
Abstract
Triple negative breast cancer (TNBC) is a devastating cancer disease characterized by its poor prognosis, distinct metastatic patterns, and aggressive biological behavior. Research indicates that the prevalence and presentation of TNBC varies among races, with Asian TNBC patients more commonly presenting with large invasive tumors, high node positivity, and high histologic grade. In this work, we applied ultra-high performance liquid chromatography-high resolution mass spectrometry (UHPLC-HRMS)-based metabolomics to discover metabolic signatures in Asian female TNBC patients. Serum samples from 31 TNBC patients and 31 healthy controls (CN) were involved in this study. A total of 2860 metabolic features were detected in the serum samples. Among them, 77 metabolites, whose levels were significantly different between TNBC with CN, were confirmed. Using multivariate statistical analysis, literature mining, metabolic network and pathway analysis, we performed an in-depth study of the metabolic alterations in the Asian TNBC population. In addition, we discovered a panel of metabolic signatures that are highly correlated with the 5-year survival rate of the TNBC patients. This metabolomic study provides a better understanding of the metabolic details of TNBC in the Asian population.
Similar content being viewed by others


Introduction
Among women, breast cancer (BC) is the most common type of cancer and also the 2nd leading cause of death worldwide1. BC can be divided into several major subtypes based on conventional immunohistochemistry detection of hormone receptors, including human estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth receptor-2 (HER2). As a highly heterogeneous disease, patients with BC have various morphological spectrum, clinical presentation, and prognostic outcomes2. Among the various subtypes, triple negative breast cancer (TNBC) uniquely lacks expression of all three hormone receptors and accounts for 15–20% of the BC cases. TNBC has attracted more attention compared with other BC subtypes as it is typically associated with high aggression, poor prognosis and a high risk of disease relapse within 5 years following diagnosis3. Women with TNBC have a high frequency of metastasis to the lung, liver and brain, and survival is generally poor. Another troubling feature associated with the disease is the disparity of presentation and survival compared with other ethnicities4,5,6,7,8,9. It is thus of great demand to study the molecular basis of TNBC in order to guide the development of promising drugs and therapies for treatment.
Metabolomics is an emerging technology for health science research, representing a more recent addition to the suite of “omics” tools. In particular, mass spectrometry (MS)-based metabolomic analysis enables the most comprehensive measurement of metabolites in a given biological system. It is thus a powerful analytical tool to identify metabolic biomarkers associated with disease or abnormal phenotypes for clinical applications4. Since metabolites are the end products of gene regulatory processes and protein activities, metabolomics has also been widely used to understand metabolic mechanisms underlying disease phenotypes in order to guide the development of better therapeutic strategies.
The prevalence of TNBC varies among different races and ethnic groups. For instance, a previous study performed in California, USA showed that Asian women have a lower lifetime risk of TNBC than white, African-American, and Hispanic counterparts5. Other studies indicate that TNBC among Asians shows trends of earlier age of onset and more aggressive biological behavior6,7,8. The metabolic signatures in TNBC are of critical biological importance for both mechanistic research and clinical application. However, most previous studies have been conducted in Western populations, and few in Asian populations. We believe that a comprehensive metabolomics study of TNBC among Asians would facilitate the discovery of new treatment-dependent metabolites and increase understanding of responses to treatment that occur in TNBC.
In this study, we collected serum samples from 31 TNBC patients and 31 healthy women in southwest China. We applied a UHPLC-HRMS platform for global metabolomic profiling, followed by univariate and multivariate statistical analyses to identify statistically significant metabolites in TNBC vs. CN. Our study discovered a total of 77 significantly altered metabolites, covering a wide range of metabolic classes, including lipid, amino acids, and carboxylic acids. Comparing to the reported BC metabolomics studies, we identified some consistent metabolic changes as well as some unique metabolic changes in Asian female TNBC patients. Finally, from archived prognostic data for the TNBC patients, we identified 6 metabolites that can stratify patients’ 5-year survival rate. This work presents the first metabolomics study of TNBC in Asian population, thus serving for a better mechanistic understanding of disease progression and prognosis.
Results
Clinical characteristics of subjects
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Chongqing Cancer Hospital. All experiments were performed in strict compliance with the requirements of the Human Ethics Procedures and Guidelines of the People’s Republic of China. Serum samples of TNBC patients were collected from hospitalized female patients with histopathologically confirmed TNBC at Chongqing Cancer Hospital (China). These patients were enrolled in this study from October 2013 to February 2015. Serum samples of the CN group were collected from the age-matched healthy participant volunteers. All serum samples were collected before any medication and surgery towards TNBC. The status of ER, PR and HER2 were negative and the cancers ranged from stages I to III. The demographic and clinical characteristics of study participants are shown in Table 1. Detailed clinical parameter for the TNBC patients are presented in Supplementary Table S1. Informed consent and ethical committee approval was obtained from every participant.
Metabolomics workflow
Figure 1 shows the schematic workflow of the global metabolomics study. Serum samples were collected from 31 TNBC patients and 31 CN volunteers. Metabolites were extracted from the serum samples and analyzed using the UHPLC-HRMS platform. Both electrospray ionization positive (ESI+) and negative (ESI−) mode-based MS analyses were performed to achieve comprehensive metabolome profiling of the serum samples. After LC-MS analysis, metabolic features were extracted from each individual sample and aligned to create a metabolite-intensity table for downstream data interpretation. In total, 2860 metabolic features were consistently detected in all serum samples, including 1856 features in ESI+ mode and 1004 features in ESI− mode, respectively. The data interpretation was conducted by a suite of bioinformatic tools. Firstly, all metabolic features and their relative MS abundance were analyzed in principal component analysis (PCA) and orthogonal partial least square-discrimination analysis (OPLS-DA) to gain a global view of the metabolic differences between TNBC and CN (Fig. 2). Further, statistically significant metabolic features were extracted using the criteria of fold change ≥ 1.2 or ≤0.83 and p-value ≤ 0.05 and visualized using volcano plot and heatmap (Fig. 3). Metabolite identification was performed by comparing retention time, accurate mass, and tandem MS spectra of the metabolic features against in-house metabolite standard library as well as HMDB9 and METLIN10. Seventy-seven statistically significant metabolites were confirmed (Table 2). These metabolites were then used to construct correlation-based metabolic networking analysis and pathway enrichment analysis to better understand their biological significance. Finally, since TNBC has poor prognostic outcome, we attempted to utilize the metabolomic data to identify metabolites that are potentially correlated with the 5-year survival rate.

Workflow of UHPLC-HRMS-based metabolomics for metabolomic profiling and data interpretation of serum samples from TNBC and CN.

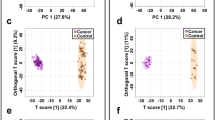
Multivariate statistical analysis results. (a) PCA score plot of the analysis in ESI (+) mode. (PC1 covers 11.6% of the variables; PC2 covers 6.4% of the variables). (b) PCA score plot of the analysis in ESI (−) mode (PC1 covers 8.9% of the variables; PC2 covers 6.1% of the variables). (c) OPLS-DA score plot of the analysis in ESI (+) mode (R2X = 0.209, R2Y = 0.99, Q2 = 0.883). (d) OPLS-DA score plot of the analysis in ESI (−) mode (R2X = 0.183, R2Y = 0.984, Q2 = 0.833).

(a) Representative Volcano plot (fold change threshold = 1.5 and p-value (FDR adjusted) in ESI (+) mode metabolomics data: 0.05; (b) Representative heatmap of top 100 significant metabolites in ESI (+) mode metabolomics data.
Global metabolic profiles of TNBC and CN serum samples
To gain an overview of the metabolic differences between TNBC and CN, we first performed principle component analysis (PCA) of all 62 serum samples. Quality control (QC) data was also included in the PCA analysis to monitor the overall instrument robustness and stability. As shown in Fig. 2a,b, the PCA score plots clearly separate the TNBC and CN groups. In addition, the QC samples were clustered together, indicating the excellent analytical reliability of the applied metabolomics platform.
We next applied OPLS-DA, a supervised multivariate statistical analysis tool, to identify the metabolic features that contribute to the metabolic differences between TNBC and CN serum samples. The OPLS-DA score plots in Fig. 2c,d respectively show very clear separations between TNBC and CN groups in both ESI+ and ESI− modes. Metabolic features with VIP scores ≥ 1.5 in the OPLS-DA analyses were extracted and retained as significant metabolic features for downstream analysis. The quality of these OPLS-DA models were evaluated via an internal cross validation, which calculates the goodness of fit parameter (R2Y) and predictive ability of the model (Q2). In both ESI+ and ESI− analyses, we observed high R2Y and Q2 values and the differences between R2Y and Q2 are smaller than 0.2, which indicates no model overfitting. Together, these results show that there are significant and valid global metabolic differences between the serum metabolome of TNBC and CN.
Discovery of metabolic signatures for TNBC
To identify and confirm high-confidence metabolic signatures that contribute to TNBC, we first calculated fold changes and p-values for all metabolic features in TNBC vs. CN. In total, 805 and 257 significant metabolic features (FD ≥1.2 or ≤0.83 and p-value ≤ 0.05) were found in ESI+ and ESI− analysis modes, respectively. We further refined the list of significant metabolic features using OPLS-DA VIP scores and only kept those with scores ≥1.5. The refined significant metabolic features were then searched against our in-house metabolite library to confirm the identity of 77 metabolites. By applying the chemical taxonomy in HMDB, we were able to categorize significant metabolites into 14 metabolite classes and 25 subclasses (Table 2). Of these 77 altered metabolites, 38 metabolites were upregulated in the TNBC samples and the other 39 metabolites were upregulated in the CN samples. A large portion of the dysregulated metabolites (45 out of the total 77) were in the class of glycerophospholipids. Notably, all altered amino acids were upregulated in the TNBC samples. Although directions of alteration for the significantly changed lipids are not consistent, we can observe some consistency within the subclasses of lipids. For example, all cardiolipin (CL) species are downregulated in TNBC samples. We also conducted literature mining of all 77 dysregulated metabolites to compare against other metabolomics studies of BC or TNBC. While most of the significant metabolites have been previously reported in literature, a few of them (e.g., N-acetyl-L-histidine, octanoylcarnitine) were uniquely discovered for the first time in this Asian female TNBC population. The table of detailed meta-analysis results is available in Supplementary Table S2.
Metabolic network and pathway enrichment analysis
To further understand the underlying biological meanings of these dysregulated metabolites, we performed correlation-based metabolic network analysis. Correlation-based network analysis was used to find the abundance correlation of two metabolites to infer potential biological interpretation. It provides convenient visualization of the potential biological relationships and underlying activity correlations of metabolites. The analysis results were visualized using the MetScape11 plugin available in Cytoscape (3.7.1)12. Figure 4 shows the metabolic network analysis results in circle layout, which locates all nodes in the network around a circle. In the circle network, each node represents the significantly altered metabolites and the edge between two nodes represents the correlation coefficient, with positive correlation in red and negative correlation in green. The darker color indicates the higher correlation coefficient. An edge is displayed only if the correlation between the two metabolites is ≥0.2 or ≤−0.2. The color labels of the metabolic nodes are based on their taxonomy classes identified from HMDB. From the molecular network results (Fig. 4) we can see that the dysregulated metabolites have strong correlations with each other, suggesting the important underlying biological meanings of the metabolites during the TNBC progression. Among these metabolic correlations, we identified 68 pairs of metabolic correlation with coefficient ≥0.4 or ≤−0.4 (Supplementary Table S3).

Correlation-based metabolic network analysis. Each node represents one metabolite and the edge connecting two nodes represents the correlation coefficient, with positive correlation in red and negative correlation in green.
To further understand the metabolic changes on the pathway level, we performed pathway enrichment analysis in MetaboAnalyst (https://www.metaboanalyst.ca). A total of 16 metabolic pathways were predicted with pathway significance p-value ≤ 0.05 Fig. 5, Supplementary Table S4) with the top three significantly altered metabolic pathways being glycerophospholipid metabolism, aminoacyl-tRNA biosynthesis, and valine, leucine and isoleucine biosynthesis.

Metabolic pathway analysis. Predicted metabolic pathways with p-value ≤ 0.05 are listed.
Metabolic signatures correlated with 5-year survival rate in TNBC patients
TNBC is known to have poor prognostic outcomes; therefore metabolites correlated with prognostic outcomes at an early stage could guide further therapeutic intervention. In this study, we focused on the 5-year survival rate, which is an important statistic that reflects cancer progress and treatment success. Out of the 31 TNBC cases, 7 patients were deceased within 5 years of treatment and the other 24 patients survived 5 years following treatment. After log transformation and auto-scaling of the dataset, we performed Receiver Operating Characteristic (ROC) analysis of all significant metabolic features using IBM SPSS statistics. We successfully discovered 6 metabolites with Area Under Curve (AUC) values larger than 0.75 (Fig. 6). These metabolites are dUMP, L-octanoylcarnitine, L-proline, lysoPC (22:1), PS (22:0/0:0), and uric acid (Table 3).

ROC curves for dUMP, L-octaoylcarnitine, L-proline, LysoPC (20:1), PS (22:0/0:0), and uric acid.
Discussion
The overall goal of this study was to delineate the unique serum metabolic signature for TNBC in the Asian female population. Upon applying a state-of-the-art metabolomics platform on serum samples from 31 well-classified TNBC patients and 31 CN volunteers, we were able to detect a large number of significantly changed metabolites that are associated with TNBC and further confirmed 78 of them. Through comprehensive data interpretation, we acquired a better understanding of the metabolic features in Asian female TNBC patients. Finally, further statistical analysis suggested that six of the metabolites were well-correlated with the 5-year TNBC survival rate. To our present knowledge, this is the first metabolomics study of TNBC in the Asian female population.
One unique feature of this study is that all of the serum samples were collected after disease diagnosis and before drug treatment. Therefore, identified metabolic changes were the direct reflection of altered cancer metabolism rather than metabolic perturbations caused by therapeutic intervention. In our study, many of these metabolic changes are consistent with previous BC or TNBC studies of blood, tissue samples of human population or cell culture-based studies. For instance, the dysregulation of glycerophospholipids, including phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylglycerol (PG), lysophosphatidylcholine (lysoPC), lysophosphatidylethanolamine (lysoPE), monoradylglycerolipid (MG), phosphatidic acid (PA), cardiolipin (CL) have been reported in many other types BC or TNBC studies13,14,15. We noted several metabolites as novel in our study, including N-acetyl-L-histidine, phosphatidylinositol (PIP), phosphatidylinositol (PI), and dehydroascorbic acid. These metabolic signatures could be sourced from the unique genetic, lifestyle, and other environmental factors, leading to the unique TNBC phenotype in Asian females, such as being diagnosed at a younger age of 40–50 and more aggressive disease behavior16. Therefore, these unique metabolic signatures could potentially be used to further investigate the intriguing mechanism of race-specific TNBC phenotypes and disease outcome.
Further metabolic network analysis (Fig. 4) suggested that the alterations of various metabolic concentrations in the serum of TNBC patients were not independent of each other. Instead, correlation-based metabolic network analysis suggested inherited correlations among the dysregulated metabolites. The most significant correlations between these metabolites are indicative of particular phenotypes or biological aberrations. While the dysregulated metabolic metabolites and metabolic pathways have been well investigated in disease phenotypes, the association between metabolites in disease conditions is not well studied. In this work, we provided a comprehensive correlation-based metabolic network for all dysregulated metabolites (Fig. 4, Supplementary Table S3). It thus provides rich information for further investigation to understand the underlying metabolic mechanisms for TNBC progression.
Beyond the alteration of individual metabolites, further pathway enrichment analysis systematically predicted the alteration of metabolic pathways (Fig. 5, Supplementary Table S4). The systems level alteration of metabolites in glycerophospholipid metabolism pathway is consistent with metabolomics studies of BC in other populations14,17. The altered lipid concentrations are important to cell membrane modeling and inflammation and have been reported to be associated with advanced metastatic BC in cell lines18, serum19, plasma20, and tissue21,22.
The consistent upregulation of amino acids in this study suggested that the pathway of aminoacyl-tRNA biosynthesis was disrupted. Aminoacyl-tRNA biosynthesis, an essential process for protein synthesis and cell viability23, conjugates amino acids to tRNA and delivers amino acids for incorporation into polypeptide chains. Cancer proliferation requires large amounts of biomass to sustain tumor growth, and the elevated concentration of amino acids in the blood is likely due to the increased demand for protein synthesis in TNBC patients.
Another unique feature of this study is that all participating patients were recruited in 2013–2015 and consistent follow-ups on prognosis outcomes were maintained. For instance, 5-year survival and cancer metastasis status are available for all patients. Determination of prognosis is the most immediate challenge in patient management and is critically important for the design of the most appropriate cancer therapy to improve survival. The development of traditional prognostic factors (e.g., lymph node metastasis, tumor size, and tumor grade) and molecular prognostic biomarkers (e.g., uPA/PAl1, Oncotype DX, and MammaPrint) has demonstrated great success for BC prognosis24. Apart from the aforementioned tissue biopsy-based analysis, the use of “liquid biopsy” from a blood sample has become an appealing expectation as it provides a non-invasive and easily accessible tool for patient stratification. From that perspective, mi-RNA, circulating tumor cells, and circulating tumor DNA-based assays have been developed and have shown promising predictive power. Since metabolites are the downstream product of gene and protein activity, metabolites can potentially be used as prognostic biomarkers.
Using this prognostic information, six metabolites (Table 3), including dUMP, L-octanoylcarnitine, L-proline, lysoPC (22:1), PS (22:0/0:0), and uric acid, were discovered as highly associated with 5-year survival rate. dUMP is the precursor of dTMP, which is necessary for DNA synthesis and repair. The conversion of dUMP to dTMP is catalyzed by thymidylate synthase. A previous study in colon cancer demonstrated that high level of thymidylate synthase is associated with lymph node metastasis and more advanced stages25. Similarly, L-octanoylcarnitine, an important metabolite in carnitine metabolism, has been shown to be a prognostic marker to differentiate between prostate cancer and benign prostatic hyperplasia26. L-proline is a non-essential amino acid in humans. Proline can feed the TCA cycle through the urea cycle and is oxidized by proline dehydrogenase to form reactive oxygen species (ROS). The potential application of proline as the biomarkers for BC diagnosis and prognosis has been previously suggested, though the mechanisms leading to its perturbance are unclear27. LysoPC is an abundant extracellular lipid that stimulates cell proliferation. Previous studies have shown that lysoPC is not only significantly altered in BC28 but can also stimulate cancer cell migration and early tumor recurrence23,29. PS is an essential component of human cells and presents mainly on the inner leaflet of the cell membrane; however, the oxidative stress in BC cells can cause the exposure of PS. We believe that the higher concentration of PS (22:0/0:0) observed in the study is correlated with higher metastasis and death rate of TNBC patients. Finally, serum uric acid has long been known as a significant risk factor for excessive cancer risk, recurrence, and mortality30
. It is therefore expected that the elevated concentration of uric acid (or called hyperuricemia) in TNBC patients could be a good indicator of poor 5-year survival rate.
Among the limitations of the present study is the relatively small to medium sample size. This is attributed to the relatively low percentage of TNBC in the overall incidence of BC. Future work is needed in a more targeted approach to validate the discovery using a larger cohorts, ultimately including samples from population-wide case-control studies.
In summary, we presented a global metabolomics study of TNBC patients in the Asian population. We observed significantly altered metabolites in TNBC serum samples and developed a metabolite-based prognostic biomarker panels for the prediction of 5-year survival rate of TNBC. The application of information-rich analytical methods provides insights into understanding metabolic signatures that are associated with TNBC. We expect that our study will facilitate the development of better treatment strategies to combat TNBC within the Asian population. It may also open up new possibilities for the development of personalized medicine for TNBC patients.
Methods
Serum sample collection
Before treatment of BC, 3–5 ml blood samples were drawn from 31 TNBC patients and 31 healthy controls, respectively. The clinical characteristics of subjects were summarized in Table 1 and detailed clinical parameters were presented in Supplementary Table 1. Blood samples were incubated at room temperature for 30 min to allow the blood to clot. To purify serum samples, the clotted blood was centrifuged for 5 min at 3000 r/min. The upper serum layer was extracted and stored in −80 °C until needed.
Metabolite extraction
Prior to LC-MS analysis, 400 μL methanol was added to 100 μL serum sample in a 1.5 ml Eppendorf tube and vortex-mixed for 30 s to precipitate serum proteins and extract serum metabolites. The sample mixture was centrifuged at 12000 rpm for 15 min at 4 °C. 200 μL of the supernatant was transferred to LC vials for LC-MS analysis. Samples were kept at 4 °C throughout the analysis31.
Metabolomic profiling by UHPLC-QTOF MS
LC-MS analysis was carried out using an Agilent 1290 Infinity ultrahigh performance liquid chromatography system coupled to an Agilent 6530 UHD and Accurate-Mass QTOF MS. An Agilent Zorbax C18 column (100 mm × 2.1 mm, 1.8 μm particle size) was used for LC separation. Mobile phase A was water in 0.1% fomic acid (FA) and mobile phase B was ACN in 0.1% FA. The flow rate was set as t = 0 min, 5% B; t = 1 min, 5% B; t = 6 min, 20% B; t = 9 min, 50% B; t = 13 min, 95% B; t = 15 min, 95% B. Additional 10 min post gradient run at 5% B was performed to re-equilibrium the column for the next analysis. The LC flow rate was 0.35 mL min−1. The column was maintained at 40 °C. Experimental sample injection order was randomized and each sample was injected 4 μL.
The Agilent QTOF MS was equipped with an electrospray ionization (ESI) source operating in either positive or negative ion mode. The ESI+ with the spray voltage set at 4,000 V, Sampling cone 3,500 V. Nitrogen was used as nebulizer gas, and nebulizer gas was delivered at a flow rate of 50 L h−1 with a source temperature of 100 °C. Extraction cone 4 V. Desolvation gas (nitrogen) was heated to 350 °C and delivered at a flow rate of 600 L h−1.
The negative ion mode with the capillary voltage set at 3 500 V, Sampling cone 5 000 V, Source temperature 100 °C, Desolvation temperature 300 °C, Cone gas flow 50 L/h, Desolvation gas flow 700 L h−1, Extraction cone 4 V, Scan time 0.03 s, Inter scan time) 0.02 s. Masses were acquired from m/z 50 to 1,000 with a scan time of 0.03 s and inter scan delay of 0.02 s over a 15 min analysis time.
The MS was operated in full scan mode. Data dependent LC-tandem MS was performed on the pooled sample to collect fragmentation spectra for metabolite identification. Leucine encephalin (100 ng/ml) was used as the lock mass ([M+H]+, m/z 556.2771 in ESI (+) and [M−H]−, m/z 554.2615 in ESI (−)) for internal mass calibration.
QC sample was prepared by taking 10 µL of each individual sample and pooled them together. QC served as “technical replicates” for the study and was analyzed in between every 8 sample injection.
Data processing, statistical analysis and metabolite identification
Agilent Mass Profiler software was used to extract metabolic features from the LC-MS data and generate a metabolite-intensity table containing the retention time, accurate mass and intensities of all metabolites found in the samples.
Prior to any statistical analysis, data transformation and data scaling were performed on metabolic features using log transformation and auto scaling (mean-centered and divided by the standard deviation of each metabolic features) Multivariate statistical analysis, including principle component analysis (PCA) and orthogonal signal correction partial least squares discrimination analysis (OPLS-DA) was performed on SIMCA-P (version 13.0). Univariate statistical analysis, including volcano plot, fold changes, and t-test statistics were performed using MetaboAnalyst 4.0 (https://www.metaboanalyst.ca/)32.
Metabolite identification was performed by matching experimental tandem MS spectra, retention time, and accurate mass of the metabolic features against in-house standard tandem MS spectra library as well as spectral databases such as METLIN and HMDB.
Correlation-based metabolic network analysis and metabolic pathway analysis
Correlation-based metabolic networking analysis was performed using the MetScape 3 plugin available on Cytoscape 3.7. After log transformation and auto scaling, the confirmed significantly changed metabolites and their MS signal intensities were used to calculate the Pearson’s correlation coefficient and debiased squared partial correlation results using the CorrelationCalculator available on the MetScape website (http://metscape.ncibi.org/calculator.html). The correlation file was then uploaded onto the MetScape plugin on Cytoscape to visualize the correlation network.
Metabolic pathway analysis was performed using the pathway analysis function available on MetaboAnalyst 4.0 (https://www.metaboanalyst.ca/)32. The 77 confirmed significant metabolites were entered as the input list. A hypergeometric test was used to evaluate the pathway significance. Homo sapiens (KEGG) was used as the pathway library for prediction analysis. The hypergeometric test and relative-betweenness centrality were selected for over representation analysis and pathway topology analysis, respectively.
Discovery and evaluation the performance of prognostic biomarkers
Diagnostic performance was evaluated on the IBM SPSS statistics 25 platform. After log transformation and auto scaling, the confirmed significantly changed metabolites and their MS signal intensities were used in SPSS for ROC analysis.
Ethical approval and informed consent
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Chongqing Cancer Hospital. All experiments were performed in strict compliance with the requirements of the Human Ethics Procedures and Guidelines of the People’s Republic of China.
References
Breast Cancer, W. H. O, https://www.who.int/cancer/prevention/diagnosis-screening/breast-cancer/en/.
Di Cosimo, S. & Baselga, J. Management of breast cancer with targeted agents: importance of heterogenicity. Nature reviews Clinical oncology 7, 139 (2010).
Anders, C. K. & Carey, L. A. Biology, Metastatic Patterns, and Treatment of Patients with Triple-Negative Breast Cancer. Clinical Breast Cancer 9, S73–S81 (2009).
Xia, J., Broadhurst, D. I., Wilson, M. & Wishart, D. S. Translational biomarker discovery in clinical metabolomics: an introductory tutorial. Metabolomics 9, 280–299 (2013).
Kurian, A. W., Fish, K., Shema, S. J. & Clarke, C. A. Lifetime risks of specific breast cancer subtypes among women in four racial/ethnic groups. Breast Cancer Research 12, R99 (2010).
Sajid, M. T. et al. Age-related frequency of triple negative breast cancer in women. J. Coll. Physicians. Surg. Pak. 24, 400–403 (2014).
Sharma, M. et al. Triple negative breast cancer in people of North East India: Critical insights gained at a regional cancer centre. Asian Pac J Cancer Prev 15, 4507–4511 (2014).
Li, C.-Y. et al. Clinicopathological and prognostic characteristics of triple-negative breast cancer (TNBC) in Chinese patients: a retrospective study. Asian Pacific Journal of Cancer Prevention 14, 3779–3784 (2013).
Wishart, D. S. et al. HMDB 4.0: the human metabolome database for 2018. Nucleic acids research 46, D608–D617 (2017).
Guijas, C. et al. METLIN: a technology platform for identifying knowns and unknowns. Analytical chemistry 90, 3156–3164 (2018).
Karnovsky, A. et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 28, 373–380 (2011).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504 (2003).
Cui, M., Wang, Q. & Chen, G. Serum metabolomics analysis reveals changes in signaling lipids in breast cancer patients. Biomedical chromatography 30, 42–47 (2016).
Cala, M. P. et al. Multiplatform plasma metabolic and lipid fingerprinting of breast cancer: A pilot control-case study in Colombian Hispanic women. PloS one 13, e0190958 (2018).
Zielinski, C., Stuller, I., Rausch, P. & Müller, C. Increased serum concentrations of cholesterol and triglycerides in the progression of breast cancer. Journal of cancer research and clinical oncology 114, 514–518 (1988).
Wang, C. et al. Triple negative breast cancer in Asia: An insider’s view. Cancer treatment reviews 62, 29–38 (2018).
Kanaan, Y. M. et al. Metabolic profile of triple-negative breast cancer in African-American women reveals potential biomarkers of aggressive disease. Cancer Genomics-Proteomics 11, 279–294 (2014).
Kim, H.-Y. et al. Comparative metabolic and lipidomic profiling of human breast cancer cells with different metastatic potentials. Oncotarget 7, 67111 (2016).
Tenori, L. et al. Exploration of serum metabolomic profiles and outcomes in women with metastatic breast cancer: a pilot study. Molecular oncology 6, 437–444 (2012).
Shen, J., Yan, L., Liu, S., Ambrosone, C. B. & Zhao, H. Plasma metabolomic profiles in breast cancer patients and healthy controls: by race and tumor receptor subtypes. Translational oncology 6, 757 (2013).
Kim, I. C. et al. Lipid profiles for HER2-positive breast cancer. Anticancer research 33, 2467–2472 (2013).
Foulkes, W. D., Smith, I. E. & Reis-Filho, J. S. Triple-negative breast cancer. New England journal of medicine 363, 1938–1948 (2010).
Gaetano, C. G. et al. Inhibition of autotaxin production or activity blocks lysophosphatidylcholine-induced migration of human breast cancer and melanoma cells. Molecular Carcinogenesis: Published in cooperation with the University of Texas MD Anderson Cancer Center 48, 801–809 (2009).
Nicolini, A., Ferrari, P. & Duffy, M. J. Prognostic and predictive biomarkers in breast cancer: Past, present and future. Semin Cancer Biol. 52, 56–73 (2018).
Garcia, J. M., Silva, J. M., Dominguez, G., Silva, J. & Bonilla, F. Heterogeneous tumor clones as an explanation of discordance between plasma DNA and tumor DNA alterations. Genes, Chromosomes and Cancer 31, 300–301 (2001).
Giskeødegård, G. F. et al. Metabolic markers in blood can separate prostate cancer from benign prostatic hyperplasia. British journal of cancer 113, 1712 (2015).
Geck, R. C. & Toker, A. Nonessential amino acid metabolism in breast cancer. Advances in biological regulation 62, 11–17 (2016).
Qiu, Y. et al. Mass spectrometry-based quantitative metabolomics revealed a distinct lipid profile in breast cancer patients. International journal of molecular sciences 14, 8047–8061 (2013).
Abdelzaher, E. & Mostafa, M. F. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) upregulation in breast carcinoma contributes to tumor progression and predicts early tumor recurrence. Tumor Biology 36, 5473–5483 (2015).
Fini, M. A., Elias, A., Johnson, R. J. & Wright, R. M. Contribution of uric acid to cancer risk, recurrence, and mortality. Clinical and translational medicine 1, 16 (2012).
Chen, Y. et al. Metabolomic profiling of human serum in lung cancer patients using liquid chromatography/hybrid quadrupole time-of-flight mass spectrometry and gas chromatography/mass spectrometry. Journal of cancer research and clinical oncology 141, 705–718 (2015).
Chong, J. et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic acids research 46, W486–W494 (2018).
Acknowledgements
This study was financially supported by the Performance Motivation and Guidance Special Project of Chongqing Science & Technology Commission (cstc2017jxj1130006), the Fundamental Research Funds for the Central Universities (2019CDYGYB022), Cancer Foundation of China (LC2016W01) and Chongqing Cancer Hospital 1110 talent project. The authors gratefully acknowledge Shanghai Sensichip Infotech Co., Ltd of China with the help of data processing guideline. Part of the statistical analysis and data visualization were enabled using the software purchased using University of British Columbia Start-up grant held by Tao Huan. We also thank David D Y Chen of University of British Columbia for the technique assistance.
Author information
Authors and Affiliations
Contributions
The author’s responsibilities were as follows: conceptualization, L.L., X.Z., W.N. and T.H.; methodology, L.L., Q.Z. and T.H.; software, L.L. and T.H.; validation, L.L., Q.Z. and T.H.; formal analysis, X.Z.; investigation, L.L., X.Z., W.N. and T.H.; resources, X.L.; data curation, L.L. and T.H.; writing-original draft preparation, L.L.; writing review and editing, N.V., T.H. and W.N.; project administration, L.L.; funding acquisition, L.L., and W.N.;” All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Zheng, X., Zhou, Q. et al. Metabolomics-Based Discovery of Molecular Signatures for Triple Negative Breast Cancer in Asian Female Population. Sci Rep 10, 370 (2020). https://doi.org/10.1038/s41598-019-57068-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-57068-5
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
