell embryos. J. Reprod. Fertil. 103, 27–32 (1995).
Acknowledgements
We thank Dr. Kyoko Uchiyama (LIVESTOCK IMPROVEMENT ASSOCIATION OF JAPAN, INC.), Dr. Yamashiro Hideaki (Graduate School of Science and Technology, Niigata University), and Dr. Akiko Takizawa (Human & Molecular Genetic Center, Medical College of Wisconsin) for invaluable advice and discussions. We are grateful to our staff: Tomoko Kondo, Yukie Haruguchi, Kiyoko Yamashita, Eri Ishida, Maki Sakaguchi, Yuka Deshimaru and Shuuji Tsuchiyama for their technical support and helpful discussions. This work was supported by grants for Research on Development of New Drugs [Project ID: 16769865] from the Japan Agency for Medical Research and Development (AMED), the Collaborative Research Project (2019-15) of Brain Research Institute, Niigata University, and Collaborative Research Project between Kumamoto University and Kyudo Co. Ltd.
Author information
Authors and Affiliations
Contributions
N.N. designed the work, wrote the initial draft of the manuscript and made figures and movie. N.N., N.M., S.N. and E.N. contributed to the acquisition of the data. N.N. and T.T. contributed to the analysis, interpretation of the data, and revision of the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nakagata, N., Mikoda, N., Nakao, S. et al. Establishment of sperm cryopreservation and in vitro fertilisation protocols for rats. Sci Rep 10, 93 (2020). https://doi.org/10.1038/s41598-019-57090-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-57090-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
